

Abstract
In vitro, cytochrome b5 modulates the rate of cytochrome P450-dependent mono-oxygenation reactions. However, the role of this enzyme in determining drug pharmacokinetics in vivo and the consequential effects on drug absorption distribution, metabolism, excretion, and toxicity are unclear. In order to resolve this issue, we have carried out the conditional deletion of microsomal cytochrome b5 in the liver to create the hepatic microsomal cytochrome b5 null mouse. These mice develop and breed normally and have no overt phenotype. In vitro studies using a range of substrates for different P450 enzymes showed that in hepatic microsomal cytochrome b5 null NADH-mediated metabolism was essentially abolished for most substrates, and the NADPH-dependent metabolism of many substrates was reduced by 50–90%. This reduction in metabolism was also reflected in the in vivo elimination profiles of several drugs, including midazolam, metoprolol, and tolbutamide. In the case of chlorzoxazone, elimination was essentially unchanged. For some drugs, the pharmacokinetics were also markedly altered; for example, when administered orally, the maximum plasma concentration for midazolam was increased by 2.5-fold, and the clearance decreased by 3.6-fold in hepatic microsomal cytochrome b5 null mice. These data indicate that microsomal cytochrome b5 can play a major role in the in vivo metabolism of certain drugs and chemicals but in a P450- and substrate-dependent manner.
Microsomal cytochrome b5 is a 15.2-kDa hemoprotein, located in the endoplasmic reticulum with its obligate electron donor, cytochrome b5 reductase. This protein is involved in fatty acid desaturation, reactivation of methemoglobin to hemoglobin, and electron transfer into the cytochrome P450 system (1–4).
The role of cytochrome b5 in cytochrome P450 monooxygenase reactions has been controversial for nearly 40 years (5, 6). Depending on the cytochrome P450 involved, the experimental conditions, and the substrate utilized, cytochrome b5 has been shown to stimulate or inhibit cytochrome P450 reactions (2, 6). Indeed, in the case of cytochrome P450 CYP2B4, activity can be both stimulated or inhibited by the presence of the enzyme, depending on the concentration of cytochrome b5 (7, 8). Cytochrome b5 has been shown to affect the in vitro activities of a wide range of human P450s, such as CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4, as well as the metabolism of an extensive number of commonly used drugs (9, 10). Although the mechanism(s) of the stimulatory effect of cytochrome b5 on P450 metabolism appear complex, it is widely accepted that the P450 catalytic cycle involves the sequential transfer of two electrons from NADPH via P450 oxidoreductase (POR).2 Hildebrandt and Estabrook (5) concluded that cytochrome b5/cytochrome b5 reductase was able to supply the second (rate-limiting) electron from NADH and possibly in a more rapid manner. However, recent data from Waskell et al. (8) suggest that cytochrome b5 and POR are capable of reducing P450s at much the same rate, albeit that overall catalysis occurs more slowly with POR, leading to speculation that the oxyferrous form of P450 may exist in different conformations with POR or cytochrome b5 (11).
A noncatalytic (i.e. allosteric) role has also been proposed for the interaction of cytochrome b5 with cytochrome P450, based on reports that apocytochrome b5 (i.e. without the heme prosthetic group) can stimulate P450 catalysis (1, 12, 13). Although these findings have been debated (14–16) and appear to depend upon the enzyme studied, the mechanism(s) underlying such allosteric effects remains uncertain. It does seem clear however, that cytochrome b5 binding can cause conformational changes to the substrate access channel and binding pocket in the P450 enzyme (1, 17). In many cases, allosteric stimulation increases P450 activity by a relatively small amount, and the physiological relevance of this effect, if any, is unclear (6).
All studies on cytochrome b5 to date have been carried out in vitro; the in vivo relevance of these findings has remained unknown (9, 10, 17–20). Although the prediction of in vivo drug pharmacokinetic data obtained from in vitro experiments rarely takes the potential contribution of cytochrome b5 into account, a recent study using recombinant human P450s did find that using cytochrome b5 yielded a more accurate estimate of intrinsic clearance (21).
In order to establish the relevance of the in vitro observations to those in vivo and to evaluate whether variability in cytochrome b5 levels may affect rates of drug metabolism, we have conditionally deleted microsomal cytochrome b5 in the liver. Using hepatic microsomal cytochrome b5 null (HBN) mice, we demonstrate that the in vitro rate of NADPH- and NADH-dependent metabolism of a range of both model substrates and probe drugs is markedly changed. Furthermore, we show that when probe drugs are administered to mice, significant changes in drug pharmacokinetics occur.
EXPERIMENTAL PROCEDURES
Chemicals—All reagents unless stated were purchased from Sigma. NADPH was obtained from Melford Laboratories (Ipswich, UK). Bufuralol, 1′-hydroxybufuralol, 7-benzyloxy-4-trifluoromethylcoumarin (BFC), 7-methoxy-4-trifluoromethylcoumarin (MFC), 7-hydroxy-4-trifluoromethylcoumarin, and hydroxytolbutamide were purchased from BD Gentest (Cowley, UK). Midazolam, 1-hydroxymidazolam, and 4-hydroxymidazolam were kind gifts from Roche Applied Science, and 1-hydroxymetoprolol and O-demethylmetoprolol were generous gifts from Astra Häsle (Mölndal, Sweden).
Generation of Hepatic Microsomal Cytochrome b5 Null Mice—A targeting vector was constructed from an 18-kb DNA fragment, produced by fusing overlapping PCR fragments generated from mouse 129/Ola genomic DNA (supplemental Fig. 1A), containing exons 2–5 of the mouse cytochrome b5 gene. A cassette, flanked by same orientation loxP sites and containing a selectable marker (neomycin), driven by the herpes simplex thymidine kinase promoter, was cloned into a BclI site in intron 1, and a third loxP site was cloned into a KpnI site in intron 5. The construct was checked by PCR and sequencing and transfected into GK129/1 embryonic stem cells by electroporation; the embryonic stem cells were subsequently cultured in 96-well plates under G418 selection. G418-resistant clones were screened for specific homologous recombination by Southern blot analysis, using BglII and an 800-bp PCR fragment generated using 5′-GGCACAACACCAATTATTTGTC-3′ and 5′-GACAGTCCTTAACACAAGCTC-3′ as forward and reverse primers, respectively. Two correctly targeted embryonic stem cell clones (Cytb +/lox5) were expanded, injected into C57BL/6 blastocysts, and transferred into pseudopregnant mice. Male chimeric mice were bred to C57BL/6 mice, and heterozygous offspring were screened by Southern blot and multiplex PCR to confirm germ line transmission of the Cytb +/lox5 genotype (supplemental Fig. 1B) using the following primer set: 1) forward primer, 5′-CCAATGGTCTCTCCTTGGTC-3′;2) lox/neomycin reverse primer, 5′-CAATAGCAGCCAGTCCCTTC-3′; 3) wild-type reverse primer, 5′-GATGGAGTTCCCCGATGAT-3′.
Mouse Breeding and Maintenance—Cytb5lox/+ mice were crossed to produce homozygous Cytb5lox/lox mice and maintained by random breeding on a 129P2 × C57BL/6 genetic background. Cytb5lox/lox mice were crossed with a transgenic mouse line expressing Cre recombinase under the control of the hepatocyte-specific rat albumin promoter (CreALB) (22) on a C57BL/6 background, and Cytb5lox/+::CreALB offspring were backcrossed with Cytb5lox/lox mice to generate liver-specific microsomal cytochrome b5 conditional knock-out mice (HBN; Cytb5lox/lox::CreALB) and control (wild-type, Cytb5lox/lox) mice. The HBN line was thereafter maintained by random intercrossing of these two lines. The presence of the CreALB transgene was determined as previously described (23).
All mice were maintained under standard animal house conditions, with free access to food and water, and a 12-h light/12-h dark cycle. All animal work was carried out on male 10-week-old mice in accordance with the Animal Scientific Procedures Act (1986) and after local ethical review.
In Vivo Drug Treatments—HBN and wild-type mice were administered the following drugs either concomitantly as a mixture or individually: chlorzoxazone (5 mg/kg), metoprolol (2 mg/kg), midazolam (5 mg/kg), phenacetin (5 mg/kg), and tolbutamide (5 mg/kg), dissolved in mixture buffer (5% ethanol, 5% DMSO, 35% polyethylene glycol 200, 40% phosphate buffered saline, and 15% water), by intravenous injection or orally by gavage.
Preparation of Microsomes—Microsomes were prepared from wild-type and HBN mouse tissues, using 0.3–0.5 g of tissue, by a modified method of Meehan et al. (24), using sonication instead of mechanical homogenization (25). Microsomal protein concentrations were determined using the Bio-Rad protein assay reagent. POR activity was estimated by NADPH-dependent cytochrome c reduction (26). Microsomes were stored at –70 °C until required.
Immunoblotting—Western blot analysis was carried out as previously described using polyclonal antisera raised against human POR (27); rat cytochrome b5; rat CYP2A1, CYP2B1, CYP2C6, CYP3A1, and CYP4A1 (28); or human full-length CYP2A4, CYP2D6, and CYP2E1 (25, 29). Polyclonal antiserum to rat cytochrome b5 reductase and a monoclonal antibody raised against rat CYP1A1 were also used.3 The polyclonal antiserum to cytochrome b5 oxidoreductase was a kind gift from Dr. Hao Zhu (Kansas University Medical Center, Kansas City, KS). Immunoreactive proteins were detected using polyclonal goat anti-rabbit, anti-mouse, or anti-sheep horseradish peroxidase immunoglobulins as secondary antibodies (Dako, Ely, UK) and visualized using Immobilon™ chemiluminescent horseradish peroxidase substrate (Millipore, Watford, UK) and a FUJIFILM LAS-3000 mini-imaging system (Fujifilm UK Ltd.). Densitometric analysis was performed using Multi Gauge version 2.2 software (Fujifilm UK Ltd.).
Generic Microsomal Incubations—Microsomal incubations were carried out in triplicate in 50 mm Hepes, pH 7.4, 30 mm MgCl2 containing mouse liver microsomes and substrate prewarmed to 37 °C before initiation of the reaction by the addition of either NADPH or NADH to a final concentration of 0.5 or 1 mm, respectively.
Fluorogenic Assay Incubations—Assays were performed in a final volume of 150 μl using white 96-well plates and the following substrate and microsome concentrations: BFC, 50 μm substrate, 20 μg of mouse liver microsomes; EFC, 40 μm substrate, 15 μg of mouse liver microsomes; MFC, 180 μm of substrate, 15 μg of mouse liver microsomes; ethoxyresorufin (ER) and benzoxyresorufin (BR), 1 μm substrate, 11.25 μg of mouse liver microsomes. Reactions were measured in real time for 3 min, using the recommended excitation and emission wavelengths for each probe using a Fluroskan Ascent FL plate reading fluorimeter (Labsystems, UK). Turnover rates were calculated using authentic metabolite standards (7-hydroxy-4-trifluoromethylcoumarin for BFC, EFC, and MFC assays and resorufin for ER and BR assays).
NADH-mediated Incubations for HPLC or Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Analysis—NADH-mediated reactions were performed in triplicate using the following conditions: bufuralol, 300 μm (final concentration), 20 μg of mouse liver microsomes in a final volume of 150 μl for 6 min; chlorzoxazone, 1 mm (final concentration) and 20 μg of mouse liver microsomes in a final volume of 150 μl for 15 min; midazolam, 50 μm (final concentration) and 25 μgof mouse liver microsomes in a final volume of 150 μl for 9 min; metoprolol, 800 μm (final concentration) and 30 μg of mouse liver microsomes in a final volume of 150 μl for 60 min; phenacetin, 200 μm (final concentration) and 20 μg of mouse liver microsomes in a final volume of 100 μl for 9 min; tolbutamide, 800 μm (final concentration) and 30 μg of mouse liver microsomes in a final volume of 150 μl for 60 min. Assays were stopped by the addition of either 0.5 assay volumes (for bufuralol, chlorzoxazone, and tolbutamide assays) or 1 assay volume (for midazolam and phenacetin assays) of ice-cold methanol and incubated on ice for 10 min.
Kinetic Determinations—Assays to determine the apparent kinetic parameters were performed in triplicate with wild-type and HBN liver microsomes under conditions of linearity for time and protein (data not shown) using the same buffer/NADPH conditions as described above, with the following concentrations of substrates: chlorzoxazone, 10–1000 μm; phenacetin, 1.7–150 μm; midazolam, 0.9–75 μm; metoprolol, 10–1000 μm; tolbutamide, 10–1000 μm (12 concentration points/determination). Metabolites were detected by LC-MS/MS as described in the supplemental materials. The data generated were analyzed by nonlinear regression using the Michaelis-Menten equation (chlorzoxazone, phenacetin, midazolam, and metoprolol), whereas the Hill equation (tolbutamide) (Equation 1) was used to determine the kinetic parameters Vmax,S50, and the Hill coefficient (n).
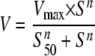 |
(Eq. 1) |
In Vivo Pharmacokinetics—Whole blood (10 μl) was taken from the tail vein at intervals after drug administration and transferred into a tube containing heparin (10 μl, 15 IU/ml). Internal standard solution (10 μl; 500 ng of caffeine and 500 ng of resorpine) was added to each tube. Protein precipitation was carried out by adding methanol (75 μl), followed by 8% sulfosalicylic acid (55 μl). Samples were mixed for 1 min and centrifuged at 13,000 rpm for 5 min, and the supernatant was analyzed by HPLC.
The range of concentrations for the standard curves was constructed for quantifying blood levels by spiking blank blood samples with known amounts of chlorzoxazone, metoprolol, midazolam, phenacetin, and tolbutamide. Extraction and protein precipitation were carried out as outlined above for the test samples.
Analysis of in Vitro and in Vivo Data—Average rates of metabolism were calculated for each triplicate incubation of mouse liver microsomes from each genotype (n = 6), and these data were then used to calculate p values using an unpaired t test (available on the World Wide Web). Pharmacokinetic parameters were calculated using WinNonLin software, version 3.1. A simple noncompartmental model was used to calculate area under the curve (AUC), terminal half-life, maximum plasma concentration (Cmax), and clearance. Details of assays and separation conditions for HPLC and LC-MS/MS are given in the supplemental materials.
RESULTS
Hepatic Microsomal Cytochrome b5 Null Mice—Mice nulled for hepatic microsomal cytochrome b5 (Cytb5lox/lox::CreALB) were born at the expected Mendelian ratio, developed normally, and displayed no overt phenotype when compared with their wild-type littermates (Cytb5lox/lox). Livers from adult HBN mice looked similar to those of wild-type animals, with no gross changes in general morphology or pathology noted. To establish whether hepatic microsomal cytochrome b5 protein had been deleted specifically, microsomes from liver, lung, kidney, and small and large intestine were immunoblotted for cytochrome b5 expression (Fig. 1A). Cytochrome b5 protein was virtually undetectable in microsomes from the livers of HBN mice; however, occasionally, a very faint band was observed in some samples, possibly originating from liver cells other than hepatocytes. There were no discernible differences in cytochrome b5 expression between the HBN and wild-type mice in the other organs examined. No cytochrome b5 protein could be detected in microsomes from the large intestine of either genotype (data not shown).
FIGURE 1.

Characterization of protein expression in microsomal cytochrome b5 hepatic null mice. A, immunoblot analysis for cytochrome b5 expression in liver, kidney, lung, and small intestine microsomes (5 μg of protein/lane) from Cytb5lox/lox::CreAlb (HBN) and Cytb5lox/lox (wild type), as detailed under “Experimental Procedures.” B, immunoblot analysis for expression of cytochrome P450-dependent monooxygenase components in liver microsomes (5 μg of protein/lane) from Cytb5lox/lox::CreAlb (HBN) and Cytb5lox/lox (wild type), as detailed under “Experimental Procedures.” C, densitometric analysis of the liver microsome blots. Wild-type expression was fixed at 100%, and percentage difference was calculated for cytochrome b5 liver null (HBN) samples. Values were normalized for loading against Ponceau Red staining.
In order to investigate whether concomitant changes in P450 expression had occurred, three representative liver microsomal samples of each genotype were analyzed for P450 enzyme profiles, POR, and cytochrome b5 reductase levels (Fig. 1B). Although the expression of some P450s was slightly elevated in the HBN mice, these changes were not statistically significant. Cytochrome b5 reductase level was slightly increased when the immunoreactive bands were quantified by densitometry (Fig. 1C). A slight reduction in Cyp2d expression was also observed. As can be seen from supplemental Fig. 2A, there was no difference observed in the amount of spectrally active P450 in the HBN as compared with wild-type liver microsomes, with both samples containing ∼0.15 nmol of P450/mg of microsomal protein. POR protein levels were also essentially unchanged in HBN compared with wild-type mice, and although a slight increase in POR activity in HBN mice was measured using cytochrome c as substrate (228 ± 65 versus 180 ± 26 nmol cytochrome c reduced/min/mg), this change was not significant.
In Vitro Cytochrome P450 Activities—Six commonly used P450 model substrates were used to establish if there were any in vitro differences in metabolism between liver microsomes from HBN and wild-type mice: BFC (CYP3A), MFC (CYP2C), EFC (CYP1A/2B), ER (CYP1A1/2, CYP1B1), BR (CYP2B/3A), and bufuralol (CYP2D) (30–36). Assays were performed at substrate concentrations of ∼5 times Km (mouse), as defined by the literature, or data were generated with recombinant mouse P450s (data not shown). With NADPH as electron donor, significantly lower turnover rates were measured in HBN mice relative to wild-type samples for all substrates tested. The NADPH-mediated rates in HBN liver microsomes, expressed relative to wild-type samples, were as follows: ER (73%), bufuralol (44%), BR (34%), BFC (19%), MFC (11%), and EFC (10%) (Fig. 2, supplemental Table 3). The addition of recombinant cytochrome b5 to HBN microsomes led to a concentration-dependent increase in BFC or chlorzoxazone metabolism, restoring activities to wild-type levels, demonstrating that the change in metabolism was due to the absence of cytochrome b5. Cytochrome b5 alone had no activity toward BFC or chlorzoxazone (supplemental Fig. 2, B and C).
FIGURE 2.

NADPH- and NADH-dependent cytochrome P450 activities in hepatic microsomes from wild-type and microsomal cytochrome b5 hepatic null mice. Assays were performed in triplicate as described under “Experimental Procedures.” Each bar represents mean ± S.D. from six mouse liver microsome preparations. Black bars represent NADPH-mediated, and white bars NADH-mediated activities, respectively. *, p ≤ 0.05; **, p ≤ 0.005; ***, p ≤ 0.001.
NADH could support monooxygenase activity for all model substrates in the wild-type samples, although the activity relative to NADPH was highly substrate-dependent, ranging from 4% (BR) to 74% (ER) (Fig. 2 and supplemental Table 3). All NADH-catalyzed reaction rates with the exception of BR were significantly lower in the HBN samples compared with wild type, with barely detectable activities using BFC (0.66%), EFC (0.79%), and MFC (0.28%) as substrates (Fig. 2 and supplemental Table 3). The metabolism of ER and bufuralol was also reduced, to 52 and 28% of wild-type values, respectively.
The NADH-mediated in vitro turnovers of the probe drugs chlorzoxazone (Cyp2e1), metoprolol (Cyp2d), midazolam (Cyp2c, Cyp3a), tolbutamide (Cyp2c), and phenacetin (Cyp1a) (37–41) were also examined with liver microsomes from both HBN and wild-type mice. NADH supported the metabolism of all substrates examined in wild-type liver microsomes, with the exception of tolbutamide. Rates observed were 601 ± 152 (chlorzoxazone 6′-hydroxylation), 128 ± 60 (metoprolol O-demethylation), 17.6 ± 0.2 (metoprolol α-hydroxylation), 17.1 ± 8.0 (midazolam 1′-hydroxylation), 9.1 ± 3.8 (midazolam 4-hydroxylation), and 244 ± 63 pmol/min/mg (phenacetin hydroxylation). These rates observed in wild-type liver microsomes were significantly lower than those observed using NADPH as co-factor, ranging from 7 to 29% (O-demethylmetoprolol and acetaminophen production, respectively) of the NADPH rate. Furthermore, the NADH-dependent turnover rates observed in HBN liver microsomes (177 ± 64 (chlorzoxazone 6′-hydroxylation), 90 ± 21 (metoprolol O-demethylation), 7.6 ± 2.2 (metoprolol α-hydroxylation), 4.3 ± 2.0 (midazolam 1′-hydroxylation), 2.1 ± 2.7 (midazolam 4-hydroxylation), and 67 ± 18 pmol/min/mg (phenacetin hydroxylation)) were significantly lower than the NADH-dependent wild-type rates (chlorzoxazone (29% reduction), metoprolol (O-demethyl 71%, α-hydroxy 44%, acid 83% reduction), midazolam (1′- and 4-hydroxy 25% reduction), and phenacetin (29% reduction)).
In Vitro Kinetics—The apparent kinetic parameters of the probe drugs using NADPH as co-factor were determined in wild-type and HBN liver microsomes, and metabolism of chlorzoxazone, phenacetin, midazolam, and metoprolol followed standard Michaelis-Menten kinetics (Table 1). The Michaelis constant for chlorzoxazone 6-hydroxylation, midazolam 1′-hydroxylation, phenacetin oxidation, and metoprolol α-hydroxylation were unchanged by the removal of microsomal cytochrome b5. Conversely, for those reactions, the Vmax was markedly reduced in the HBN samples, ranging from a 2-fold (phenacetin oxidation) to a 6-fold (metoprolol α-hydroxylation) decrease. Midazolam 4-hydroxylation and metoprolol O-demethylation reactions again showed decreased Vmax values for the HBN samples (20- and 3-fold, respectively); however, the Km values were marginally affected, with the HBN samples exhibiting a 1.7-fold reduction in Km for midazolam 4-hydroxylation and a 1.8-fold increase in Km for metoprolol O-demethylation as compared with wild type. The data obtained for tolbutamide hydroxylation could not be fitted using the Michaelis-Menten equation but did conform to the Hill equation. Interestingly, the HBN sample exhibited 68-fold higher Km for tolbutamide as well as a 5-fold reduction in Vmax as compared with wild type. The Hill coefficient for both wild type and HBN were similar (0.38 versus 0.29) and indicative of tolbutamide binding in a negatively cooperative manner. The alternative of a 2 Km,2 Vmax fit was considered for these data; however, when plotted in such a manner, the Km1 parameter for both wild type and HBN was subject to such a large error as to render it meaningless.
TABLE 1.
Kinetic analyses of the metabolism of probe drugs by hepatic microsomes from HBN mice and wild-type controls
Assays were performed in triplicate for each concentration of substrate. S.D. values given are from the fit of the curve as calculated using the Michaelis-Menten equation (GraFit version 5 (Erithacus Software, Horley, UK)). For tolbutamide, the Hill equation (GraFit version 5) was used to fit the data, giving Hill coefficients (n) of 0.38 and 0.29 for wild type and HBN, respectively.
|
Substrate
|
Metabolite
|
Wild type
|
HBN
|
||
|---|---|---|---|---|---|
| Km | Vmax | Km | Vmax | ||
| μm | pmol/min/mg | μm | pmol/min/mg | ||
| Chlorzoxazone | 6-Hydroxychlorzoxazone | 47 ± 9 | 1930 ± 100 | 53 ± 6 | 387 ± 11 |
| Midazolam | 1′-Hydroxymidazolam | 2.1 ± 0.3 | 389 ± 15 | 2.0 ± 0.3 | 116 ± 4 |
| Midazolam | 4-Hydroxymidazolam | 13.3 ± 0.4 | 146 ± 2 | 7.7 ± 0.9 | 7.3 ± 0.3 |
| Phenacetin | Acetaminophen | 5.6 ± 1.0 | 133 ± 6 | 4.1 ± 0.7 | 77 ± 3 |
| Metoprolol | α-Hydroxymetoprolol | 84 ± 7 | 483 ± 14 | 99 ± 9 | 76 ± 3 |
| Metoprolol | O-Desmethylmetoprolol | 86 ± 9 | 1650 ± 2 | 157 ± 14 | 531 ± 21 |
| Tolbutamide | Hydroxytolbutamide | 11200 ± 3400 | 211 ± 17 | 165 ± 48 | 44 ± 165 |
To ensure that the observed changes in P450 activity were not a consequence of perturbations of the phospholipid bilayer, fatty acid profile analysis was carried out on endoplasmic reticulum membrane fractions from the livers of both wild type and HBN mice. No significant changes in the levels of fatty acids were observed. In addition, hepatic lipid levels were unchanged, as determined by Oil Red O staining (supplemental Fig. 3).
In Vivo Pharmacokinetics of Probe Drugs in Wild-type and HBN Mice—We next investigated the effect of the absence of hepatic microsomal cytochrome b5 on the in vivo pharmacokinetics of the probe drugs in wild-type and HBN mice. Initially, mice were administered chlorzoxazone, metoprolol, midazolam, tolbutamide, and phenacetin either as an intravenous or oral mixture. Previous work with these drugs in wild-type mice, individually and as a mixture, had demonstrated that simultaneous delivery did not result in altered pharmacokinetics as compared with single administration (not shown). Following dosing of the drug cassette to wild-type and HBN mice, quantification of individual drugs and their metabolites was carried out by LC-MS/MS. The complete set of pharmacokinetic parameters for each drug is summarized in supplemental Tables 4 (intravenous) and 5 (oral), and the elimination profiles of the drugs are shown in Fig. 3 (intravenous) and supplemental Fig. 4 (oral).
FIGURE 3.

Pharmacokinetic disposition of an intravenously administered P450 drug mixture in wild-type and hepatic microsomal cytochrome b5 null mice. A P450 drug mixture containing chlorzoxazone (5 mg/kg), metoprolol (2 mg/kg), midazolam (5 mg/kg), phenacetin (5 mg/kg), and tolbutamide (5 mg/kg) was administered intravenously at the indicated doses to wild-type (black circles) or HBN (open circles) mice, and blood samples were taken at timed intervals to determine the in vivo pharmacokinetic parameters of the parent compounds. *, p ≤ 0.05; **, p ≤ 0.005; ***, p ≤ 0.001, where n = 5.
Intravenous Pharmacokinetics—Following intravenous administration of the drug mixture, wild-type mice showed a higher concentration of parent drug in the blood at the early time points (0–60 min) compared with the HBN mice for all drugs in the mixture. In contrast, from 2 h, the average concentration of parent drug was statistically much lower in the wild-type than in HBN mice for most of the compounds and remained so for the duration of the experiment. Pharmacokinetic analysis for each drug showed that the clearance was consistently faster, and the AUC was consistently lower (except in the case of midazolam) in wild-type compared with HBN mice. Furthermore, the terminal half-life for phenacetin, tolbutamide, chlorzoxazone, and midazolam was statistically different between the two lines, being significantly longer in HBN mice, increasing from 53 to 168 min, 197 to 321 min, 92 to 257 min, and 83 to 329 min, respectively (Table 2, Fig. 3, and supplemental Table 4). For metoprolol, although a 2.7-fold increase of the terminal half-life was found in HBN mice, this did not reach statistical significance (p = 0.1).
TABLE 2.
In vivo pharmacokinetic data from HBN mice and wild-type controls dosed with a drug mixture
Adult male HBN and WT mice were administered a drug mixture intravenously; serial blood samples were collected over an 8-h period and analyzed for plasma drug concentrations by LC-MS/MS and key pharmacokinetic parameters calculated. Absolute oral bioavailability was calculated by dividing AUC (0 → ∞, oral) by AUC (0 → ∞, intravenous) (supplemental Tables 4 and 5).
|
Drug
|
Terminal half-life (intravenous)
|
Bioavailability (%)
|
||
|---|---|---|---|---|
| Wild type | HBN | Wild type | HBN | |
| Midazolam | 83 ± 15 | 329 ± 132 | 14 | 55 |
| Phenacetin | 53 ± 11 | 168 ± 56 | 0.6 | 0.8 |
| Metoprolol | 96 ± 13 | 260 ± 65 | 0.3 | 0.3 |
| Chlorzoxazone | 92 ± 19 | 257 ± 76 | 3.7 | 1.9 |
| Tolbutamide | 197 ± 37 | 321 ± 25 | 95 (154)a | 47 (135)a |
Comparative data where tolbutamide was dosed as a single agent.
The major metabolites of metoprolol (O-demethylmetoprolol), phenacetin (acetaminophen), and midazolam (1-OH midazolam) were also measured in the serum of HBN and wild-type mice after intravenous administration; the results are presented in supplemental Fig. 5A. Interestingly, the overall profiles of metabolite production in HBN mice were shifted to the right, consistent with a delayed metabolism of the parent compound (Fig. 3 and supplemental Fig. 5A). Tolbutamide and chlorzoxazone metabolites were below the limit of detection at all time points.
Oral Pharmacokinetics—Following oral administration of the drug mixture, differences in the elimination profiles of all of the drugs were observed between wild-type and HBN mice. In the case of midazolam, phenacetin, and metoprolol, AUC and Cmax were consistently higher in HBN mice, whereas the clearance was slower (supplemental Fig. 4, supplemental Table 5). For midazolam, AUC and Cmax were significantly increased from 34 to 124 min·μg/ml and from 0.4 to 1.0 μg/ml, respectively, whereas clearance was decreased from 149 to 41 ml/min/kg (supplemental Table 5). The phenacetin elimination profile also showed significant changes, with Cmax increasing from 0.06 to 0.09 μg/ml in HBN and clearance decreasing from 688 to 550 ml/min/kg. Although the phenacetin AUC increased from 1.5 to 1.8 min·μg/ml, this change was not significant. In the case of metoprolol, similar trends were observed in Cmax and AUC; however, these did not reach statistical significance. In contrast to the observations above, the alterations in the pharmacokinetic parameters for tolbutamide gave significantly lower AUC and Cmax values and a (nonsignificant) higher clearance rate, relative to wild-type animals (supplemental Fig. 4 and supplemental Table 5). This unexpected effect appeared to be a consequence of using the drug mixture (see below). Interestingly, although the terminal half-life for chlorzoxazone was significantly decreased in HBN mice from 194.0 to 46 min, the decreased AUC and Cmax and increased clearance in HBN mice were not significantly different (supplemental Table 5). Additionally, there were no significant differences between HBN and wild-type mice in the plasma concentration of chlorzoxazone at any of the time points analyzed (supplemental Fig. 4).
Administration of the probe drugs individually to HBN and wild-type mice yielded essentially the same elimination profiles for phenacetin, metoprolol, midazolam, and chlorzoxazone, compared with those obtained from use of the drug mixture (data not shown). However, when tolbutamide was administered singly (supplemental Fig. 4A, inset), the elimination profiles were found to be reversed in comparison with that obtained from the drug mixture (i.e. significantly increased AUC and Cmax and significantly decreased clearance in HBN mice). Similar to data obtained with the drug mixture administered intravenously, the pattern of metabolite production in the HBN animals showed a delay in maximal levels of circulating metabolites (supplemental Fig. 5).
Using the oral and intravenous dosing data, percentage bioavailability was calculated (Table 2). HBN animals showed a 4-fold increase in midazolam bioavailability, whereas for phenacetin, metoprolol, and chlorzoxazone, bioavailability was very low in the wild-type animals (<4%) and unaltered in the nulls. When dosed as part of the drug mixture, tolbutamide bioavailability in the HBN mice was decreased by ∼50% as compared with wild type; however, when orally dosed as a single agent and recalculated, both wild-type and HBN animals showed >100% bioavailability.
DISCUSSION
All previously published work on the effects of cytochrome b5 on NADPH-dependent P450 activity has been carried out in vitro. In order to establish the in vivo role of this hemoprotein in drug disposition, we have generated a mouse where this enzyme has been conditionally inactivated in the liver.
To exclude the possibility that a global deletion of microsomal cytochrome b5 would be lethal, a conditional targeting strategy was used. Mice deleted for hepatic microsomal cytochrome b5 developed normally, with the Cytb5lox/lox::CreALB genotypes being found in Mendelian proportions. No alterations in liver morphology, hepatic lipid content, or hepatic serum markers were observed.
The hepatic deletion of microsomal cytochrome b5 was shown to be both specific and essentially complete by Western blotting (Fig. 1A) and, importantly, unlike the hepatic deletion of cytochrome P450 reductase (23, 42), did not result in significantly increased expression of hepatic P450 proteins (Fig. 1, B and C). These data indicate that at the time points studied, a reduction in P450 activity, as a consequence of microsomal cytochrome b5 deletion, is not compensated by an increase in P450 expression. The use of a range of both model substrates and probe drugs clearly demonstrated that the absence of microsomal cytochrome b5 significantly attenuated P450-mediated metabolism in vitro (Fig. 2 and Table 1). The extent of suppression of enzyme activity varied markedly between substrates, as has been previously reported (2, 6). For all fluorocoumarin substrates employed, there was a highly significant decrease in NADPH-mediated P450 metabolism in liver microsomes from the HBN mice (as high as 90% for EFC), whereas for the two resorufin analogues and bufuralol, the change was still significant (at least 50% decrease for each compound) but less marked. This variability in cytochrome b5 effects was further illustrated by the use of a series of probe drugs, where the kinetic parameters defining the in vitro metabolism were markedly changed (Table 1). For all substrates, with the exception of tolbutamide, the data followed Michaelis-Menten kinetics, and Km values were essentially unchanged in the HBN samples. This suggests that cytochrome b5 binding does not cause a conformational change in the structure of the P450s responsible for this metabolism. In contrast, in both wild type and HBN samples, the data for tolbutamide oxidation did not fit Michaelis-Menten kinetics, exhibiting negatively cooperative binding. Furthermore, the removal of cytochrome b5 had a pronounced effect on tolbutamide binding, decreasing the Km by 68-fold, indicating that the presence of cytochrome b5 does affect tolbutamide binding.
In all cases, Vmax values were substantially reduced by the removal of cytochrome b5 (range 1.7–20-fold lower for acetaminophen and 4-hydroxymidazolam production, respectively), implying that the presence of cytochrome b5 is critical for optimal turnover in the P450s investigated. Interestingly, the effects of cytochrome b5 were different dependent on the site of metabolism (e.g. with midazolam, the Vmax for oxidation at the 1- versus 4-hydroxy sites was 29 versus 5% of the control, respectively). A similar effect was also seen for metoprolol oxidation (Table 1), indicating that cytochrome b5 effects show specificity for different sites of oxidation.
These findings provide strong genetic evidence that the modulatory effects of cytochrome b5 are both substrate- and P450-dependent and that the activities of many P450 enzymes are affected by this protein. Similar reductions in P450 activities (60–70%) have also been demonstrated using antibodies to cytochrome b5, both in microsomal fractions and with purified protein (43, 44). Furthermore, the addition of recombinant membrane-bound cytochrome b5 to HBN liver microsomes led to a concentration-dependent increase of catalytic activities toward BFC and chlorzoxazone, indicating that the loss of activity in HBN samples was indeed due to the absence of cytochrome b5 (supplemental Fig. 2, B and C). The in vitro data generated from the probe drugs suggests that in general, the absence of cytochrome b5 results in reduced rates of P450-mediated metabolism.
It is interesting to note that in some cases the fluorocoumarin substrates and tolbutamide NADH-mediated metabolism were essentially abolished in HBN mice, whereas for the other compounds, residual activity remained (e.g. in the case of ER or BR), indicating the existence of an alternative metabolic pathway involving NADH but independent of cytochrome b5 (Fig. 2). One possible explanation for the residual activity could be the presence of an alternate electron donor, such as the recently described outer mitochondrial cytochrome b5, which is derived from a gene locus on chromosome 8 (45, 46) and encodes for a protein of 16.3 kDa, slightly larger than microsomal cytochrome b5 at 15.2 kDa and ∼45% identical at the amino acid level. However, to date, there is no evidence for the interaction of outer mitochondrial cytochrome b5 with the microsomal P450 system.
Recently, a novel, soluble, NAD(P)H reductase (NAD(P)H cytochrome b5 oxidoreductase), located in the lumen of the endoplasmic reticulum, was reported (47), which contains both cytochrome b5 and cytochrome b5 reductase domains. In order to confirm that this enzyme was not induced in the HBN mice, and potentially compensating for the loss of microsomal cytochrome b5, we obtained antiserum to NAD(P)H cytochrome b5 oxidoreductase; immunoblotting liver samples from wild-type and HBN mice found no difference in expression levels (data not shown). Li and Porter (48) have also described a second microsomal reductase, which is active with squalene monooxygenase and apparently capable of supporting up to 40% of the activity of this enzyme, although the identity of this reductase and its interactions with other enzymes remain unclear.
In addition, due to its role in fatty acid desaturation, it is possible that changes in the phospholipid bilayer, as observed in stearoyl-CoA-desaturase null mice (49), are responsible for the observed effects of cytochrome b5 on P450 metabolism. However, no changes in the fatty acid composition of endoplasmic reticulum membranes from wild-type and HBN mice were found (supplemental Fig. 3).
In order to establish the in vivo effects of the hepatic cytochrome b5 deletion, we administered a series of probe drugs by both intravenous and oral routes to control and HBN mice. These drugs were chosen because they have been shown to be metabolized in a number of species by different cytochrome P450s. The interpretation of such experiments is inevitably complex, particularly because in the mouse there is little background historical information on the key factors that define their pharmacokinetic parameters. However, consistent with the in vitro studies, in general, it was evident that the deletion of hepatic cytochrome b5 caused marked changes in drug pharmacokinetics independent of the route of administration.
When administered intravenously, the terminal half-life was increased in HBN mice for all drugs used (Table 2). This increase is consistent with decreased liver metabolism as a consequence of hepatic microsomal cytochrome b5 deficiency and provides the first evidence that strongly supports the conclusion that cytochrome b5 plays a role in in vivo P450-mediated drug metabolism. In addition, the profiles of metabolite production for midazolam, phenacetin, and metoprolol were different between HBN and wild-type mice; for example, maximal metabolite levels of 1′-hydroxymidazolam occurred 3 times more slowly in HBN mice than in wild type (120 versus 40 min) when dosed intravenously (supplemental Fig. 5). These differences in metabolite production are consistent with the in vitro kinetic data shown in Table 1. For example, the Vmax values for 1′-hydroxymidazolam were ∼3 times higher in wild type as compared with HBN. No major differences in the formation of the demethyl metabolite of metoprolol were observed at the early time points between HBN and wild-type mice, despite major in vitro differences (supplemental Fig. 5 and Table 1).
Following oral administration of the drug mixture, the interpretation of pharmacokinetic parameters is more complex due to factors such as the absorption across gastrointestinal tract and hepatic blood flow. This added complexity was manifest in the fact that for some drugs, such as midazolam, microsomal cytochrome b5 deficiency and the consequent reduction in hepatic metabolism can lead to increased bioavailability (Table 2) as well as a decreased rate of elimination. These alterations in pharmacokinetics could therefore potentially lead to an exaggerated response to the drug. However, for the other drugs, first pass liver metabolism does not appear to be important in determining bioavailability, which is presumably dependent primarily on efficiency of gastrointestinal absorption or metabolism. When dosed orally, the Cmax appears to be the most reliable indicator of a change in metabolizing capacity and acts as a valuable corollary to the increased half-life observed following intravenous administration.
In summary, mice with a hepatic deletion of microsomal cytochrome b5 display a significant change in the metabolism of a range of substrates by a number of cytochrome P450 enzymes in vitro and, more importantly, in vivo. The data presented in this report suggest that the level and/or activity of hepatic microsomal cytochrome b5 can play a significant role in the metabolism, disposition, and therapeutic effectiveness of clinically used drugs. The extrapolation of in vitro data to in vivo and the relative importance to drug pharmacokinetics and disposition are very complex and involve a wide range of parameters that could include the cytochrome P450/cytochrome b5 ratio, the cytochrome P450 enzyme involved, the substrate concentration, the P450 turnover number, and the overall role of cytochrome P450 metabolism as a rate-limiting factor in drug disposition.
Supplementary Material
Acknowledgments
We thank Catherine Hughes and Susanne van Schelven for excellent technical assistance.
Author's Choice—Final version full access.
This work was supported by Cancer Research UK Programme Grant C4639/A5661 (to C. R. W.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–5 and Tables 1–5.
Footnotes
The abbreviations used are: POR, cytochrome P450 oxidoreductase; AUC, area under the curve; BFC, 7-benzyloxy-4-trifluoromethylcoumarin; BR, benzyloxyresorufin; Cmax, maximum concentration of drug achieved in the blood after dosing; EFC, 7-ethoxy-4-trifluoromethylcoumarin; ER, ethoxyresorufin; HBN, hepatic microsomal cytochrome b5 null; HPLC, high performance liquid chromatography; MFC, 7-methoxy-4-trifluoromethylcoumarin; LC-MS/MS, liquid chromatography-tandem mass spectrometry.
C. R. Wolf, unpublished results.
References
- 1.Loughran, P. A., Roman, L. J., Miller, R. T., and Masters, B. S. (2001) Arch. Biochem. Biophys. 385 311–321 [DOI] [PubMed] [Google Scholar]
- 2.Schenkman, J. B., and Jansson, I. (2003) Pharmacol. Ther. 97 139–152 [DOI] [PubMed] [Google Scholar]
- 3.Shet, M. S., Faulkner, K. M., Holmans, P. L., Fisher, C. W., and Estabrook, R. W. (1995) Arch. Biochem. Biophys. 318 314–321 [DOI] [PubMed] [Google Scholar]
- 4.Vergeres, G., and Waskell, L. (1995) Biochimie (Paris) 77 604–620 [DOI] [PubMed] [Google Scholar]
- 5.Hildebrandt, A., and Estabrook, R. W. (1971) Arch. Biochem. Biophys. 143 66–79 [DOI] [PubMed] [Google Scholar]
- 6.Porter, T. D. (2002) J. Biochem. Mol. Toxicol. 16 311–316 [DOI] [PubMed] [Google Scholar]
- 7.Zhang, H., Hamdane, D., Im, S. C., and Waskell, L. (2008) J. Biol. Chem. 283 5217–5225 [DOI] [PubMed] [Google Scholar]
- 8.Zhang, H., Im, S. C., and Waskell, L. (2007) J. Biol. Chem. 282 29766–29776 [DOI] [PubMed] [Google Scholar]
- 9.Aoyama, T., Nagata, K., Yamazoe, Y., Kato, R., Matsunaga, E., Gelboin, H. V., and Gonzalez, F. J. (1990) Proc. Natl. Acad. Sci. U. S. A. 87 5425–5429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamazaki, H., Nakamura, M., Komatsu, T., Ohyama, K., Hatanaka, N., Asahi, S., Shimada, N., Guengerich, F. P., Shimada, T., Nakajima, M., and Yokoi, T. (2002) Protein Expression Purif. 24 329–337 [DOI] [PubMed] [Google Scholar]
- 11.Zhang, H., Gruenke, L., Arscott, D., Shen, A., Kasper, C., Harris, D. L., Glavanovich, M., Johnson, R., and Waskell, L. (2003) Biochemistry 42 11594–11603 [DOI] [PubMed] [Google Scholar]
- 12.Auchus, R. J., Lee, T. C., and Miller, W. L. (1998) J. Biol. Chem. 273 3158–3165 [DOI] [PubMed] [Google Scholar]
- 13.Yamazaki, H., Johnson, W. W., Ueng, Y. F., Shimada, T., and Guengerich, F. P. (1996) J. Biol. Chem. 271 27438–27444 [DOI] [PubMed] [Google Scholar]
- 14.Guryev, O. L., Gilep, A. A., Usanov, S. A., and Estabrook, R. W. (2001) Biochemistry 40 5018–5031 [DOI] [PubMed] [Google Scholar]
- 15.Hlavica, P., and Lewis, D. F. (2001) Eur. J. Biochem. 268 4817–4832 [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki, H., Shimada, T., Martin, M. V., and Guengerich, F. P. (2001) J. Biol. Chem. 276 30885–30891 [DOI] [PubMed] [Google Scholar]
- 17.Yamazaki, H., Gillam, E. M., Dong, M. S., Johnson, W. W., Guengerich, F. P., and Shimada, T. (1997) Arch. Biochem. Biophys. 342 329–337 [DOI] [PubMed] [Google Scholar]
- 18.Cooper, M. T., and Porter, T. D. (2001) Mutat. Res. 484 61–68 [DOI] [PubMed] [Google Scholar]
- 19.Mokashi, V., Li, L., and Porter, T. D. (2003) Arch. Biochem. Biophys. 412 147–152 [DOI] [PubMed] [Google Scholar]
- 20.Voice, M. W., Zhang, Y., Wolf, C. R., Burchell, B., and Friedberg, T. (1999) Arch. Biochem. Biophys. 366 116–124 [DOI] [PubMed] [Google Scholar]
- 21.Emoto, C., and Iwasaki, K. (2007) Xenobiotica 37 986–999 [DOI] [PubMed] [Google Scholar]
- 22.Postic, C., Shiota, M., Niswender, K. D., Jetton, T. L., Chen, Y., Moates, J. M., Shelton, K. D., Lindner, J., Cherrington, A. D., and Magnuson, M. A. (1999) J. Biol. Chem. 274 305–315 [DOI] [PubMed] [Google Scholar]
- 23.Henderson, C. J., Otto, D. M., Carrie, D., Magnuson, M. A., McLaren, A. W., Rosewell, I., and Wolf, C. R. (2003) J. Biol. Chem. 278 13480–13486 [DOI] [PubMed] [Google Scholar]
- 24.Meehan, R. R., Forrester, L. M., Stevenson, K., Hastie, N. D., Buchmann, A., Kunz, H. W., and Wolf, C. R. (1988) Biochem. J. 254 789–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pritchard, M. P., Glancey, M. J., Blake, J. A., Gilham, D. E., Burchell, B., Wolf, C. R., and Friedberg, T. (1998) Pharmacogenetics 8 33–42 [DOI] [PubMed] [Google Scholar]
- 26.Strobel, H. W., and Dignam, J. D. (1978) Methods Enzymol. 52 89–96 [DOI] [PubMed] [Google Scholar]
- 27.Smith, G. C., Tew, D. G., and Wolf, C. R. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 8710–8714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forrester, L. M., Henderson, C. J., Glancey, M. J., Back, D. J., Park, B. K., Ball, S. E., Kitteringham, N. R., McLaren, A. W., Miles, J. S., Skett, P., and Wolf, C. R. (1992) Biochem. J. 281 359–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pritchard, M. P., Ossetian, R., Li, D. N., Henderson, C. J., Burchell, B., Wolf, C. R., and Friedberg, T. (1997) Arch. Biochem. Biophys. 345 342–354 [DOI] [PubMed] [Google Scholar]
- 30.Burke, M. D., Thompson, S., Weaver, R. J., Wolf, C. R., and Mayer, R. T. (1994) Biochem. Pharmacol. 48 923–936 [DOI] [PubMed] [Google Scholar]
- 31.Cannady, E. A., Dyer, C. A., Christian, P. J., Sipes, I. G., and Hoyer, P. B. (2003) Toxicol. Sci. 73 423–430 [DOI] [PubMed] [Google Scholar]
- 32.Donato, M. T., Jimenez, N., Castell, J. V., and Gomez-Lechon, M. J. (2004) Drug Metab. Dispos. 32 699–706 [DOI] [PubMed] [Google Scholar]
- 33.Goosen, T. C., Mills, D. E., and Hollenberg, P. F. (2001) J. Pharmacol. Exp. Ther. 296 198–206 [PubMed] [Google Scholar]
- 34.McLaughlin, L. A., Dickmann, L. J., Wolf, C. R., and Henderson, C. J. (2008) Drug Metab. Dispos. 36 1322–1331 [DOI] [PubMed] [Google Scholar]
- 35.Nerurkar, P. V., Park, S. S., Thomas, P. E., Nims, R. W., and Lubet, R. A. (1993) Biochem. Pharmacol. 46 933–943 [DOI] [PubMed] [Google Scholar]
- 36.Ueng, Y. F., Jan, W. C., Lin, L. C., Chen, T. L., Guengerich, F. P., and Chen, C. F. (2002) Drug Metab. Dispos. 30 349–353 [DOI] [PubMed] [Google Scholar]
- 37.Court, M. H., Von Moltke, L. L., Shader, R. I., and Greenblatt, D. J. (1997) Biopharm. Drug Dispos. 18 213–226 [DOI] [PubMed] [Google Scholar]
- 38.DeLozier, T. C., Tsao, C. C., Coulter, S. J., Foley, J., Bradbury, J. A., Zeldin, D. C., and Goldstein, J. A. (2004) J. Pharmacol. Exp. Ther. 310 845–854 [DOI] [PubMed] [Google Scholar]
- 39.Lofgren, S., Hagbjork, A. L., Ekman, S., Fransson-Steen, R., and Terelius, Y. (2004) Xenobiotica 34 811–834 [DOI] [PubMed] [Google Scholar]
- 40.Masubuchi, Y., Iwasa, T., Hosokawa, S., Suzuki, T., Horie, T., Imaoka, S., Funae, Y., and Narimatsu, S. (1997) J. Pharmacol. Exp. Ther. 282 1435–1441 [PubMed] [Google Scholar]
- 41.Perloff, M. D., von Moltke, L. L., Court, M. H., Kotegawa, T., Shader, R. I., and Greenblatt, D. J. (2000) J. Pharmacol. Exp. Ther. 292 618–628 [PubMed] [Google Scholar]
- 42.Gu, J., Weng, Y., Zhang, Q. Y., Cui, H., Behr, M., Wu, L., Yang, W., Zhang, L., and Ding, X. (2003) J. Biol. Chem. 278 25895–25901 [DOI] [PubMed] [Google Scholar]
- 43.Eberhart, D. C., and Parkinson, A. (1991) Arch. Biochem. Biophys. 291 231–240 [DOI] [PubMed] [Google Scholar]
- 44.Yang, M. X., and Cederbaum, A. I. (1995) Arch. Biochem. Biophys. 324 282–292 [DOI] [PubMed] [Google Scholar]
- 45.Altuve, A., Silchenko, S., Lee, K. H., Kuczera, K., Terzyan, S., Zhang, X., Benson, D. R., and Rivera, M. (2001) Biochemistry 40 9469–9483 [DOI] [PubMed] [Google Scholar]
- 46.Altuve, A., Wang, L., Benson, D. R., and Rivera, M. (2004) Biochem. Biophys. Res. Commun. 314 602–609 [DOI] [PubMed] [Google Scholar]
- 47.Zhu, H., Larade, K., Jackson, T. A., Xie, J., Ladoux, A., Acker, H., Berchner-Pfannschmidt, U., Fandrey, J., Cross, A. R., Lukat-Rodgers, G. S., Rodgers, K. R., and Bunn, H. F. (2004) J. Biol. Chem. 279 30316–30325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li, L., and Porter, T. D. (2007) Arch. Biochem. Biophys. 461 76–84 [DOI] [PubMed] [Google Scholar]
- 49.Miyazaki, M., Kim, Y. C., Gray-Keller, M. P., Attie, A. D., and Ntambi, J. M. (2000) J. Biol. Chem. 275 30132–30138 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.