

Abstract
TRPV1 receptors are polymodal cation channels that open in response to diverse stimuli including noxious heat, capsaicin, and protons. Because Ca2+ is vital for TRPV1 signaling, we sought to precisely measure its contribution to TRPV1 responses and discovered that the Ca2+ current was tuned by the mode of activation. Using patch clamp photometry, we found that the fraction of the total current carried by Ca2+ (called the Pf%) was significantly smaller for TRPV1 currents evoked by protons than for those evoked by capsaicin. Using site-directed mutagenesis, we discovered that the smaller Pf% was due to protonation of three acidic amino acids (Asp646, Glu648, and Glu651) that are located in the mouth of the pore. Thus, in keeping with recent reports of time-dependent changes in the ionic permeability of some ligand-gated ion channels, we now show for the first time that the physiologically important Ca2+ current of the TRPV1 receptor is also dynamic and depends on the mode of activation. This current is significantly smaller when the receptor is activated by a change in pH, owing to atomic scale interactions of H+ and Ca2+ with the fixed negative charge of side chains in the pore.
The TRPV1 receptor is a member of the vanilloid subclass of the transient receptor potential family (TRP)2 of ion channel proteins (1). They are non-selective cation channels activated by a range of stimuli including capsaicin, protons, noxious heat, polyamines, anandamide, camphor, (N-vanillyl)-9-oleamide, and spider venom toxins (2). Although the channel discriminates poorly among monovalent cations, it exhibits a high divalent selectivity, and activation produces a significant flux of Ca2+ into cells (3). The resulting increase in [Ca2+]i in turn helps to trigger a number of important physiological and pathophysiological responses including thermal and chemical sensation, neurogenic inflammation, presynaptic regulation of transmitter release, and itch (4–6).
Given the importance of TRPV1 receptors for pain and sensory transduction, it is surprising that the molecular basis of the Ca2+ current is poorly understood. Some of the confusion reflects the inconsistent data available from reversal potential-based studies of relative ionic permeability (1, 7–9), which may be explained by the recent discovery that the permeability of the channel changes during receptor activation in a time-dependent manner (10). Moreover, although the relative Ca2+ permeability of the TRPV1 receptor is the subject of several studies (3, 10), there is no systematic investigation of the structural basis of the more physiologically relevant parameter, the agonist-evoked Ca2+ current. In this study, we used a fluorimetric flux technique (11) to calculate the fraction of the TRPV1-mediated current that is carried by Ca2+ (Pf%) under conditions where holding potential and extracellular ionic balance are within a normal physiological range. These conditions are noteworthy because the more common method of judging the contribution of Ca2+ relies on reversal potential measurements that use an unphysiologically high [Ca2+]o (usually 112 mm) and estimate relative Ca2+ permeability at a membrane potential equal to the Erev of the agonist-gated current (usually ≥0 mV). We report the serendipitous discovery that proton activation of TRPV1 receptors evokes a current with a smaller Pf% than that evoked by capsaicin and that the difference results from titration of fixed negative charge in the pore.
EXPERIMENTAL PROCEDURES
Molecular Biology and Cell Culture—We used wild-type and mutant rat TRPV1 receptors that were made and expressed using conventional techniques. Point mutations were introduced with the QuikChange II site-directed mutagenesis kit (Stratagene, La Jolla, CA) and verified by automated DNA sequencing (Retrogen Inc., San Diego, CA). Human embryonic kidney-293 cells (HEK-293; ATCC, Manassas, VA) were co-transfected with cDNAs encoding TRPV1 channels and a fluorescent reporter using Effectene (Qiagen, Valencia, CA). These cells were subsequently replated at low density onto poly-l-lysine-coated glass coverslips the night before the experiment.
Patch Clamp Photometry—We used the fluorimetric flux technique as described previously in detail (12, 13). Briefly, the Pf% was determined by simultaneously measuring total membrane current and fluorescence in cells loaded with a high concentration (2 mm) of the calcium-sensitive dye, K5fura-2 (Invitrogen). Membrane current was recorded from single cells held at –60 mV. The intracellular solution contained (in mm): 140 CsCl, 10 tetraethylammonium Cl, 10 HEPES, 2 K5fura-2, 4.8 CsOH, pH 7.35. Light emitted from by fura-2 (380 nm excitation, 510 nm emission) was gathered by a microscope objective and directed to the input of a Model 714 photomultiplier detection system (Photontechnology International, South Brunswick, NJ). To account for the day-to-day variation in the sensitivity of the microscope and photomultiplier tube, the fura-2 signal was normalized to a “bead unit” (BU). One BU equaled the average fluorescence of seven Fluoresbrite carboxy BB 4.6-μm microspheres (Polysciences, Warrington, PA) measured one-at-a-time on the morning of that day's experiment (14, 15). The extracellular bath solution was (in mm): 140 NaCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, titrated to pH 7.4 with 4 NaOH. HEPES was replaced by MES in experiments performed using low pH (pH 5.0–6.0) solutions. In experiments on glutamatergic NR1/NR2A receptors, the extracellular solution also contained 100 μm glycine and 0 mm Mg2+. Control and test solutions were applied using triple-barreled theta glass and a SF-77 Perfusion Fast-Step System (Warner Instruments, Hamden, CT).
The Pf% was calculated as follows.
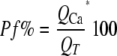 |
(Eq. 1) |
QT is total charge and equal to the integral of the agonist-gated transmembrane current. QCa is the part of QT carried by Ca2+ and equal to ΔF380 divided by the calibration factor Fmax. Fmax was determined in a separate set of experiments as described previously (12).
Single Channel Experiments—Unitary currents were measured in excised membrane patches from HEK-293 cells expressing wild-type or mutant TRPV1 receptors. Currents were recorded at –60 mV in an extracellular solution containing (in mm): 154 NaCl, 10 glucose, 10 HEPES, 2 CaCl2 (pH 7.4). Traces were sampled at 10 kHz and filtered at 1 kHz. Chord conductance (g) was calculated as I/V where I was the average single channel current determined from Gaussian fits of all-points current amplitude histograms obtained from 10–30 s of activity.
Data Analysis—All data are presented as the mean ± S.E. Significant differences among groups were determined using InStat (GraphPad, San Diego, CA) by one-way analysis of variance with Tukey's post hoc. The p values of ≤0.01 were considered significant, unless noted otherwise.
RESULTS
Agonist-dependent Ca2+ Currents through TRPV1 Channels—We measured Ca2+ current through TRPV1 receptors using the dye overload method pioneered by Neher and co-workers (14) and Dani and co-workers (16) and the technique of choice to measure the contribution of Ca2+ to ion channel responses under physiological conditions (17). We voltage-clamped HEK-293 cells transiently expressing the wild-type TRPV1 receptor at –60 mV using recording pipettes containing 2 mm K5fura-2. We allowed the fura-2 to enter the cell from the patch pipette and equilibrate for a period of 10 min and then activated the TRPV1 receptors for 0.5–2 s with capsaicin or protons (pH 5.0). By applying drugs for ≤2 s, we circumvented the time-dependent change in ionic permeability seen during longer (>10 s) drug applications (10). In all cases, activation of TRPV1 channels produced inward membrane currents and decreases in the fura-2 emission (510 nm) when the dye was excited by light (380 nm). A decrease in fluorescence indicates an increase in [Ca2+]i. As expected, the time courses of ΔF380 and QT superimposed, indicating that the increase in [Ca2+]i is due entirely to direct Ca2+ entry through the TRPV1 channel. Examples are shown in Fig. 1, A–F. Here, we applied capsaicin and protons to cells while recording membrane current (Fig. 1, A and D) and fura-2 fluorescence (Fig. 1, B and E), and in each case, the time course of ΔF380 matched the time course of QT (Fig. 1, B and E). Likewise, the time courses of the Ca2+ currents, determined by calibrating and differentiating the ΔF380 values, matched the time courses of the TRPV1-mediated whole-cell currents (Fig. 1, C and F). We calculated the Pf% values of the agonist-evoked currents and were surprised to find that the amount of Ca2+ that enters the cell depended on the mode of receptor activation. We discovered that whole-cell currents evoked by capsaicin had an average Pf%of 9.9 ± 0.4% (n = 17). By comparison, the Pf% of the proton-gated current was significantly smaller (6.6 ± 0.4%; n = 10). Although Fig. 1 shows experiments from separate cells, we measured an identical difference in the Pf% values of the proton- and capsaicingated currents recorded from the single cells exposed to both agonists. In these experiments, the magnitude of the difference in Pf% was the same regardless of the order of agonist application (i.e. protons followed by capsaicin, or vice versa).
FIGURE 1.

The Pf% of TRPV1 currents. A–F, capsaicin and protons activate inward currents (A and D) and ΔF380 values (blue traces of B and E) in cells expressing TRPV1 receptors. Note that the time course of ΔF380 mimics that of QT (red traces of B and E). We differentiated and smoothed the calibrated F380 signal to get an estimate of the size and time course of the agonist-evoked Ca2+ currents (C and E). The figure shows that the Ca2+ component of the capsaicin current (C) is significantly larger than that of the proton current (F). In panels G–K, HEK-293 cells were transfected with plasmids encoding α4β2 nAChR (G), NR1/NR2A glutamate (H), P2X1 purinergic (I), TRPV1 thermosensitive (J), and purinergic P2X2 (K) receptors. The left-hand panels show a schematic representation of a single subunit of each receptor. The putative pore-forming domains are shown in red. The adjacent panels, from left to right, show the whole-cell current, integrated whole-cell current (QT), and ΔF380 evoked by selective agonists. G, α4β2 nicotinic receptors stimulated with 100 μm ACh (red) and 100 μm nicotine (blue). H, NR1/NR2A glutamatergic receptors stimulated with 100 μm glutamate (red) and 100 μm aspartate (blue). I, P2X1 receptors stimulated with 1 μm ATP (blue) or 1 μm α,β-methylene-ATP (red). J, TRPV1 receptors stimulated with 10 μm capsaicin (blue) and pH 5 (red). The yellow and red stars highlight the difference in the orders of magnitude of QT and ΔF380. K, P2X2 receptors stimulated with 300μm ATP at two different pHs (pH 7.4, blue; pH5, red). L, pooled Pf% data (n > 6) for each receptor studied. abMeATP, α,β-methylene-ATP.
We considered the possibility that the divergent values resulted from alternative sources of intracellular Ca2+, interference by Mg2+, or activation of other proton-gated currents; however, additional experiments suggest that these are not responsible (see supplemental data). Thus, we show for the first time that the amount of Ca2+ that flows through TRPV1 channels depends in part on the identity of the agonist, with capsaicin evoking a Ca2+ current that is almost 50% larger than that caused by protons.
Other Ligand-gated Ion Channels Do Not Demonstrate Agonist-specific Pf% Values—Might the agonist-specific Ca2+ flux of the TRPV1 receptor be a general feature shared by other types of ligand-gated ion channels? We considered this question because purinergic (18, 19), glutamatergic (20, 21), and TRPV1 (10) channels show activity-dependent changes in permeability, and thus, it seemed reasonable to ask whether their Ca2+ currents changed in an agonist-dependent manner. We transiently expressed nicotinic α4β2, glutamatergic NR1/NR2A, and ATP-gated P2X1 and P2X2 receptors in separate HEK-293 cells and then measured Pf% using different agonists to evoke transmembrane currents. We found that activating nicotinic receptors with either 100 μm nicotine or 100 μm acetylcholine evoked inward currents with nearly identical Pf% values (Fig. 1, G and L). In a similar fashion, stimulation of glutamatergic receptors with 100 μm glutamate or 100 μm aspartate (Fig. 1H) or P2X1 receptors with 1 μm ATP or 1 μm α,β-methylene ATP (Fig. 1I) also gave currents showing agonist-independent Pf% values (Fig. 1L). These results are in sharp contrast to those obtained using TRPV1 channels, as clearly pictured in Fig. 1J. In this experiment, we show results from a single cell in which the whole-cell current evoked by capsaicin is smaller than that evoked by protons. However, despite the fact that QT is smaller for the capsaicin-evoked current, the ΔF380 is larger. This result is unexpected if Ca2+ current is agonist-independent.
We considered the possibility that increasing [H+]o may increase [H+]i, perhaps due to protons leaking through the patch seal. In so doing, this might decrease the affinity of fura-2 for Ca2+ and interfere with our accurate determination of Pf%. To test this hypothesis, we measured the Pf% of ATP-gated currents through rat P2X2 receptors at pH 5.0 and 7.4 (Fig. 1K) and found no difference in the calculated values (Fig. 1L). We conclude that agonist-dependent modulation of Ca2+ flux is not a general property of ligand-gated ion channels, but rather, within the limitation of the channels we examined, is specific to polymodal TRPV1 receptors.
Neutralizing the Charge on Acidic Side Chains Reduced Pf%—Why is the Pf% of the proton-gated current smaller than that of the capsaicin-gated current? The most plausible explanation is that protons simultaneously activate the receptor and titrate a source of negative charge involved in facilitating Ca2+ permeation in the pore. We studied three acidic residues (Asp646, Glu648, and Glu651) located just extracellular to the putative selectivity filter of TRPV1 (Fig. 2A) by generating the following mutant receptors: D646N, E648Q, and E651Q. In each case, the fixed negative charge of the side chain was replaced by a neutral amide. We saw no obvious differences in the size or shape of the capsaicin-evoked currents of the mutant receptors when compared with the wild-type receptor except that, in all three cases, they displayed significantly reduced Pf% values equal to 4.7 ± 0.4 (n = 5), 4.4 ± 0.6 (n = 7), and 5.5 ± 0.5% (n = 8), respectively (Fig. 2, B and C). The Pf% values of the proton-activated currents were 4.6 ± 0.4 (n = 6), 3.1 ± 0.5 (n = 5), and 4.6 ± 0.5% (n = 4), respectively, which are also smaller than those of the wild-type receptor and not significantly different from the Pf% values of the capsaicin-evoked currents measured from the same mutants. Thus, it appears that the structure responsible for the agonist-specific modulation of Pf%is absent in the charge-neutralized TRPV1 mutants.
FIGURE 2.

The effect of mutagenesis on Pf%. A, the putative pore loop, TM5, and TM6 domains of TRPV1 receptors, assuming a domain structure and organization similar to the KcsA potassium channel (3, 35). B, the fluorescence signals were smoothed, converted to QCa (where QCa = F380/(Fmax*BU)), normalized to peak QT, and then multiplied by 100%. The peak value of each trace is equal to its Pf%. C, pooled Pf% data (n > 5) for wild-type (WT) and mutant TRPV1 receptors. The blue and red bars indicate Pf% values of capsaicin (Cap)- and proton-evoked currents, respectively. a indicates values different from the capsaicin-evoked current of the wild-type receptor. b indicates values different from the proton-activated current of the wild-type receptor. Panels D–G show single channel currents. Each patch contained more than one TRPV1 ion channel. The raw data traces show currents evoked by 1 μm capsaicin. The right-hand panels show all-point current amplitude histograms (right) generated from the same 4-s traces.
Asp646 is known to play a role in divalent permeability in the TRPV1 and TRPV4 receptors (10, 22, 23), and the experiments described above show that it also regulates Ca2+ current through the TRPV1 pore. The fact that neutralizing the charges of Glu648 and Glu651 reduces Pf% was less expected because mutations of these sites do not reduce relative divalent permeability (22, 24). Although substituting glutamine for glutamate is considered “safe” mutagenesis (25), we worried that the reduced Pf% measured in E648Q and E651Q was due to an unintended change in the structure of the pore and not due to the loss of charge. To test this hypothesis, we generated additional mutants (E648D and E651D) in which the carboxyl side chains of Glu648 and Glu651 were replaced by the carboxyl side chain of aspartate. E648D and E651D displayed capsaicin-evoked currents with Pf% values (9.3 ± 0.7%, n = 12; and 8.1 ± 0.3%, n = 10, respectively) that were not significantly different from the wild-type receptor (Fig. 2C). In contrast, the Pf% values of the proton-evoked currents mediated by E648D and E651D were significantly reduced when compared with their respective capsaicin-evoked currents (5.4 ± 0.9%, n = 8; and 5.3 ± 0.7%, n = 6, respectively). Thus, retaining negative charge at these positions recovered the capsaicin-evoked Ca2+ flux to normal values and recovered its pH sensitivity. These results show that the effect of mutagenesis at Glu648 and Glu651 depends on the nature of the substitution and that maintenance of Pf% requires the presence of formal charge at discreet sites.
The Single Channel Conductance of the Monovalent Current Is Unchanged—Because Pf% is a relative function, a decrease in this ratio does not necessarily mean that the channel is conducting less Ca2+; we would see the same result if monovalent current increased and the Ca2+ current remained unchanged. TRPV1 receptors are permeable to both Na+ and H3O+, and given the magnitude of our measured changes in Pf%, the currents carried by these ions would have to double to account for the agonist-dependent differences in Pf% of the wild-type receptor and the lower Pf% values of the mutant receptors. Such a change should be easy to measure as a change in the size of the single channel current. Thus, to determine whether the monovalent currents of wild-type and mutant TRPV1 channels are different, we measured single channel currents in excised outside-out membrane patches of HEK-293 cells held at –60 mV (Fig. 2, D–G). The chord conductance of the capsaicin-evoked wild-type TRPV1 receptor current was 41 ± 1.4 pS (n = 11), in good keeping with previously published results (26). To measure the single channel conductance of the proton-evoked current through the wild-type receptor, we stimulated the patch with a pH 6.0 solution because at pH 5.0, the open probability of TRPV1 receptors in the patch was too high for unitary currents to be resolved. The chord conductance (38 ± 2.1 pS; n = 5) of the proton-gated current of the wild-type TRPV1 receptor was not significantly different from the capsaicin-evoked current. Finally, we measured the single channel conductance of the capsaicin-evoked currents through the three mutant receptors, D646N, E648Q, and E651Q, and found that at 38 ± 0.8 pS (n = 5), 38 ± 0.7 pS (n = 5), and 40 ± 1.5 pS (n = 6), respectively, they also were not significantly different from the wild-type receptor. These data show that the lower Pf% of the proton-evoked current of the wild-type TRPV1 receptor and the reduced Pf%ofthe mutant receptors are not caused by an increase in monovalent current.
DISCUSSION
Ca2+ entry through TRPV1 receptors is large enough to cause a self-directed negative feedback inhibition of receptor function, down-regulation of voltage-operated Ca2+ channels, enhance transmitter release, and stimulate the production of nitric oxide (27, 28). Nevertheless, little is known about the structural determinants underlying Ca2+ current through the channel. We now show that the Pf% of the TRPV1 receptor depends on the mode of activation, and we identify three acidic residues that facilitate Ca2+ flux through the pore.
In an early study, Zeilhofer et al. (29) observed that the capsaicin-evoked current of dorsal root ganglion neurons carried a larger Pf% than the proton-evoked current and concluded that capsaicin and protons activated separate populations of receptors. Since then, the TRPV family has been cloned, and we now know that dorsal root ganglia cells express at least four subtypes (TRPV1–TRPV4) (2). Of these, the only member activated by both capsaicin and protons is TRPV1. We now show that protons do stimulate a current with a reduced Pf% when compared with capsaicin through the same receptor. Although it is possible that the different responses measured by Zeilhofer reflect heteromerization (30) and/or the contribution of modulatory proteins, our work suggests the possibility that neurons in the dorsal root ganglia express a homogenous population of TRPV1 receptors that respond to capsaicin and protons in an agonist-dependent manner.
In a similar vein, the recent report by Caterina and co-workers (10) presents compelling evidence that the cation permeability of TRPV1 channel changes during prolonged stimulation by agonists. If current flow through the TRPV1 channel follows the assumptions of the Goldman-Hodgkin-Katz constant-field equations, then the measured changes in relative permeability should produce equivalent changes in ionic current (31). Many channels deviate from Goldman-Hodgkin-Katz behavior, and changes in relative Ca2+ permeability do not necessarily reflect changes in Ca2+ current (32). We now provide direct evidence for the hypothesis of Caterina and co-workers (10) of dynamic changes in the nature of the TRPV1 current, using physiological concentrations of extracellular ions and a realistic membrane potential, by showing that the size of the Ca2+ current is tuned by the identity of the agonist. However, in contrast to the time-dependent changes in ionic permeability, the agonist-dependence is immediate, suggesting that the composition of the ionic current depends on the nature of the agonist for even the briefest of agonist applications.
Is modulation of Ca2+ entry through TRPV1 receptors relevant? Intracellular Ca2+ homeostasis is tightly regulated, and even small changes in Ca2+ entry through an ion channel can have physiologically significant effects on cell signaling. For example, cells can subtly alter the Ca2+ permeability of glutamate-gated N-methyl-d-aspartate receptors both by a direct cAMP-dependent protein kinase-dependent mechanism (20) and by an indirect mechanism involving up-regulation of receptor subunits that confer a higher degree of Ca2+ flux through the assembled receptor (21). In both cases, the change in Ca2+ entry changes the behavior of neuronal circuits by modulating either long term potentiation (20) or long term depression (21). Modulation of the Ca2+ current through TRPV1 receptors could potentially affect numerous Ca2+-dependent processes associated with TRPV1 receptor activation, including TRPV1 receptor desensitization and the regulation of voltage-operated Ca2+ channels (27, 28). The physiological relevance of the tunable TRPV1 receptor-mediated Ca2+ current constitutes an exciting avenue for further investigation.
What is the structural basis of the dynamic changes in TRPV1 receptor-mediated Ca2+ flux reported here? Asp646 occupies the analogous position within the TRPV1 pore loop as the aspartate in the TVGYGD amino acid motif of the KcsA selectivity filter (3). Our data show that neutralizing the side chain of Asp646 significantly reduced Ca2+ current through the TRPV1 receptor, which agrees with previous studies implicating this residue in regulating divalent permeability (10, 22, 24). Furthermore, our data provide the first evidence that Glu648 and Glu651 do in fact influence Ca2+ flux through the pore despite reversal potential-based measurements that suggest the opposite (22, 24). Neutralizing the charge on either Glu648 or Glu651 significantly reduced the Pf% of the capsaicin-evoked current, whereas maintaining the negative charge by substitution with aspartate had no effect. For the three mutants, D646N, E648Q, and E651Q, the Pf% of the proton-evoked current was not significantly different from that evoked by capsaicin, which is consistent with the prediction that protons evoke currents of reduced Pf% by screening a source of negative charge in the mouth of the channel pore. Other agonists are also charged; examples include the polyamine, spermine+, and the multivalent cations, Ca2+ and Gd3+. All of these interact with Glu648 (33, 34), and further studies may reveal that these agonists also attenuate Ca2+ conductance through TRPV1 receptors.
How does the fixed charge of acidic side chains influence Ca2+ current? Asp646, Glu648, and Glu651 could enhance Ca2+ entry in two ways. First, these residues may create a local surface potential that serves to concentrate Ca2+ in the mouth of the pore. Second, they may facilitate transport by dehydrating Ca2+ in a narrow part of the pore. Which of these mechanisms produces the results shown here is presently unknown.
The experiments presented in this study do not exclude the possibility that additional amino acids may contribute to Ca2+ flux, including acidic and neutral side chains located deeper within the pore. Indeed, site-directed mutagenesis of Tyr671 of the TM6 domain has been shown to severely disrupt Ca2+ permeability, supporting the possibility that other loci are involved in Ca2+ permeation (7). Nonetheless, we show that the acidic residues, Asp646, Glu648, and Glu651, make a significant contribution to the novel, agonist-dependent Ca2+ flux measured in TRPV1 receptors.
Deciphering the role of TRPV1 receptors in sensory neurons is a major focus of pain research, and the emerging role of TRPV1 receptors in regulating vascular tone may open new avenues for the treatment of blood pressure-related illnesses. Thus, the possibility that the size of the Ca2+ current is dynamically regulated by the mode of activation in TRPV1 receptors could provoke a new perspective on drug design, which has hitherto focused on chemicals that alter the gating properties of ion channels rather than their ionic selectivity.
Supplementary Material
Acknowledgments
We thank Steven Harris, Meredith Hoge, and Zhiyuan Li for ancillary assistance. We also thank Drs. David Julius (University of California, San Francisco), Henry Lester (California Institute of Technology), and Thomas Küner (Max-Planck-Institut für medizinische Forschung) for supplying the rat TRPV1 and P2X2, chick nicotinic α4β2, and rat glutamatergic NR1/NR2A cDNAs.
This work was supported, in whole or in part, by National Institutes of Health grants (to T. M. E. and B. S. K.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental text and three supplemental figures.
Footnotes
The abbreviations used are: TRP, transient receptor potential family; BU, bead unit; HEK, human embryonic kidney; MES, 4-morpholineethanesulfonic acid; pS, picosiemens.
References
- 1.Caterina, M. J., Schumacher, M. A., Tominaga, M., Rosen, T. A., Levine, J. D., and Julius, D. (1997) Nature 389 816–824 [DOI] [PubMed] [Google Scholar]
- 2.Venkatachalam, K., and Montell, C. (2007) Annu. Rev. Biochem. 76 387–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Owsianik, G., Talavera, K., Voets, T., and Nilius, B. (2006) Annu. Rev. Physiol. 68 685–717 [DOI] [PubMed] [Google Scholar]
- 4.Nilius, B., Owsianik, G., Voets, T., and Peters, J. A. (2007) Physiol. Rev. 87 165–217 [DOI] [PubMed] [Google Scholar]
- 5.Paus, R., Schmelz, M., Biro, T., and Steinhoff, M. (2006) J. Clin. Investig. 116 1174–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorostkar, M. M., and Boehm, S. (2008) Handb. Exp. Pharmacol. 184 479–527 [DOI] [PubMed] [Google Scholar]
- 7.Mohapatra, D. P., Wang, S. Y., Wang, G. K., and Nau, C. (2003) Mol. Cell. Neurosci. 23 314–324 [DOI] [PubMed] [Google Scholar]
- 8.Oh, U., Hwang, S. W., and Kim, D. (1996) J. Neurosci. 16 1659–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagy, I., and Rang, H. P. (1999) J. Neurosci. 19 10647–10655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung, M. K., Guler, A. D., and Caterina, M. J. (2008) Nat. Neurosci. 11 555–564 [DOI] [PubMed] [Google Scholar]
- 11.Neher, E. (1995) Neuropharmacology 34 1423–1442 [DOI] [PubMed] [Google Scholar]
- 12.Egan, T. M., and Khakh, B. S. (2004) J. Neurosci. 24 3413–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samways, D. S., and Egan, T. M. (2007) J. Gen. Physiol. 129 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneggenburger, R., Zhou, Z., Konnerth, A., and Neher, E. (1993) Neuron 11 133–143 [DOI] [PubMed] [Google Scholar]
- 15.Frings, S., Hackos, D. H., Dzeja, C., Ohyama, T., Hagen, V., Kaupp, U. B., and Korenbrot, J. I. (2000) Methods Enzymol. 315 797–817 [DOI] [PubMed] [Google Scholar]
- 16.Vernino, S., Rogers, M., Radcliffe, K. A., and Dani, J. A. (1994) J. Neurosci. 14 5514–5524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jatzke, C., Watanabe, J., and Wollmuth, L. P. (2002) J. Physiol. (Lond.) 538 25–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khakh, B. S., Bao, X. R., Labarca, C., and Lester, H. A. (1999) Nat. Neurosci. 2 322–330 [DOI] [PubMed] [Google Scholar]
- 19.Virginio, C., MacKenzie, A., Rassendren, F. A., North, R. A., and Surprenant, A. (1999) Nat. Neurosci. 2 315–321 [DOI] [PubMed] [Google Scholar]
- 20.Skeberdis, V. A., Chevaleyre, V., Lau, C. G., Goldberg, J. H., Pettit, D. L., Suadicani, S. O., Lin, Y., Bennett, M. V., Yuste, R., Castillo, P. E., and Zukin, R. S. (2006) Nat. Neurosci. 9 501–510 [DOI] [PubMed] [Google Scholar]
- 21.Sobczyk, A., and Svoboda, K. (2007) Neuron 53 17–24 [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Martinez, C., Morenilla-Palao, C., Planells-Cases, R., Merino, J. M., and Ferrer-Montiel, A. (2000) J. Biol. Chem. 275 32552–32558 [DOI] [PubMed] [Google Scholar]
- 23.Voets, T., Prenen, J., Vriens, J., Watanabe, H., Janssens, A., Wissenbach, U., Bodding, M., Droogmans, G., and Nilius, B. (2002) J. Biol. Chem. 277 33704–33710 [DOI] [PubMed] [Google Scholar]
- 24.Welch, J. M., Simon, S. A., and Reinhart, P. H. (2000) Proc. Natl. Acad. Sci. U. S. A. 97 13889–13894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bordo, D., and Argos, P. (1991) J. Mol. Biol. 217 721–729 [DOI] [PubMed] [Google Scholar]
- 26.Premkumar, L. S., Agarwal, S., and Steffen, D. (2002) J. Physiol. (Lond.) 545 107–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nilius, B. (2007) Biochim. Biophys. Acta. 1772 805–812 [DOI] [PubMed] [Google Scholar]
- 28.Wu, Z. Z., Chen, S. R., and Pan, H. L. (2005) J. Biol. Chem. 280 18142–18151 [DOI] [PubMed] [Google Scholar]
- 29.Zeilhofer, H. U., Kress, M., and Swandulla, D. (1997) J. Physiol. (Lond.) 503 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng, W., Yang, F., Takanishi, C. L., and Zheng, J. (2007) J. Gen. Physiol. 129 191–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hille, B. (2004) Ionic Channels of Excitable Membranes, pp. 445–450, 3rd Ed., Sinauer Associates, Inc., Sunderland, MA
- 32.Jatzke, C., Hernandez, M., and Wollmuth, L. P. (2003) J. Physiol. (Lond.) 549 439–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahern, G. P., Wang, X., and Miyares, R. L. (2006) J. Biol. Chem. 281 8991–8995 [DOI] [PubMed] [Google Scholar]
- 34.Ahern, G. P., Brooks, I. M., Miyares, R. L., and Wang, X. B. (2005) J. Neurosci. 25 5109–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen, S. L., Chait, B. T., and MacKinnon, R. (1998) Science 280 69–77 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.