

Abstract
One of the most intriguing examples of cross talk between signaling systems is the interrelationship between G protein-coupled receptor and growth factor receptor pathways leading to activation of the ERK/MAP kinase phosphorylation cascade. This review focuses on the mechanism of this cross talk, denoting primarily signaling components known to occur in the G protein-coupled receptor branch of the MAP kinase pathways in neural cells. Recent evidence is presented on the existence of a plethora of pathways, due to the multiplicity of G protein-coupled receptors, their differential interaction with heterotrimeric G protein isoforms, various effectors and second messengers. In light of this rich diversity, the review will discuss different points of convergence of G protein-coupled receptor and growth factor receptor pathways that may feature a requirement for growth factor receptor transactivation, receptor internalization and scaffolds to assemble receptor, adaptor and anchoring proteins into multiprotein complexes.
Keywords: G protein-coupled receptor, Extracellular signal-regulated kinase, MAP kinase, G protein, Calcium/calmodulin, Phosphoinositide, Receptor transactivation, Receptor endocytosis, Scaffold, Growth factor
Introduction
Since their discovery in the early 1980s, the extracellular signal-regulated kinase (ERK) subfamily of MAP kinases has had considerable impact on the signal transduction field [1, 2]. ERK/MAP kinases appear to be ubiquitous in eucaryotes and regulate many fundamental cellular processes including proliferation, differentiation, migration, survival, growth, growth arrest and apoptosis. ERK/MAP kinases not only activate transcriptional factors but they modulate many other proteins as well [3]. Their unique ability to integrate diverse signaling pathways is exemplified in the nervous system. In neurons the MAP kinases play a role in memory, plasticity, long-term potentiation and in glia, survival and differentiation [4–8].
One of the most intriguing examples of cross talk between signaling systems is the interrelationship between growth factor receptor, referred to as receptor tyrosine kinase (RTK), and G protein-coupled receptor (GPCR) pathways to activate the ERK/MAP kinase phosphorylation cascade [9–13, for reviews see 12, 13]. GPCRs are one of the largest superfamilies of proteins found in nature. This review focuses on the mechanism of this cross talk, specifying the key signaling components in the heterologous pathways in neural cells. A plethora of such pathways exists, in part due to the diversity of the GPCRs. GPCRs interact differentially with heterotrimeric G protein isoforms, effectors and second messengers. Here we will emphasize signaling components that occur in the GPCR branch of the MAP kinase pathways as the steps downstream of the RTK are less variable and better understood (fig. 1). An important factor that is clearly responsible for multiple signaling responses of GPCRs is cell type. Correlations of neural cell types with various signaling mechanisms will be discussed. The role of protein scaffolds, to which diverse signaling components of these intracellular pathways are known to dock, insulating them possibly to confer another order of specificity, will also be examined.
Fig. 1.
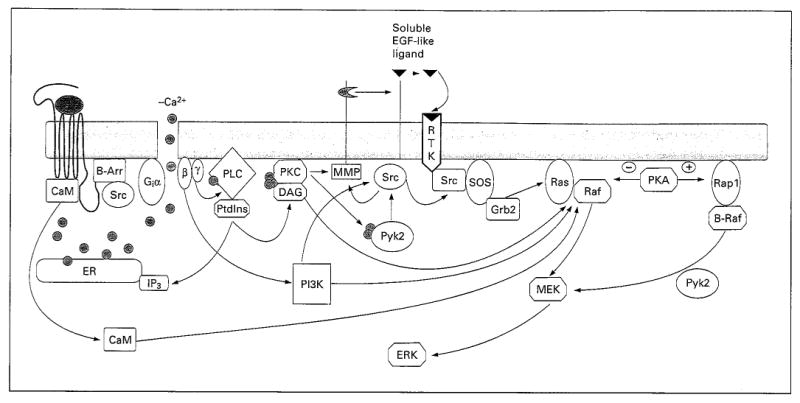
Diversity of GPCR signaling pathways to ERK/MAP kinase. The various signaling components are shaped according to their function as follows: G proteins as vertical ellipses, nonreceptor Tyr kinases as horizontal ellipses, Ser-Thr kinases as larger octagons, lipids as small octagons, adaptor proteins as rounded rectangles, Ca2+ as circles. B-Arr = β-Arrestin; IP3 = inositol triphosphate; MMP = matrix metalloproteases; PtdIns = phosphatidylinositide.
G Proteins
Most mechanisms of GPCR stimulation of ERK phosphorylation entail transduction by heterotrimeric G proteins. Gi-derived βγ subunits or Gαq are the most common subunits interacting with effectors identified to date [9–26, for reviews see 12, 13]. Gi-mediated ERK phosphorylation by GPCR agonists has been primarily implicated by sensitivity to pertussis toxin (PTX) that abolishes Gi-coupled signaling. In most pathways studied, βγ subunits of the Gi family rather than Gαi have been involved in the transduction mechanism [9–11]. However, a constitutively active Gi, gip2, has been reported to activate ERK via different pathways (see below).
Various approaches have been adopted to implicate Gβγ. They include stimulation of GPCR signaling to ERK by recombinant βγ subunits, the failure of constitutively active mutants of α subunits to activate ERK and blockade of heterologous signaling by overexpression of proteins that sequester βγ subunits. Because of the abundance of Gi proteins, their βγ subunits are thought to be a major source of the βγ dimers used for transduction in heterologous pathways [for a review see 19]. The β subunits of heterotrimeric G proteins are 35-kD proteins that contain repetitive Trp-Asp (WD) sequences that occur at about 40-amino acid intervals (WD40 repeat) [19]. Gβ interacts noncovalently with γ subunits of about 8 kD which are isoprenylated, thereby affording a hydrophobic residue to anchor the dimer to cell membranes. There are numerous examples of the interaction of βγ subunits with pleckstrin homology domains of various target proteins.
The isoforms of β and γ subunits are GPCR-specific and they are selective with respect to the isozymes of effectors with which they interact. Classically, Gi-derived βγ subunits couple with PLCβ2 to initiate phosphoinositide hydrolysis and inositol phosphate turnover in brain [14, 15]. In contrast, several GPCR (muscarinic acetylcholine, dopamine, endothelin and serotonin) interact with PLCβ1, PLCβ3 or PLCβ4 via Gαq which is PTX-insensitive. However, PTX insensitivity of GPCR activation of ERK does not automatically denote that the signaling pathway is transduced by α subunits of the Gq family of G proteins. For example, there is evidence to suggest that Gαz and Gα12 transduce chronic μ opioid receptor (MOR) inhibition of EGF-activated ERK in COS-7 cells [16]. Gαz and Gα12 resemble Gαq in that they lack cysteine residues in the C-terminus consensus site that is ADP-ribosylated by PTX. In contrast, acute opioid stimulation Of ERK is mediated by βγ subunits in both COS-7 and rat C6 glioma cells [7, 17, 18].
Go protein, which constitutes 1% of brain membrane protein, is also a transducer in this heterologous pathway. Gαo activates ERK via a protein kinase C (PKC)-dependent mechanism which is Ras-independent, raising the possibility of the existence of an alternative pathway, e.g. via Rap1/B-Raf, or implying convergence of M1 muscarinic acetylcholine receptor and RTK pathways downstream of Ras in transfected CHO cells [21]. Since it was subsequently shown that constitutively active Gαo potentiates RTK and B-Raf-activated ERK phosphorylation in the same cell line, the former Ras-independent route pertains [22]. Gαo can also modulate ERK phosphorylation by interacting directly with the GTPase-activating protein of a small geranylgeranylated G protein, Rap1 [23]. Rap1 is a relative of Ras that can either inhibit or stimulate ERK activation by several different mechanisms including one that entails forming a complex with B-Raf in neuronal cells (see below, fig. 1) [for an overview see 24].
Although Gs has not been shown to be involved as a direct transducer of the GPCR branch of the heterologous pathway to ERK, it may have an indirect effect. Upon Gs-mediated protein kinase A (PKA) phosphorylation of the β-adrenergic receptor, this GPCR switches from Gs to Gi coupling which stimulates the MAP kinase phosphorylation cascade via β-arrestin and the nonreceptor protein tyrosine kinase, Src [25, 26].
Effectors and Second Messengers
Consistent with the multiplicity of GPCRs and G proteins, many effectors and second messengers have also been implicated in heterologous GPCR signaling pathways. Here we will discuss studies where the second messengers or effectors are thought to be acting directly as opposed to indirectly, such as by feedback mechanisms. It is assumed that GPCR activation can elicit similar mechanisms of ERK phosphorylation as the second messenger or effector alone does. However, this does not occur in all cases [20, 27]. One of the first second messengers to be recognized as a modulator of ERK phosphorylation was cAMP. In 1993, five independent groups reported that cAMP inhibited either GPCR or RTK-stimulated ERK phosphorylation, involving diverse cells, RTKs and GPCRs [27–31]. Subsequently, evidence for both cAMP-dependent stimulation and inhibition of ERK phosphorylation was reported [5, 28, 32–34]. For example, in primary neuronal cells or neuronal model systems such as PC12 cells, cAMP induces ERK phosphorylation via the small GTPase, Rap1, which is activated by PKA and interacts with B-Raf [35–37]. In contrast, in astrocytes and astrocytoma cells, which do not possess B-Raf, cAMP inhibits ERK phosphorylation by the classical Ras/Raf-1 pathway. Thus, PKA phosphorylation acts on Rap1 that suppresses the Ras/Raf-1 pathway [36]. Accordingly, if B-Raf is transfected into astrocytoma cells, cAMP stimulates ERK phosphorylation as it does in B-Raf-containing neuronal cells. Such experiments reveal a limitation of overexpression of signaling components in cells that do not possess the endogenous form of the proteins.
ERK can also be activated via PKA and Rap1/B-Raf pathway upon Ca2+ influx into neurons [37, 38]. Yet another cAMP pathway to ERK and ultimately CREB phosphorylation exists in striatal neurons in which PKA causes an increase of intracellular Ca2+ stores [39]. Intracellular Ca2+ then activates Ca2+-dependent proteins, PKC and Pyk2 and the Rap1/B-Raf complex that results in ERK phosphorylation. Since ERK activation induces CREB phosphorylation, this pathway offers an alternative route to the classical pathway involving nuclear import of the catalytic subunit of PKA and its direct phosphorylation of CREB. Several neurotransmitter systems utilize PKA-mediated ERK activation via the Rap1/B-Raf complex and some have been implicated in long-term facilitation [40]. The existence of cross talk between PKA and intracellular Ca2+ that induces ERK phosphorylation via Rap1 provides yet another alternative mechanism to Ca2+-dependent phosphoinositide turnover that is mediated by the Ras/Raf complex in glia (see below and fig. 1). Nevertheless, Rap1 is directly activated by Gαo or Gαi via their specific GAPs and by Gαq via phosphoinositide turnover [23, 39, 41].
Ras and Rap pathways may differentially mediate acute and chronic stimulation of ERK by growth factors. It has been known for some time that epidermal growth factor (EGF) induces an acute activation of ERK that results in PC12 proliferation, whereas nerve growth factor (NGF) elicits a sustained activation of ERK that results in cellular differentiation [for a review see 34]. The Ras- and Rap-dependent pathways to ERK in PC12 cells influence the kinetics of this activation process [34]. Interestingly, it was found that both NGF and EGF transiently stimulate Ras [42]. Moreover, EGF acutely potentiates ERK via Ras, whereas NGF promotes prolonged Rap1 activation to account for 90% of the long-term effect on ERK.
Ca2+-dependent mechanisms of heterologous signaling, in which intracellular levels of this ion are elevated, can be initiated by opening Ca2+ channels in addition to GPCR-mediated phosphoinositide hydrolysis [7, 19, 20, 43–47]. Intracellular Ca2+ mobilization-dependent heterologous signaling is frequently found in both neuronal and glial model systems [43–46]. Inhibition of either Ca2+ channels or inositol phosphate-mediated Ca2+ release from intracellular stores blocks GPCR signaling to ERK suggesting that both are required for full activity. Inositol phosphate turnover, PKC and Pyk2 have often been implicated in such mechanisms, but PKC can also be activated directly by RTKs via, for example, PLCγ or phosphatidylinositol-3-kinase (PI3K) and participate in ERK activation exclusive of GPCR signaling [3, 34].
Pyk2 is a Ca2+-dependent, proline-rich, nonreceptor protein tyrosine kinase that is a member of the focal adhesion kinase family. In neuronal model systems, it is a key signaling component in the GPCR-ERK heterologous pathway [43, 45]. Although its intermediary role in signaling has also been established in many other cell types [46, 47], the role of Pyk2 in astrocytes is less clear. Endothelins and opioids acting via their GPCRs have been shown to stimulate the phosphorylation of Pyk2 and ERK in primary cultures of astrocytes and C6 glioma cells, an astrocytoma cell line [6, 7]. However, in neither case has Pyk2 been implicated in ERK activation unequivocally.
The precise location of Pyk2 in the GPCR signaling branch is unknown. As seen in figure 1, it was originally proposed to bridge the GPCR and RTK pathways [43, 44]. However, Satoh et al. [46] recently reported that statins, which are selective inhibitors of 3-hydroxy-3-methylglutaryl CoA, block both angiotension II-stimulated Pyk2 and ERK phosphorylation in pulmonary vein endothelial cells. They present evidence to support the notion that the Ca2+-dependent activation by angiotensin II requires geranylgeranylation of Rap1 to facilitate plasma membrane anchoring of this small G protein, thereby explaining the statin sensitivity. If this hypothesis is correct, Pyk2 may possibly be an intermediate relegated to the RTK branch of the heterologous pathway downstream of Rap1 in endothelial cells (fig. 1).
The study of the relationship of Pyk2 and Src and their requirement in heterologous signaling using embryonic fibroblasts from Pyk2-, Src- or EGF receptor (EGFR)-deficient mice have produced interesting findings of the redundancies inherent to signaling systems, a consistent theme emerging from transgenic mice studies [48]. Pyk2, Src and EGFR have been implicated in GPCR-mediated ERK activation in a number of cell types including neurons [44; for a review see 13]. A Pyk2-dominant negative mutant that inhibits GPCR-mediated ERK activation is incapable of complexing with Src [44]. Nevertheless, lysophosphatidic acid (LPA)-induced ERK activation was shown to lead to ERK phosphorylation in embryonic fibroblasts from Pyk2-, Src- or EGFR-deficient mice. These results reveal that in such cells, alternative Pyk2-, Src- or EGFR-independent pathways from GPCRs to ERK exist. Early studies with protein tyrosine kinase inhibitors such as genistein and herbimycin A suggested a role for Src in GPCR heterologous signaling [20, 49, 50]. These were confirmed by the cellular expression of a dominant negative kinase-inactive mutant of c-Src that inhibited Gβγ-mediated LPA and α2A-adrenergic activation of ERK [51]. Subsequently, Src was implicated in multiprotein complexes or signaling scaffolds together with β-arrestin and GPCRs as discussed below [52–57, for a review see 55]. It was also shown that Src plays a role in Pyk2 activation and both Src and Pyk2 are required for EGFR phosphorylation in some cells [45, 51]. However, Src has independent roles in EGF signaling as well [50, 55]. The plethora of Src signaling sites is inexplicable and emphasizes the multiplicity of heterologous pathways. The occurrence of mutations in proteins that do not have multiple roles in transformed cell types would augur well for successful development of targeted inhibitors to treat such forms of cancer. If a targeted inhibitor is selective for a signaling component that is responsible for proliferation of the transformed cell but is not required at other sites, it will have a major advantage over most of the presently used chemotherapeutic agents which feature broad toxicities and thereby multiple side effects. Moreover, in cells with the inhibited signaling pathway, redundant but regulated pathways can compensate for the blockade.
PI3K is a phosphoinositide kinase that can mediate GPCR heterologous signaling in certain cells and it is found in virtually all growth factor pathways studied [58–66]. However, PI3K is not always involved in ERK signaling. Initial evidence for its implication in heterologous signaling arose from studies on GPCR agonist-induced ERK phosphorylation examined in cultured Swiss 3T3 cells and human myeloid-derived cells [58, 59]. Subsequently, Hawes et al. [60] used two inhibitors of PI3K activity, wortmannin and LY294002, and a dominant negative mutant of the p85 subunit of PI3K (p85) to attenuate ERK phosphorylation by LPA and the α2-adrenergic receptor agonist UK-14304 in COS-7 and CHO-K1 cells. These results suggest that, in COS-7 cells, a PI3K step occurs in the GPCR branch of the pathway (fig. 1). Nevertheless, signaling downstream of Gβγ-induced PI3Kγ that activates ERK appears to require a PTK, Shc, Grb2, Sos, Ras and Raf [61]. Interestingly, ERK phosphorylation was only sensitive to wortmannin at low levels of EGF in COS-7 cells [63] and at low levels of PDGF in Swiss 3T3 cells [65]. These and other results suggest the existence of a PI3K-independent, redundant pathway to ERK when larger quantities of receptor molecules are activated [65]. Opioid stimulation of ERK is wortmannin-sensitive in COS-7 but not in C6 glioma cells, wherein MOR is overexpressed [18, Belcheva et al., unpubl. observations]. In the C6 studies, cells were grown under conditions, wherein they express an astrocytoma phenotype. In contrast, GH-releasing hormone activates ERK via PI3K in pituitary cells [62], which arise from the same parent cells as astrocytes again illustrating the diversity of heterologous signaling.
A potential mechanism of activation by PI3K entails its production of lipids that interact with pleckstrin homology domains of Sos, facilitating its attachment to the plasma membrane and colocalization with Ras [63]. Additional mechanisms linking PI3K with Ras were discovered recently [64]. Moreover, a correlation between PI3K activity and both RTK and GPCR receptor endocytosis has been found. This mechanism of action of PI3K involves the generation of phosphatidylinositol-3-phosphate that binds to an early endosomal antigen and facilitates its interaction with Rab5. Rab5 is required for endosome fusion that has been implicated in ERK activation as discussed below [34, 67]. Interestingly, PI3K can also promote β-adrenergic receptor internalization by an agonist-induced mechanism [66]. Cytosolic PI3K interacts with β-adrenergic receptor kinase I and translocates to the plasma membrane where it docks on the receptor. Inhibition of PI3K activity attenuates β-adrenergic receptor sequestration.
The ability of calmodulin (CaM) to undergo diverse interactions with proteins involved in signal transduction by GPCRs indicates it may play a role in heterologous signaling [68–74]. There exist CaM-responsive signaling components such as adenylate cyclases [36, 37], G protein β subunits [70], GPCR kinases [71], PI3K [72] and protein tyrosine phosphatase α [73]. Thus, it is clear that Ca2+/CaM strongly influences ERK signaling, but the mechanisms involved are poorly understood. A family of CaM-dependent kinases has been found in neural and other cells. However, there is little evidence that the CaM kinase family plays a role in ERK activation. Although CaM kinase phosphorylates CREB [68], ERK does not appear to be involved in the pathway(s) leading to phosphoCREB in HEK293 cells [69]. Moreover, inhibition of Ca2+/CaM-dependent kinases and phosphodiesterases did not interfere with serotonin 5-HT1A receptor-mediated ERK activation [47] or bradykinin-induced EGFR transactivation [74, also see below] suggesting a different mechanism is entailed.
CaM antagonists (W7, calmidazolium, fiuphenazine) have been used to detect several sites of CaM dependency in Gi/o- and Gq-coupled receptor-mediated Ras-dependent ERK activation in neuronal model cell lines and HEK293 cells [47, 75–77]. Although these drugs may not be highly selective, they suggest that CaM affects GPCR-mediated ERK phosphorylation at: (1) points downstream of RTK, (2) at steps involved in transactivation of RTKs, and (3) within the GPCR branch of the pathway. Ca2+/CaM can modulate Src activity and directly or indirectly affect Ras activity or signaling elements downstream of Ras (fig. 1). Ca2+/CaM activates Ras by binding to guanine nucleotide exchange factors in primary cultures of rat cortical neurons [78, 79]. In addition, CaM inhibitors block EGF stimulation of ERK phosphorylation at an undefined site downstream of Src and Ras but upstream of Raf and MEK in HEK293 cells [75]. CaM antagonists also abolish wild-type Raf kinase activity and CaM-Sepharose precipitates some but not all isoforms of Raf in PC12 cell lysates [80].
The first GPCR shown to interact with CaM was a metabotropic glutamate receptor [81]. Subsequently, evidence for direct binding of CaM to MOR emerged. These findings raised the possibility that CaM may play a role as a second messenger [82] and possibly mediate G protein-independent mechanisms [83]. CaM binding sites are located on the i3 intracellular loop of MOR, δ opioid and D2-dopamine receptors, and on the C-terminal of the metabotropic glutamate receptor [81–84]. Wang et al. [82] proposed that receptor peptide motifs for binding to Gα and CaM are similar to each other, raising the possibility that GPCR-CaM interactions could represent a common phenomenon. There have been studies on GPCR heterologous signaling that compare wild-type MOR and a mutant K273A-MOR that binds CaM poorly but couples normally to G proteins [85]. The data show that opioid agonist stimulation of the K273A-MOR generates 50% less ERK phosphorylation than wild-type MOR. Stimulation of MOR results in ERK phosphorylation by a mechanism that entails EGFR transactivation in HEK293 cells and rat astrocytes [85, Belcheva et al., unpubl. observations]. Wild-type MOR also stimulated EGFR Tyr-phosphorylation 3-fold more than K273A-MOR, indicating that direct CaM-MOR interaction plays a key role in transactivation (fig. 1). This novel pathway of CaM-dependent ERK activation and EGFR transactivation may be shared by other GPCRs shown to interact with CaM.
Sites of Convergence of GPCR and RTK Pathways
In light of the diverse GPCR pathways that signal to ERK, it is not surprising that different points of convergence with RTK signaling components have been detected (fig. 1). Two of the best-documented sites are at the Ras-Raf complex and ‘upstream’ of Ras-Raf at the growth factor receptor. Earlier findings on this subject revealed that cAMP-mediated ERK inhibition via PKA was accompanied by reduction of Ras-Raf complex activation [28, 31]. Simultaneously, it was found that PKC-α can activate Raf-1 by direct phosphorylation in NIH3T3 cells [86], but PKC activation of ERK appeared to be Ras-independent, because it was not blocked by a dominant negative mutant of Ras [87]. Subsequently, Marais et al. [88] demonstrated that Raf-1 mutants that prevent association with Ras cannot be activated by PKC in COS cells. These results indicate that PKC activation of Raf-1 requires a Ras-GTP-Raf-1 complex. Dominant negative mutants of Ras that complex with Raf-1 will not block PKC potentiation of Raf-1.
Recently, it has been discovered that some heterologous GPCR signaling to ERK occurs via Tyr phosphorylation of the RTK itself [45, 51, 55, 76, 85, 89–93]. Transactivation of EGFR rapidly ensues upon stimulation by a broad range of GPCRs including LPA, muscarinic cholinergic, α- and β-adrenergic, angiotensin, thrombin, bradykinin and MOR. It is cell type-specific. For example, LPA stimulation is EGFR transactivation-dependent in Rat-1 cells but not in PC12 cells [50]. In HEK293 cells, LPA and α2-adrenergic receptor-mediated activation is partially dependent on EGFR transactivation.
Transactivation appears to occur via undefined plasma membrane-bound metalloproteases involved in processing of EGF-like precursor molecules anchored on the cell surface [92, 93]. In some pathways Src may activate the metalloproteases [54], in others, PKC and various other related signaling components [55]. A similar EGFR transactivation mechanism involving an autocrine metalloprotease-dependent release of heparin-binding (HB)-EGF resulting from IGF stimulation has also been proposed [94]. These data reveal that cross talk exists between different growth factor receptor signaling pathways (fig. 1).
CaM may inhibit the RTK transactivation process. CaM inhibitors have been shown to promote shedding of membrane-bound growth factors via matrix metalloproteases [95, 96]. Accordingly, CaM antagonists were found to stimulate the cleavage of several membrane proteins including EGFR binding ligands in CHO and human epithelial cells, and this process was PKC-independent. In addition, Bosch et al. [97] reported that the CaM inhibitor, W13, alone induced an increase in Ras, Raf and ERK activation in cultured NIH3T3 or NRK cells. The stimulation of ERK by W13 alone may be explained by the induction of the release of endogenous plasma membrane-bound EGFR binding ligands leading to the activation of this receptor and subsequent activation of Ras-Raf. In accordance with these results, the CaM inhibitor W7 attenuated DAMGO stimulation of EGFR phosphorylation in wild-type MOR cells [85, Belcheva et al., unpubl. observations]. However, in cells expressing mutant K273A MOR that binds to CaM poorly, the major CaM-dependent pathway is not operative and W7 has no effect. Therefore, a minor pathway of ERK activation exists in mutant K273A MOR cells, which appears to be CaM-independent.
Evidence for GPCR transactivation of RTKs other than EGFR has been reported. These include angiotensin-induced PDGFR tyrosine phosphorylation [98] in vascular smooth muscle cells, LPA-stimulated PDGFR tyrosine phosphorylation in L cells by a PKC-independent mechanism [99] and opioid-stimulated FGFR activation in rat C6 glioma cells [100]. In these studies, a blockade of RTK tyrosine phosphorylation results in attenuation of ERK phosphorylation. In contrast, IGFR phosphorylation by thrombin has been reported to stimulate DNA synthesis but not ERK phosphorylation in vascular smooth muscle cells [101]. The mechanism of transactivation has not been elaborated on for PDGFR and FGFR to the extent that it has been for EGFR, although some evidence for a metalloproteinase requirement has been obtained in the FGFR studies in C6 cells [100].
Role of Receptor Endocytosis in GPCR Signaling to ERK
An important relationship exists between receptor signaling and intracellular trafficking [102–115, for reviews see 13, 102, 111]. The occurrence of intracellular receptors has been recognized for some time and initially it was thought that they were either newly synthesized en route to the cell surface or internalized receptors undergoing trafficking to an intracellular site for degradation. Subsequently, receptor endocytosis was shown to be also important for the resensitization of receptors that underwent inactivation on the plasma membrane. For example, GPCRs are inactivated upon being phosphorylated by GPCR kinases, PKA and PKC. As a result, they are thought to internalize generally by docking with β-arrestin in clathrin-coated pits. Upon invagination, clathrin-coated vesicles become acidic endosomes in which the GPCR is dephosphorylated back to an active form by the action of phosphatases. Endosomes then recycle the GPCR back to the plasma membrane unless regulatory factors dictate degradation by lysosomal targeting.
Recently, Vieira et al. [103] reported that activation of ERK1/2 via EGFR is suppressed in cells transfected with dynamin-mutant K44A that is defective in its GTPase activity. Since dynamin has been implicated in the process in which clathrin-coated pits are pinched off of the plasma membrane, the mutant blocked receptor endocytosis by this mechanism. Thus, the data suggested that endocytic trafficking of EGFR appears to be important for full activation of ERK. Heterologous signaling studies with dynamin mutant K44A and other inhibitors of receptor endocytosis revealed that in addition to a reduction in ERK phosphorylation, GPCR internalization was also blocked, suggesting a requirement for intracellular GPCRs and RTKs [8, 51, 52, 104–111, for a review see 111]. Subsequently, it was learned that dynamin plays additional roles in signaling so that dynamin K44A mutant blocks other processes in addition to receptor endocytosis [112]. Thus, the challenge has been to find inhibitors of receptor endocytosis that are selective. As many as five mechanistically distinct inhibitors of clathrin-mediated endocytosis have been used to attenuate ERK activation by GPCR agonists indicating that receptor endocytosis is required for ERK activity. Nevertheless, it soon became apparent that in numerous GPCR heterologous signaling pathways ERK phosphorylation is not affected by inhibitors of receptor endocytosis [8, 112–115; for a review see 55]. Recently, Pierce et al. [110] used both selective inhibitors and confocal microscopy to explain these discrepancies. They showed that endocytosis of RTK, but not GPCR, was required. Moreover, blockade of RTK endocytosis impacted GPCR heterologous signaling only in EGFR transactivation-dependent mechanisms. The question remains whether RTK internalization into clathrin-coated pits or vesicles is required in heterologous signaling and whether an RTK scaffold is assembled at the site [55, 91] (see below).
The same group reported additional support for this model by using cocultures of two types of COS-7 cells [54]. Donor cells were transfected with α2A-adrenergic receptors, whereas acceptor cells lacked this receptor. In donor cells α2A-adrenergic receptor-mediated stimulation of ERK phosphorylation via transactivation-independent and transactivation-dependent pathways was detected. Acceptor cells possessed only an EGFR pathway that responded to HB-EGF shedding by the α2A-adrenergic receptor-activated donor cells to stimulate ERK phosphorylation by a paracrine mechanism. Dynamin K44A had no effect on donor cell ERK activation, as it contained an EGFR transactivation-independent pathway. However, K44A mutant attenuated ERK phosphorylation in the acceptor cell, which was only stimulated via an EGFR transactivation-dependent mechanism.
Scaffold Proteins
One of the messages of this review is that a diversity and redundancy of ERK signaling pathways exist in cells (fig. 1). It is fitting then to conclude the review with scaffold proteins as they offer a possible mechanism of specificity to cope with the redundancies and thereby prevent cross talk between different GPCR mitogenic signaling pathways. Multiprotein complexes comprised of different components of heterologous signaling pathways have been identified in many cells [for reviews see 116–118]. The fact that diverse pathways to ERK in the same cells are initiated by overlapping stimuli suggests specificity may be achieved via scaffolds. The first MAP kinase scaffolds identified were found in yeast and were comprised of functionally different types of proteins [117]. In addition to kinases and other enzymes, such scaffolds bind simultaneously adaptor and anchoring proteins to tether them to specific membranes in the cell, thereby providing compartmentation [for a review describing the various types of protein functions, the motifs in their binding domains that allow proteins to interact with one another and the sundry mechanisms by which substrates are brought to their enzymes or enzymes are transported to their substrates during signaling see 116].
The first yeast scaffold protein identified, Ste5, formed a high-molecular-weight complex, consisting of a MAP kinase signaling module [117]. MAP kinase kinase kinase was associated with MAP kinase kinase (MEK), MAP kinase and a pheromone-activated G protein. Subsequently, an additional three MAP kinase modules containing combinations of different kinase isoforms were found on other scaffolds in yeast. These modules were dedicated to processes such as filamentation and osmotic shock rather than mating. Similar modules have been found in mammalian cells with low-molecular-weight GTP binding proteins (i.e., Ras, Rac, Rap) that immediately precede the Raf-1-MEK-ERK troika. Mammalian scaffolds for modules containing MAP kinase isoforms such as c-jun N-terminal kinase (JNK), P38 as well as ERK have been documented [117, 118]. The evidence for two mammalian scaffold candidates, KSR and MP1, for ERK modules has been compelling [118–122]. Focal adhesion kinases, Pyk2/FAK, have raised the possibility of integrin-based scaffolds in GPCR heterologous signaling to ERK [13, 43, 45, 55, 74, 90, 123].
Recently β-arrestin, an adaptor protein that binds phosphorylated GPCRs and targets them for endocytosis, has been shown to be a candidate for a multifunctional scaffold protein acting with both JNK and ERK modules [55–57, 124–126]. This was accomplished by combinations of yeast two-hybrid screening, coimmunoprecipitation, gel filtration and immunofluorescence confocal microscopy analyses. A novel facet of the β-arrestin scaffold is its ability to assemble MAP kinase modules with a GPCR and target it to the endosome of the cell. This combination of GPCR with the MAP kinase module may afford the specificity to differentiate overlapping mitogenic signals from distinct GPCRs.
Conclusion
It is now amply clear that GPCR signaling does not consist of solitary, linear pathways that lead to acute enzyme regulation or long-term gene expression. Instead, cross talk between GPCR and growth factor signaling is so rampant that the current, compelling question relates to how specificity is achieved with the existence of a myriad of overlapping mitogenic stimuli and seemingly redundant pathways with MAP kinases playing an integral role (fig. 1). Given the limitations of previous studies that are dependent on overexpression of signaling components and cell-type variants, it is necessary that future research focus on endogenous signaling components, primary cultures of homogeneous cells and in vivo studies with transgenic mice.
Acknowledgments
This study was supported in part by research grants DA05412 and DA13475 from the National Institute on Drug Abuse.
References
- 1.Cooper JA, Sefton BM, Hunter T. Diverse mitogenic agents induce the phosphorylation of two related 42,000-dalton proteins on tyrosine in quiescent chick cells. Mol Cell Biol. 1984;4:30–37. doi: 10.1128/mcb.4.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kohno M. Diverse mitogenic agents induce rapid phosphorylation of a common set of cellular proteins at tyrosine in quiescent mammalian cells. J Biol Chem. 1985;260:1771–1779. [PubMed] [Google Scholar]
- 3.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 4.Impey S, Obrietan K, Storm DR. Making new connections: Role of ERK MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- 5.Grewal SS, York RD, Stork PJS. Extracellular-signal-regulated kinase signalling in neurons. Curr Opin Neurobiol. 1999;9:544–553. doi: 10.1016/S0959-4388(99)00010-0. [DOI] [PubMed] [Google Scholar]
- 6.Cazaubon S, Chaverot N, Romero IA, Girault JA, Adamson P, Strosberg AD, Couraud PO. Growth factor activity of endothelin-1 in primary astrocytes mediated by adhesion-dependent and -independent pathways. J Neurosci. 1997;17:6203–6212. doi: 10.1523/JNEUROSCI.17-16-06203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bohn LM, Belcheva MM, Coscia CJ. Mitogenic signaling via endogenous kappa-opioid receptors in C6 glioma cells: Evidence for the involvement of protein kinase C and the mitogen-activated protein kinase signaling cascade. J Neurochem. 2000;74:564–573. doi: 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bohn LM, Belcheva MM, Coscia CJ. Mu-opioid agonist inhibition of kappa-opioid receptor-stimulated extracellular signal-regulated kinase phosphorylation is dynamin-dependent in C6 glioma cells. J Neurochem. 2000;74:574–581. doi: 10.1046/j.1471-4159.2000.740574.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koch WJ, Hawes BE, Allen LF, Lefkowitz RJ. Direct evidence that G(i)-coupled receptor stimulation of mitogen-activated protein kinase is mediated by G(beta-gamma) activation of p21(Ras) Proc Natl Acad Sci USA. 1994;91:12706–12710. doi: 10.1073/pnas.91.26.12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crespo P, Xu N, Simonds WF, Gutkind JS. Ras-dependent activation of MAP kinase pathway mediated by G-protein beta gamma subunits. Nature. 1994;369:418–420. doi: 10.1038/369418a0. [DOI] [PubMed] [Google Scholar]
- 11.Faure M, Voynoyasenetskaya TA, Bourne HR. cAMP and beta-gamma subunits of heterotrimeric G proteins stimulate the mitogen-activated protein kinase pathway in COS-7 cells. J Biol Chem. 1994;269:7851–7854. [PubMed] [Google Scholar]
- 12.Marinissen MJ, Gutkind JS. G-protein-coupled receptors and signaling networks: Emerging paradigms. Trends Pharmacol Sci. 2001;22:368–376. doi: 10.1016/s0165-6147(00)01678-3. [DOI] [PubMed] [Google Scholar]
- 13.Luttrell LM, Daaka Y, Lefkowitz RJ. Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol. 1999;11:177–183. doi: 10.1016/s0955-0674(99)80023-4. [DOI] [PubMed] [Google Scholar]
- 14.Watanabe M, Nakamura M, Sato K, Kano M, Simon MI, Inoue Y. Patterns of expression for the mRNA corresponding to the four isoforms of phospholipase C-beta in mouse brain. Eur J Neurosci. 1998;10:2016–2025. doi: 10.1046/j.1460-9568.1998.00213.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang SY, Coso OA, Collins R, Gutkind JS, Simonds WF. A C-terminal mutant of the G protein beta subunit deficient in the activation of phospholipase C-beta. J Biol Chem. 1996;271:20208–20212. doi: 10.1074/jbc.271.33.20208. [DOI] [PubMed] [Google Scholar]
- 16.Belcheva MM, Wong YH, Coscia CJ. Evidence for transduction of mu but not kappa opioid modulation of extracellular signal-regulated kinase activity by G(z) and G(12) proteins. Cell Signal. 2000;12:481–489. doi: 10.1016/s0898-6568(00)00095-4. [DOI] [PubMed] [Google Scholar]
- 17.Belcheva MM, Vogel Z, Ignatova E, Avidorreiss T, Zippel R, Levy R, Young EC, Barg J, Coscia CJ. Opioid modulation of extracellular signal-regulated protein kinase activity is Ras-dependent and involves G (beta-gamma) subunits. J Neurochem. 1998;70:635–645. doi: 10.1046/j.1471-4159.1998.70020635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coscia CJ, Bohn LM, Belcheva MM. Mechanism of kappa and mu opioid modulation of C6 glioma cell proliferation. Soc Neurosci Abstr. 1999;25:1035. [Google Scholar]
- 19.Inglese J, Koch WJ, Touhara K, Lefkowitz RJ. G-beta-gamma interactions with pH domains and Ras-MAPK signaling pathways. Trends Biochem Sci. 1995;20:151–156. doi: 10.1016/s0968-0004(00)88992-6. [DOI] [PubMed] [Google Scholar]
- 20.Hawes BE, Vanbiesen T, Koch WJ, Luttrell LM, Lefkowitz RJ. Distinct pathways of G(i)- and G(q)-mediated mitogen-activated protein kinase activation. J Biol Chem. 1995;270:17148–17153. doi: 10.1074/jbc.270.29.17148. [DOI] [PubMed] [Google Scholar]
- 21.van Biesen T, Hawes BE, Raymond JR, Luttrell LM, Koch WJ, Lefkowitz RJ. G(o)-protein alpha-subunits activate mitogen-activated protein kinase via a novel protein kinase C-dependent mechanism. J Biol Chem. 1996;271:1266–1269. doi: 10.1074/jbc.271.3.1266. [DOI] [PubMed] [Google Scholar]
- 22.Antonelli V, Bernasconi F, Wong YH, Vallar L. Activation of B-Raf and regulation of the mitogen-activated protein kinase pathway by the G(o) alpha chain. Mol Biol Cell. 2000;11:1129–1142. doi: 10.1091/mbc.11.4.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jordan JD, Carey KD, Stork PJS, Iyengar R. Modulation of Rap activity by direct interaction of G alpha(o) with Rapl GTPase-activating protein. J Biol Chem. 1999;274:21507–21510. doi: 10.1074/jbc.274.31.21507. [DOI] [PubMed] [Google Scholar]
- 24.Bos JL, Zwartkruis FJT. Rhapsody in G proteins. Nature. 1999;400:820–821. doi: 10.1038/23594. [DOI] [PubMed] [Google Scholar]
- 25.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the beta(2)-adrenergic receptor to different G proteins by protein kinase A. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 26.Zou Y, Komuro I, Yamazaki T, Kudoh S, Uozumi H, Kadowaki T, Yazaki Y. Both Gs and Gi proteins are critically involved in isoproterenol-induced cardiomyocyte hypertrophy. J Biol Chem. 1999;274:9760–9770. doi: 10.1074/jbc.274.14.9760. [DOI] [PubMed] [Google Scholar]
- 27.Burgering BMT, Pronk GJ, Vanweeren PC, Chardin P, Bos JL. cAMP antagonizes p21(Ras)-directed activation of extracellular signal-regulated kinase-2 and phosphorylation of mSos nucleotide exchange factor. EMBO J. 1993;12:4211–4220. doi: 10.1002/j.1460-2075.1993.tb06105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cook SJ, McCormick F. Inhibition by cAMP of Ras-dependent activation of Raf. Science. 1993;262:1069–1072. doi: 10.1126/science.7694367. [DOI] [PubMed] [Google Scholar]
- 29.Graves LM, Bornfeldt KE, Raines EW, Potts BC, Macdonald SG, Ross R, Krebs EG. Protein kinase-A antagonizes platelet-derived growth factor-induced signaling by mitogen-activated protein kinase in human arterial smooth muscle cells. Proc Natl Acad Sci USA. 1993;90:10300–10304. doi: 10.1073/pnas.90.21.10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sevetson BR, Kong XM, Lawrence JC. Increasing cAMP attenuates activation of mitogen-activated protein kinase. Proc Natl Acad Sci USA. 1993;90:10305–10309. doi: 10.1073/pnas.90.21.10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu J, Dent P, Jelinek T, Wolfman A, Weber MJ, Sturgill TW. Inhibition of the EGF-activated MAP kinase signaling pathway by adenosine 3′,5′-monophosphate. Science. 1993;262:1065–1069. doi: 10.1126/science.7694366. [DOI] [PubMed] [Google Scholar]
- 32.Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Cyclic AMP inhibits activation of mitogen-activated protein kinase and cell proliferation in response to growth factors in cultured rat cortical astrocytes. J Neurochem. 1996;67:2246–2255. doi: 10.1046/j.1471-4159.1996.67062246.x. [DOI] [PubMed] [Google Scholar]
- 33.Schmitt JM, Stork PJS. Beta(2)-adrenergic receptor activates extracellular signal-regulated kinases (ERKs) via the small G protein Rap1 and the serine/threonine kinase b-Raf. J Biol Chem. 2000;275:25342–25350. doi: 10.1074/jbc.M003213200. [DOI] [PubMed] [Google Scholar]
- 34.York RD, Molliver DC, Grewal SS, Stenberg PE, McCleskey EW, Stork PJS. Role of phosphoinositide 3-kinase and endocytosis in nerve growth factor-induced extracellular signal-regulated kinase activation via Ras and Rap1. Mol Cell Biol. 2000;20:8069–8083. doi: 10.1128/mcb.20.21.8069-8083.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJS. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 36.Dugan LL, Kim JS, Zhang YJ, Bart RD, Sun YL, Holtzman DM, Gutmann DH. Differential effects of cAMP in neurons and astrocytes. Role of b-Raf. J Biol Chem. 1999;274:25842–25848. doi: 10.1074/jbc.274.36.25842. [DOI] [PubMed] [Google Scholar]
- 37.Grewal SS, Fass DM, Yao H, Ellig CL, Goodman RH, Stork PJS. Calcium and cAMP signals differentially regulate cAMP-responsive element-binding protein function via a Rap1-extracellular signal-regulated kinase pathway. J Biol Chem. 2000;275:34433–34441. doi: 10.1074/jbc.M004728200. [DOI] [PubMed] [Google Scholar]
- 38.Grewal SS, Horgan AM, York RD, Withers GS, Banker GA, Stork PJS. Neuronal calcium activates a Rap1 and B-Raf signaling pathway via the cyclic adenosine monophosphate-dependent protein kinase. J Biol Chem. 2000;275:3722–3728. doi: 10.1074/jbc.275.5.3722. [DOI] [PubMed] [Google Scholar]
- 39.Zanassi P, Paolillo M, Feliciello A, Avvedimento EV, Gallo V, Schinelli S. cAMP-dependent protein kinase induces cAMP-response element-binding protein phosphorylation via an intracellular calcium release/ERK-dependent pathway in striatal neurons. J Biol Chem. 2001;276:11487–11495. doi: 10.1074/jbc.M007631200. [DOI] [PubMed] [Google Scholar]
- 40.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu HX, Kandel ER. MAP kinase translocates into the nucleus of he presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 41.Mochizuki N, Ohba Y, Kiyokawa E, Kurata T, Murakami T, Ozaki T, Kitabatake A, Nagashima K, Matsuda M. Activation of the ERK/MAPK pathway by an isoform of Rap1GAP associated with G alpha(i) Nature. 1999;400:891–894. doi: 10.1038/23738. [DOI] [PubMed] [Google Scholar]
- 42.Kao SC, Jaiswal RK, Kolch W, Landreth GE. Identification of the mechanisms regulating the differential activation of the MAPK cascade by epidermal growth factor and nerve growth factor in PC12 cells. J Biol Chem. 2001;276:18169–18177. doi: 10.1074/jbc.M008870200. [DOI] [PubMed] [Google Scholar]
- 43.Lev S, Moreno H, Martinez R, Canoll P, Peles E, Musacchio JM, Plowman GD, Rudy B, Schlessinger J. Protein tyrosine kinase Pyk2 involved in Ca2+-induced regulation of ion channel and MAP kinase functions. Nature. 1995;376:737–745. doi: 10.1038/376737a0. [DOI] [PubMed] [Google Scholar]
- 44.Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 45.Soltoff SP. Related adhesion focal tyrosine kinase and the epidermal growth factor receptor mediate the stimulation of mitogen-activated protein kinase by the G-protein-coupled P-2Y2 receptor. J Biol Chem. 1998;273:23110–23117. doi: 10.1074/jbc.273.36.23110. [DOI] [PubMed] [Google Scholar]
- 46.Satoh K, Ichihara K, Landon EJ, Inagami T, Tang H. 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitors block calcium-dependent tyrosine kinase Pyk2 activation by angiotensin II in vascular endothelial cells – Involvement of geranylgeranylation of small G protein Rap1. J Biol Chem. 2001;276:15761–15767. doi: 10.1074/jbc.M009165200. [DOI] [PubMed] [Google Scholar]
- 47.Della Rocca GJ, van Biesen T, Daaka Y, Luttrell DK, Luttrell LM, Lefkowitz RJ. Ras-dependent mitogen-activated protein kinase activation by G protein-coupled receptors. Convergence of G(i)- and G(q)-mediated pathways on calcium/calmodulin, Pyk2, and Src kinase. J Biol Chem. 1997;272:19125–19132. doi: 10.1074/jbc.272.31.19125. [DOI] [PubMed] [Google Scholar]
- 48.Andreev J, Galisteo ML, Kranenburg O, Logan SK, Chiu ES, Okigaki M, Cary LA, Moolenaar WH, Schlessinger J. Src and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade. J Biol Chem. 2001;276:20130–20135. doi: 10.1074/jbc.M102307200. [DOI] [PubMed] [Google Scholar]
- 49.van Corven EJ, Hordijk PL, Medema RH, Bos JL, Moolenaar WH. Pertussis toxin-sensitive activation of p21ras by G protein-coupled receptor agonists in fibroblasts. Proc Natl Acad Sci USA. 1993;90:1257–1261. doi: 10.1073/pnas.90.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Della Rocca GJ, Maudsley S, Daaka Y, Lefkowitz RJ, Luttrell LM. Pleiotropic coupling of G protein-coupled receptors to the mitogen-activated protein kinase cascade. Role of focal adhesions and receptor tyrosine kinases. J Biol Chem. 1999;274:13978–13984. doi: 10.1074/jbc.274.20.13978. [DOI] [PubMed] [Google Scholar]
- 51.Luttrell LM, Daaka Y, Della Rocca GJ, Lefkowitz RJ. G protein-coupled receptors mediate two functionally distinct pathways of tyrosine phosphorylation in Rat 1a fibroblasts. Shc phosphorylation and receptor endocytosis correlate with activation of ERK kinases. J Biol Chem. 1997;272:31648–31656. doi: 10.1074/jbc.272.50.31648. [DOI] [PubMed] [Google Scholar]
- 52.Luttrell LM, Ferguson SSG, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin FT, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta(2) adrenergic receptor Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 53.Cao WH, Lutrell LM, Medvedev AV, Pierce KL, Daniel KW, Dixon TM, Lefkowitz RJ, Collins S. Direct binding of activated c-Src to the beta(3)-adrenergic receptor is required for MAP kinase activation. J Biol Chem. 2000;275:38131–38134. doi: 10.1074/jbc.C000592200. [DOI] [PubMed] [Google Scholar]
- 54.Pierce KL, Tohgo A, Ahn S, Field ME, Luttrell LM, Lefkowitz RJ. Epidermal growth factor (EGF) receptor-dependent ERK activation by G protein-coupled receptors. A co-culture system for identifying intermediates upstream and downstream of heparin-binding EGF shedding. J Biol Chem. 2001;276:23155–23160. doi: 10.1074/jbc.M101303200. [DOI] [PubMed] [Google Scholar]
- 55.Pierce KL, Luttrell LM, Lefkowitz RJ. New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene. 2001;20:1532–1539. doi: 10.1038/sj.onc.1204184. [DOI] [PubMed] [Google Scholar]
- 56.DeFea KA, Vaughn ZD, O'Bryan EM, Nishijima D, Dery O, Bunnett NW. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a beta-arrestin-dependent scaffolding complex. Proc Natl Acad Sci USA. 2000;97:11086–11091. doi: 10.1073/pnas.190276697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. Beta-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stephens L, Eguinoa A, Corey S, Jackson T, Hawkins PT. Receptor stimulated accumulation of phosphatidylinositol (3,4,5)-trisphosphate by G-protein mediated pathways in human myeloid derived cells. EMBO J. 1993;12:2265–2273. doi: 10.1002/j.1460-2075.1993.tb05880.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumagai N, Morii N, Fujisawa K, Nemoto Y, Narumiya S. ADP-ribosylation of Rho p21 inhibits lysophosphatidic acid-induced protein tyrosine phosphorylation and phosphatidylinositol 3-kinase activation in cultured Swiss 3T3 cells. J Biol Chem. 1993;268:24535–24538. [PubMed] [Google Scholar]
- 60.Hawes BE, Luttrell LM, van Biesen T, Lefkowitz RJ. Phosphatidylinositol 3-kinase is an early intermediate in the G-beta-gamma-mediated mitogen-activated protein kinase signaling pathway. J Biol Chem. 1996;271:12133–12136. doi: 10.1074/jbc.271.21.12133. [DOI] [PubMed] [Google Scholar]
- 61.Lopezi-Ilasaca M, Crespo P, Pellici PG, Gutkind JS, Wetzker R. Linkage of G protein-coupled receptors to the MAPK signaling pathway through PI3-kinase gamma. Science. 1997;275:394–397. doi: 10.1126/science.275.5298.394. [DOI] [PubMed] [Google Scholar]
- 62.Pombo CM, Zalvide J, Gaylinn BD, Dieguez C. Growth hormone-releasing hormone stimulates mitogen-activated protein kinase. Endocrinology. 2000;141:2113–2119. doi: 10.1210/endo.141.6.7513. [DOI] [PubMed] [Google Scholar]
- 63.Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kranenburg O, Moolenaar WH. Ras-MAP kinase signaling by lysophosphatidic acid and other G protein-coupled receptor agonists. Oncogene. 2001;20:1540–1546. doi: 10.1038/sj.onc.1204187. [DOI] [PubMed] [Google Scholar]
- 65.Duckworth BC, Cantley LC. Conditional inhibition of the mitogen-activated protein kinase cascade by wortmannin. Dependence of signal strength. J Biol Chem. 1997;272:27665–27670. doi: 10.1074/jbc.272.44.27665. [DOI] [PubMed] [Google Scholar]
- 66.Prasad SVN, Barak LS, Rapacciuolo A, Caron MG, Rockman HA. Agonist-dependent recruitment of phosphoinositide 3-kinase to the membrane by beta-adrenergic receptor kinase 1. A role in receptor sequestration. J Biol Chem. 2001;276:18953–18959. doi: 10.1074/jbc.M102376200. [DOI] [PubMed] [Google Scholar]
- 67.Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H. EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature. 1998;394:494–498. doi: 10.1038/28879. [DOI] [PubMed] [Google Scholar]
- 68.Enslen H, Tokumitsu H, Stork PJS, Davis RJ, Soderling TR. Regulation of mitogen-activated protein kinases by a calcium/calmodulin-dependent protein kinase cascade. Proc Natl Acad Sci USA. 1996;93:10803–10808. doi: 10.1073/pnas.93.20.10803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang DX, Tolbert LM, Carlson KW, Sadee W. Nuclear Ca2+/calmodulin translocation activated by mu-opioid (OP3) receptor. J Neurochem. 2000;74:1418–1425. doi: 10.1046/j.1471-4159.2000.0741418.x. [DOI] [PubMed] [Google Scholar]
- 70.Liu MY, Yu B, Nabanishi O, Wieland T, Simon M. The Ca2+-dependent binding of calmodulin to an N-terminal motif of the heterotrimeric G protein beta subunit. J Biol Chem. 1997;272:18801–18807. doi: 10.1074/jbc.272.30.18801. [DOI] [PubMed] [Google Scholar]
- 71.Chuang TT, Paolucci L, Deblasi A. Inhibition of G protein-coupled receptor kinase subtypes by Ca2+/calmodulin. J Biol Chem. 1996;271:28691–28696. doi: 10.1074/jbc.271.45.28691. [DOI] [PubMed] [Google Scholar]
- 72.Joyal JL, Burks DJ, Pons S, Matter WF, Vlahos CJ, White WF, Sacks DB. Calmodulin activates phosphatidylinositol 3-kinase. J Biol Chem. 1997;272:28183–28186. doi: 10.1074/jbc.272.45.28183. [DOI] [PubMed] [Google Scholar]
- 73.Liang L, Leong K, Seow KT, Ng CH, Pallen CJ. Calmodulin binds to and inhibits the activity of the membrane distal catalytic domain of receptor protein-tyrosine phosphatase alpha. J Biol Chem. 2000;275:30075–30081. doi: 10.1074/jbc.M004843200. [DOI] [PubMed] [Google Scholar]
- 74.Zwick E, Wallasch C, Daub H, Ullrich A. Distinct calcium-dependent pathways of epidermal growth factor receptor transactivation and Pyk2 tyrosine phosphorylation in PC12 cells. J Biol Chem. 1999;274:20989–20996. doi: 10.1074/jbc.274.30.20989. [DOI] [PubMed] [Google Scholar]
- 75.Della Rocca GJ, Mukhin YV, Gamovskaya MN, Daaka Y, Clark GJ, Luttrell LM, Lefkowitz RJ, Raymond JR. Serotonin 5-HT1A receptor-mediated ERK activation requires calcium/calmodulin-dependent receptor endocytosis. J Biol Chem. 1999;274:4749–4753. doi: 10.1074/jbc.274.8.4749. [DOI] [PubMed] [Google Scholar]
- 76.Eguchi S, Matsumoto T, Motley ED, Utsunomiya H, Inagami T. Identification of an essential signaling cascade for mitogen-activated protein kinase activation by angiotensin II in cultured rat vascular smooth muscle cells. Possible requirement of G(q)-mediated p21(Ras) activation coupled to a Ca2+/calmodulin-sensitive tyrosine kinase. J Biol Chem. 1996;271:14169–14175. doi: 10.1074/jbc.271.24.14169. [DOI] [PubMed] [Google Scholar]
- 77.Egea J, Espinet C, Comella JX. Calcium influx activates extracellular-regulated kinase/mitogen-activated protein kinase pathway through a calmodulin-sensitive mechanism in PC12 cells. J Biol Chem. 1999;274:75–85. doi: 10.1074/jbc.274.1.75. [DOI] [PubMed] [Google Scholar]
- 78.Farnsworth CL, Freshney NW, Rosen LB, Ghosh A, Greenberg ME, Feig LA. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nature. 1995;376:524–527. doi: 10.1038/376524a0. [DOI] [PubMed] [Google Scholar]
- 79.Ebinu JO, Bottorff DA, Chan EYW, Stang SL, Dunn RJ, Stone JC. RasGRP, a Ras guanyl nucleotide-releasing protein with calcium- and diacylglycerol-binding motifs. Science. 1998;280:1082–1086. doi: 10.1126/science.280.5366.1082. [DOI] [PubMed] [Google Scholar]
- 80.Egea J, Espinet C, Soler RM, Peiro S, Rocamora N, Cornelia JX. Nerve growth factor activation of the extracellular signal-regulated kinase pathway is modulated by Ca2+ and calmodulin. Mol Cell Biol. 2000;20:1931–1946. doi: 10.1128/mcb.20.6.1931-1946.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Minakami R, Jinnai N, Sugiyama H. Phosphorylation and calmodulin binding of the metabotropic glutamate receptor subtype 5 (mGlur5) are antagonistic in vitro. J Biol Chem. 1997;272:20291–20298. doi: 10.1074/jbc.272.32.20291. [DOI] [PubMed] [Google Scholar]
- 82.Wang DX, Sadee W, Quillan JM. Calmodulin binding to G protein-coupling domain of opioid receptors. J Biol Chem. 1999;274:22081–22088. doi: 10.1074/jbc.274.31.22081. [DOI] [PubMed] [Google Scholar]
- 83.Heuss C, Gerber U. G-protein-independent signaling by G-protein-coupled receptors. Trends Neurosci. 2000;23:469–475. doi: 10.1016/s0166-2236(00)01643-x. [DOI] [PubMed] [Google Scholar]
- 84.Bofill-Cardona E, Kudlacek O, Yang Q, Ahorn H, Freissmuth M, Nanoff C. Binding of calmodulin to the D-2-dopamine receptor reduces receptor signaling by arresting the G protein activation switch. J Biol Chem. 2000;275:32672–32680. doi: 10.1074/jbc.M002780200. [DOI] [PubMed] [Google Scholar]
- 85.Belcheva MM, Szùcs M, Wang D, Sadee W, Coscia CJ. Mu-opioid receptor-mediated ERK-activation involves calmodulin-dependent EGF receptor transactivation. J Biol Chem. 2001;276:33847–33853. doi: 10.1074/jbc.M101535200. [DOI] [PubMed] [Google Scholar]
- 86.Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme D, Rapp UR. Protein kinase-C-alpha activates Raf-1 by direct phosphorylation. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- 87.Ueda Y, Hirai S, Osada S, Suzuki A, Mizuno K, Ohno S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem. 1996;271:23512–23519. doi: 10.1074/jbc.271.38.23512. [DOI] [PubMed] [Google Scholar]
- 88.Marais R, Light Y, Mason C, Paterson H, Olson MF, Marshall CJ. Requirement of Ras-GTP-Raf complexes for activation of Raf-1 by protein kinase C. Science. 1998;280:109–112. doi: 10.1126/science.280.5360.109. [DOI] [PubMed] [Google Scholar]
- 89.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 90.Keely SJ, Calandrella SO, Barrett KE. Carbachol-stimulated transactivation of epidermal growth factor receptor and mitogen-activated protein kinase in T-84 cells is mediated by intracellular Ca2+, PYK-2, and p60 (Src) J Biol Chem. 2000;275:12691–12625. doi: 10.1074/jbc.275.17.12619. [DOI] [PubMed] [Google Scholar]
- 91.Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, Lefkowitz RJ, Luttrell LM. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem. 2000;275:9572–9580. doi: 10.1074/jbc.275.13.9572. [DOI] [PubMed] [Google Scholar]
- 92.Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- 93.Carpenter G. EGF receptor transactivation mediated by the proteolytic production of EGF-like agonists. STKE. 2000;15:1–3. doi: 10.1126/stke.2000.15.pe1. Science's STKE: http://stke.sciencemag.org/cgi/content/full/OC_sigtrans;2001/15/pel. [DOI] [PubMed]
- 94.Roudabush FL, Pierce KL, Maudsley S, Khan KD, Luttrell LM. Transactivation of the EGF receptor mediates IGF-1-stimulated She phosphorylation and ERK1/2 activation in COS-7 cells. J Biol Chem. 2000;275:22583–22589. doi: 10.1074/jbc.M002915200. [DOI] [PubMed] [Google Scholar]
- 95.Dong JY, Wiley HS. Trafficking and proteolytic release of epidermal growth factor receptor ligands are modulated by their membrane-anchoring domains. J Biol Chem. 2000;275:557–564. doi: 10.1074/jbc.275.1.557. [DOI] [PubMed] [Google Scholar]
- 96.Diaz-Rodriguez F, Esparis-Ogando A, Montero JC, Yuste L, Pandiella A. Stimulation of cleavage of membrane proteins by calmodulin inhibitors. Biochem J. 2000;346:359–367. [PMC free article] [PubMed] [Google Scholar]
- 97.Bosch M, Gil J, Bachs O, Agell N. Calmodulin inhibitor W13 induces sustained activation of ERK2 and expression of p21(Cip1) J Biol Chem. 1998;273:22145–22150. doi: 10.1074/jbc.273.34.22145. [DOI] [PubMed] [Google Scholar]
- 98.Linseman DA, Benjamin CW, Jones DA. Convergence of angiotensin ii and platelet-derived growth factor receptor signaling cascades in vascular smooth muscle cells. J Biol Chem. 1995;270:12563–12568. doi: 10.1074/jbc.270.21.12563. [DOI] [PubMed] [Google Scholar]
- 99.Herrlich A, Daub H, Knebel A, Herrlich P, Ullrich A, Schultz G, Gudermann T. Ligand-independent activation of platelet-derived growth factor receptor is a necessary intermediate in lysophosphatidic, acid-stimulated mitogenic activity in L cells. Proc Natl Acad Sci USA. 1998;95:8985–8990. doi: 10.1073/pnas.95.15.8985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tan Y, Belcheva MM, Haas P, Coscia CJ. Delineation of the mechanism of ERK regulation by mu opioids in an astrocytic model system. Soc Neurosci Abstr. 2001;27:39. [Google Scholar]
- 101.Rao GN, Delafontaine P, Runge MS. Thrombin stimulates phosphorylation of insulin-like growth factor-1 receptor, insulin receptor substrate-1, and phospholipase C-gamma-1 in rat aortic smooth muscle cells. J Biol Chem. 1995;270:27871–27875. doi: 10.1074/jbc.270.46.27871. [DOI] [PubMed] [Google Scholar]
- 102.Roth BL, Willins DL, Kroeze WK. G protein-coupled receptor (GPCR) trafficking in the central nervous system. Relevance for drugs of abuse. Drug Alcohol Depend. 1998;51:73–85. doi: 10.1016/s0376-8716(98)00067-2. [DOI] [PubMed] [Google Scholar]
- 103.Vieira AV, Lamaze C, Schmid SL. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science. 1996;274:2086–2089. doi: 10.1126/science.274.5295.2086. [DOI] [PubMed] [Google Scholar]
- 104.Luttrell LM, Della Rocca GJ, van Biesen T, Luttrell DK, Lefkowitz RJ. G-beta-gamma subunits src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G protein-coupled receptor-mediated Ras activation. J Biol Chem. 1997;272:4637–4644. doi: 10.1074/jbc.272.7.4637. [DOI] [PubMed] [Google Scholar]
- 105.Daaka Y, Luttrell LM, Ahn S, Della Rocca GJ, Ferguson SSG, Caron MG, Lefkowitz RJ. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–688. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- 106.Ignatova EG, Belcheva MM, Bohn LM, Neuman MC, Coscia CJ. Requirement of receptor internalization for opioid stimulation of mitogen-activated protein kinase: Biochemical and immunofluorescence confocal microscopic evidence. J Neurosci. 1999;19:56–63. doi: 10.1523/JNEUROSCI.19-01-00056.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ahn S, Maudsley S, Luttrell LM, Lefkowitz RJ, Daaka Y. Src-mediated tyrosine phosphorylation of dynamin is required for beta2-adrenergic receptor internalization and mitogen-activated protein kinase signaling. J Biol Chem. 1999;274:1185–1188. doi: 10.1074/jbc.274.3.1185. [DOI] [PubMed] [Google Scholar]
- 108.Vogler O, Nolte B, Voss M, Schmidt M, Jakobs KH, van Koppen CJ. Regulation of muscarinic acetylcholine receptor sequestration and function by beta-arrestin. J Biol Chem. 1999;274:12333–12338. doi: 10.1074/jbc.274.18.12333. [DOI] [PubMed] [Google Scholar]
- 109.Kramer HK, Andria ML, Esposito DH, Simon EJ. Tyrosine phosphorylation of the delta-opioid receptor. Evidence for its role in mitogen-activated protein kinase activation and receptor internalization. Biochem Pharmacol. 2000;60:781–792. doi: 10.1016/s0006-2952(00)00400-7. [DOI] [PubMed] [Google Scholar]
- 110.Pierce KL, Maudsley S, Daaka Y, Luttrell LM, Lefkowitz RJ. Role-of-endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc Natl Acad Sci USA. 2000;97:1489–1494. doi: 10.1073/pnas.97.4.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ferguson SSG. Evolving concepts in G protein-coupled receptor endocytosis: The role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 112.Whistler JL, von Zastrow M. Dissociation of functional roles of dynamin in receptor-mediated endocytosis and mitogenic signal transduction. J Biol Chem. 1999;274:24575–24578. doi: 10.1074/jbc.274.35.24575. [DOI] [PubMed] [Google Scholar]
- 113.Schramm NL, Limbird LE. Stimulation of mitogen-activated protein kinase by G protein-coupled alpha(2)-adrenergic receptors does not require agonist-elicited endocytosis. J Biol Chem. 1999;274:24935–24940. doi: 10.1074/jbc.274.35.24935. [DOI] [PubMed] [Google Scholar]
- 114.Li JG, Luo LY, Krupnick JG, Benovic JL, Liu-Chen LY. U50, 488H-induced internalization of the human kappa opioid receptor involves a beta-arrestin- and dynamin-dependent mechanism. Kappa receptor internalization is not required for mitogen-activated protein kinase activation. J Biol Chem. 1999;274:12087–12094. doi: 10.1074/jbc.274.17.12087. [DOI] [PubMed] [Google Scholar]
- 115.DeGraff JL, Gagnon AW, Benovic JL, Orsini MJ. Role of arrestins in endocytosis and signaling of alpha(2)-adrenergic receptor subtypes. J Biol Chem. 1999;274:11253–11259. doi: 10.1074/jbc.274.16.11253. [DOI] [PubMed] [Google Scholar]
- 116.Pawson T, Scott JD. Signaling through scaffold, anchoring, and adaptor proteins. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 117.Whitmarsh AJ, Davis RJ. Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem Sci. 1998;23:481–485. doi: 10.1016/s0968-0004(98)01309-7. [DOI] [PubMed] [Google Scholar]
- 118.Burack WR, Shaw AS. Signal transduction: Hanging on a scaffold. Curr Opin Cell Biol. 2000;12:211–216. doi: 10.1016/s0955-0674(99)00078-2. [DOI] [PubMed] [Google Scholar]
- 119.Schaeffer HJ, Catling AD, Eblen ST, Collier LS, Krauss A, Weber MJ. MP1: A MEK binding partner that enhances enzymatic activation of the MAP kinase cascade. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- 120.Xing HR, Lozano J, Kolesnick R. Epidermal growth factor treatment enhances the kinase activity of kinase suppressor of Ras. J Biol Chem. 2000;275:17276–17280. doi: 10.1074/jbc.C900989199. [DOI] [PubMed] [Google Scholar]
- 121.Morrison DK. KSR: A MAPK scaffold of the Ras pathway? J Cell Sci. 2001;114:1609–1612. doi: 10.1242/jcs.114.9.1609. [DOI] [PubMed] [Google Scholar]
- 122.Wunderlich W, Fialka I, Teis D, Alpi A, Pfeifer A, Parton RG, Lottspeich F, Huber LA. A novel 14-kilodalton protein interacts with the mitogen-activated protein kinase scaffold MP1 on a late endosomal/lysosomal compartment. J Cell Biol. 2001;152:765–776. doi: 10.1083/jcb.152.4.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schlaepfer DD, Hunter T. Signal transduction from the extracellular matrix – A role for the focal adhesion protein-tyrosine kinase FAK. Cell Struct Funct. 1996;21:445–450. doi: 10.1247/csf.21.445. [DOI] [PubMed] [Google Scholar]
- 124.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. Beta-arrestin 2: A receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 125.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA. 2001;98:2449–2454. doi: 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Miller WE, Lefkowitz RJ. Expanding roles for beta-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr Opin Cell Biol. 2001;13:139–145. doi: 10.1016/s0955-0674(00)00190-3. [DOI] [PubMed] [Google Scholar]