

Abstract
Eukaryotic Okazaki fragments are initiated by a RNA/DNA primer, which is removed before the fragments are joined. Polymerase δ displaces the primer into a flap for processing. Dna2 nuclease/helicase and flap endonuclease 1 (FEN1) are proposed to cleave the flap. The single-stranded DNA-binding protein, replication protein A (RPA), governs cleavage activity. Flap-bound RPA inhibits FEN1. This necessitates cleavage by Dna2, which is stimulated by RPA. FEN1 then cuts the remaining RPA-free flap to create a nick for ligation. Cleavage by Dna2 requires that it enter the 5′-end and track down the flap. Because Dna2 cleaves the RPA-bound flap, we investigated the mechanism by which Dna2 accesses the protein-coated flap for cleavage. Using a nuclease-defective Dna2 mutant, we showed that just binding of Dna2 dissociates the flap-bound RPA. Facile dissociation is specific to substrates with a genuine flap, and will not occur with an RPA-coated single strand. We also compared the cleavage patterns of Dna2 with and without RPA to better define RPA stimulation of Dna2. Stimulation derived from removal of DNA folding in the flap. Apparently, coordinated with its dissociation, RPA relinquishes the flap to Dna2 for tracking in a way that does not allow flap structure to reform. We also found that RPA strand melting activity promotes excessive flap elongation, but it is suppressed by Dna2-promoted RPA dissociation. Overall, results indicate that Dna2 and RPA coordinate their functions for efficient flap cleavage and preparation for FEN1.
Eukaryotic DNA replication involves synthesis of a leading strand in the direction of parental DNA unwinding, and because of the anti-parallel structure of DNA, a lagging strand in the opposite direction of unwinding. Although leading strand synthesis occurs continuously, the lagging strand must be synthesized in a discontinuous fashion. Lagging strand synthesis is initiated by the polymerase (pol)3 α-primase complex, which synthesizes 8–12 nucleotides (nt) of RNA followed by ∼20-nt of DNA (1, 2). The pol α-primase complex is then replaced by a complex of the toroidal sliding clamp proliferating cell nuclear antigen and pol δ, which is loaded onto the DNA by the clamp loader replication factor C. This complex synthesizes another 100–150 nt of DNA before the primer terminus encounters the previously synthesized downstream segment. These segments, known as Okazaki fragments, must then be processed to create a continuous strand of DNA. Because pol α does not possess proofreading activity and synthesizes with relatively low fidelity compared with pol δ, it is proposed that the entire RNA/DNA primer, laid down by pol α, is removed to maintain genome integrity (3). Processing of the RNA/DNA primer is initiated when strand displacement synthesis, by pol δ, raises the primer into a single-stranded (ss) flap intermediate (4–6). Removal of this flap intermediate followed by fragment joining is known as Okazaki fragment processing.
Several modes of Okazaki fragment processing have been proposed to occur in Saccharomyces cerevisiae, based on biochemical and genetic data. One involves complete flap removal by flap endonuclease 1 (FEN1) (4, 7). FEN1 is a structure-specific nuclease that cleaves at the base of 5′ flap intermediates to create a nick (8). In this mode, pol δ displaces the RNA/DNA primer into short flap intermediates. These flaps are efficiently cleaved by FEN1. The resultant nicked product is then ligated by DNA ligase I.
In another proposed mode, cleavage by the nuclease/helicase Dna2 precedes FEN1 cleavage to create a nicked product (9, 10). When flaps reach a length of 20–30-nt, they are stably bound by replication protein A (RPA) (11–13). RPA binding prevents cleavage by FEN1 while stimulating Dna2 cleavage, necessitating initial flap cleavage by Dna2 (9). Dna2, unlike FEN1, cannot create a nick product for ligation. Instead, it leaves a short, ∼5-nt flap devoid of RPA. FEN1 can then cleave this short flap, creating a nick for ligation by DNA ligase I.
Recent work from our group suggests that both pathways are employed during flap processing (14). By reconstructing Okazaki fragment processing in vitro, we recently demonstrated that pol δ displaces mostly short flaps (<10-nt), which are readily cleaved by FEN1. In addition to the short flaps, a minor subset of longer flaps (10–30-nt) arises, which can be bound by RPA, requiring the Dna2/FEN1 pathway. These results suggest that the two pathways work in parallel to resolve Okazaki fragments during DNA replication.
S. cerevisiae Dna2 was identified during a screen for mutants defective in DNA replication (15, 16). It is an essential protein possessing both ssDNA endonuclease activity and 5′ to 3′ ATP-dependent helicase activity (15, 17). In addition, it physically and genetically interacts with FEN1, further implicating it in lagging strand replication (18). Recently, Dna2 was shown to possess strand annealing and strand exchange activities, which may aid in the formation of a 5′ flap intermediate during flap equilibration (19). Dna2 has subsequently been shown to interact with other proteins involved in DNA replication, including Pol32, RNase H2, Sgs1, and Exo1 (20). Furthermore, RPA was found to interact with Dna2 to stimulate both its nuclease and helicase activities, and the overexpression of RPA overcame the temperature-sensitive growth of several Dna2 mutant alleles (9, 21, 22).
S. cerevisiae RPA is a heterotrimeric protein consisting of subunits Rpa1–3, each of which is essential in yeast (23–25). In humans, RPA is also necessary for the reconstitution of SV40 DNA replication (26). Its primary role is to stabilize ssDNA, preventing it from re-annealing or forming secondary structure during DNA replication, repair, and recombination (27–29). Dna2 was shown to interact specifically with Rpa1, the large subunit of RPA (22). Contacts between the two proteins were found to occur at both the N- and C-terminal domains of both Dna2 and Rpa1. Additionally, wild type Dna2 and RPA were shown to form a complex on a DNA flap substrate (9).
We previously reported that to exhibit nuclease activity, Dna2 must enter the free 5′-end of a flap, and then track down the flap toward the base for cleavage (30). Accordingly, the tracking requirement of Dna2 necessitates that it encounter bound RPA molecules as it moves for successive cleavages. Because Dna2 is stimulated for cleavage on flaps coated by RPA, we were prompted to ask why the presence of RPA does not interfere with the tracking motion of Dna2. This question is particularly interesting because Dna2 cleavage is inhibited on flaps coated by other ssDNA-binding proteins (Escherichia coli single-stranded binding protein and human RPA) (9, 31). In the current study, we explored the mechanism by which Dna2 cleaves on RPA-coated flaps, and the manner by which RPA stimulates Dna2 cleavage activity.
EXPERIMENTAL PROCEDURES
Materials—Synthetic oligonucleotides, including the 5′-biotin and 3′-biotin conjugations, were produced by Integrated DNA Technologies. Radionucleotides [α-32P]dCTP and [γ-32P]ATP were obtained from PerkinElmer Life Sciences. The Klenow fragment of E. coli DNA polymerase I, polynucleotide kinase, streptavidin, and ATP were from Roche Applied Science. All other reagents were the best available commercial grade.
Enzyme Expression and Purification—S. cerevisiae Dna2 was cloned into the Sf9 baculovirus expression vector (Invitrogen). S. cerevisiae Dna2 E675A was created by site-directed mutagenesis as described (32). Both Dna2 wild type and E675A were then expressed and purified as described (32), except that High Five cells were utilized. S. cerevisiae RPA (33) and human RPA (13) were overexpressed and purified as previously described. Oligonucleotides—Substrate primer sequences are listed in Table 1. Downstream primers were labeled at either the 3′ terminus or the 5′ terminus with [32P], as indicated in the figures. [α-32P]dCTP was incorporated into the 3′ terminus by the Klenow enzyme and [γ-32P]ATP was incorporated at the 5′ terminus using polynucleotide kinase. After labeling, substrates were purified as described previously (34). DNA flap substrates were then annealed in 50 mm Tris-HCl, pH 8.0, 50 mm NaCl, and 1 mm dithiothreitol and in a 1:2:4 ratio of downstream to template to upstream primer. The ΔG calculations for the flap structure were done by entering the unannealed portion of the primer sequence into mfold, for analysis under reaction conditions (35, 36).
TABLE 1.
Oligonucleotides (5′-3′)
| Primer | Length (nt) | Sequence |
|---|---|---|
| Downstreama,b | ||
| D1 | 76 | GTA CCG AGC TCG AAT TCG CCC GTT TCA CGC CTG TTA GTT AAT TCA CTG GCC GTC GTT TTA CAA CGA CGT GAC TGG G |
| D2 | 76 | GTA CCG AGC TCG AAT TCG CCC GTT TCA CGC CTG TTA GTT AAT TCA CTG GCC GTC GTT TTA CAA CGA CGT GAC TGG G |
| D3 | 50 | GTA CCG AGC TCG AAT TCG CCC GTT TCA CGC CTG TTA GTT AAT TCA CTG GC |
| D4 | 53 | TTC ACG CCT GTT AGT TAA TTC ACT GGC CGT CGT TTT ACA ACG ACG TGA CTG GG |
| D5 | 53 | TTC ACG CCT GTT AGT TAA TTC ACT GGC CGT AGT TTT ACA ACG ATG TGA CTA AA |
| D6 | 53 | TTT TTT TTT TTT TTT TTT TTT TTT TCC ACC CGT CCA CCC GAC GCC ACC TCC TG |
| D7 | 55 | AGG TCT CGA CTA ACT CTA GTC GTT GTT CCA CCC GTC CAC CCG ACG CCA CCT CCT G |
| D8 | 97 | TTT TTT TTT TTT AGG TCT CGA GGC CTG CTC TAT TAT GAG CAG GCC TCG AGA CCT ACG TAG AGC TGT TTC CCA CCC GTC CAC CCG ACG CCA CCT CCT G |
| Upstream | ||
| U1 | 28 | CGC CCA GGG TTT TCC CAG GTC ACG GAC A |
| U2 | 26 | CGA CCG TGC CAG CCT AAA TTT CAA TA |
| Template | ||
| T1 | 49 | GCC CAG TCA CGT CGT TGT AAA ACG GGT CGT GAC TGG GAA AAC CCT GGC G |
| T2 | 49 | GTT TAG TCA CAT CGT TGT AAA ACT GGT CGT GAC TGG GAA AAC CCT GGC G |
| T3 | 54 | GCA GGA GGT GGC GTC GGG TGG ACG GGT GGA TTG AAA TTT AGG CTG GCA CGG TCG |
Bolded nucleotides are biotinylated.
Underline indicates foldback region.
Gel Shift Assay—The reaction volume was 20 μl, which contained 5 fmol of labeled substrate and various amounts of protein, as indicated. The reaction buffer consisted of 50 mm Tris-HCl, pH 8.0, 2 mm dithiothreitol, 30 mm NaCl, 0.1 mg/ml bovine serum albumin, 5% glycerol, 2 mm MgCl2, and 1 mm ATP, unless otherwise stated. RPA was pre-bound for 5 min at room temperature followed by addition of Dna2 E675A for 5 min at room temperature. In Figs. 3 and 4, the streptavidin was preincubated with the substrate for 10 min at 37 °C. For measurement of the dissociation constants in Table 2, the substrate concentration was lowered to 1 fmol (50 pm). All reactions were then loaded on pre-run 5% polyacrylamide gels in 0.5× TBE (Invitrogen). Gels were then subjected to electrophoresis at 150 V for 30–45 min.
FIGURE 3.

Dna2 tracking mechanism is not required for RPA dissociation. Gel shift analysis was performed with 100 fmol of RPA, 5 fmol of the 53-nt flap substrate (D2:U1:T1), and increasing amounts of Dna2 E675A (100, 200, and 500 fmol). The substrate was incubated with streptavidin (250 fmol) prior to pre-binding with RPA as indicated under “Experimental Procedures.” Dna2 was then added into the reaction (lanes 4–6). Lanes 1–3 are the substrate alone, substrate with Dna2 E675A (500 fmol) alone, and substrate with RPA (100 fmol) alone, respectively. The substrate is depicted above the gel. The asterisk indicates the site of the 3′-32P radiolabel, and B indicates the 5′ biotin.
FIGURE 4.

ssDNA alters Dna2 dissociation of RPA. Gel shift analysis was performed with 100 fmol of RPA, 5 fmol of the 50-nt ssDNA substrate (D3), and increasing amounts of Dna2 E675A (50, 100, 200, and 500 fmol). In lanes 8–14, the substrate was incubated with streptavidin (250 fmol) prior to pre-binding with RPA as indicated under “Experimental Procedures.” Dna2 was then added into the reaction (lanes 3–6 and 10–13). Lanes 1, 2, and 7 are the substrate control alone, substrate with Dna2 E675A (500 fmol) alone, and substrate with RPA (100 fmol) alone, respectively, without streptavidin. Lanes 8, 9, and 14 are the substrate alone, substrate with Dna2 E675A (500 fmol) alone, and substrate with RPA (100 fmol) alone, respectively, with streptavidin. The substrate is depicted above the gel. The asterisk indicates the site of the 5′-32P radiolabel, and B indicates the 3′ biotin.
TABLE 2.
Relative binding constants
| Protein | Kda |
|---|---|
| nm | |
| RPA | 0.35 ± 0.10 |
| Dna2 WT | 5.5 ± 0.77 |
| Dna2 E675A | 3.4 ± 0.50 |
Apparent dissociation constants measured by gel shift analysis. Results are an average of two independent experiments.
Nuclease Assay—Reactions were performed at 37 °C for 10 min. The reaction volume was 20 μl, which contained 5 fmol of labeled substrate and various amounts of protein, as indicated. The reaction buffer consisted of 50 mm Tris-HCl, pH 8.0, 2 mm dithiothreitol, 30 mm NaCl, 0.1 mg/ml bovine serum albumin, 2 mm MgCl2, and 1 mm ATP, unless otherwise stated in the figure legends. The reactions were stopped by addition of 2× termination dye, consisting of 90% formamide (v/v), 10 mm EDTA, 0.01% bromphenol blue, and 0.01% xylene cyanol. A 15% polyacrylamide gel, containing 7 m urea, was then used to separate the reactions.
Strand Melting Assay—RPA strand melting was measured using native gel electrophoresis. The reaction buffer consisted of 50 mm Tris-HCl, pH 8.0, 2 mm dithiothreitol, 30 mm NaCl, 0.1 mg/ml bovine serum albumin, and 5% glycerol with or without 1 mm ATP and 2 mm MgCl2, as indicated in the figure legends. Substrates (5 fmol) were incubated with various amounts of RPA at 37 °C for 15 min. Reactions were then stopped by addition of a 6 × helicase dye, containing 30% glycerol, 50 mm EDTA, 0.9% SDS, 0.25% bromphenol blue, and 0.25% xylene cyanole. Reactions were then loaded onto 5% polyacrylamide gels in 0.5× TBE (Invitrogen). Gels were then subjected to electrophoresis at 150 V for 30 min.
Gel Analysis—Experiments were done in at least duplicate and representative gels are shown. After running conditions, gels were transferred to filter paper (Whatman) and dried on a gel dryer (Bio-Rad) with vacuum (Savant). Gels were then exposed to a phosphorscreen and analyzed by phosphorimaging (GE Healthcare). Analysis of the image was then performed using ImageQuantMac, version 1.2.
Calculation of Dissociation Constants—After gel shift analysis, curves were fit using nonlinear least squares regression of the hyperbolic equation,
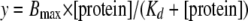 |
where y is the percent of oligonucleotide bound, [Protein] is the concentration of protein in nm, Bmax is the maximum binding, and Kd is the equilibrium dissociation constant.
RESULTS
Dna2 Removes Flap-bound RPA—Because Dna2 is necessary to cleave flaps bound by RPA, we planned to determine the mechanism by which Dna2 gains access to the flap. Previously, it had been proposed that RPA dissociates as a consequence of flap cleavage by Dna2 (9). However, another possibility is that Dna2 dissociates RPA from the flap to gain access to binding sites for tracking and cleavage. To distinguish these hypotheses, we tested the ability of the nuclease-defective mutant Dna2 E675A to dissociate RPA. This mutant Dna2 would allow us to establish whether RPA dissociation depends on cleavage. Reactions employed a double flap substrate with a 53-nt flap. RPA was pre-bound to the substrate prior to the addition of Dna2 E675A (see “Experimental Procedures”). Pre-binding of RPA allowed us to observe the subsequent effects of Dna2 addition on flap-bound RPA. Saturating amounts of RPA were pre-bound to the flap so that we could assess Dna2 binding to RPA-bound flaps, without having to simultaneously interpret results of Dna2 binding to naked flaps. Gel shift analysis was then carried out to determine whether Dna2 and/or RPA were bound to the labeled flap substrate (Fig. 1).
FIGURE 1.

Dna2 dissociates RPA from flap substrates. Gel shift analysis was performed with 100 fmol of RPA, 5 fmol of a 53-nt flap substrate (D2:U1:T1), and increasing amounts of Dna2 E675A (50, 100, 200, and 500 fmol). Lanes 1–7 contain no ATP and lanes 8–14 contain 1 mm ATP. RPA was pre-bound to the substrate as indicated under “Experimental Procedures.” Dna2 was then added into the reaction (lanes 3–6 and 10–13). Lane 1 and 8 are the substrate alone. Lanes 2 and 9 are substrate with RPA (100 fmol) alone and lanes 7 and 14 are substrate with Dna2 (500 fmol) alone. The substrate used is depicted above the gel and the asterisk indicates the site of the 3′-32P radiolabel.
We previously demonstrated that although the Dna2 E675A mutant is devoid of nuclease activity, it retains both substrate binding and helicase activities (34). These properties of the Dna2 E675A allowed us to also assess the effects of helicase function on Dna2 binding to the RPA-coated substrate. To accomplish this assessment, binding reactions were carried out in the presence or absence of ATP, which is required for Dna2 helicase function.
With RPA alone (Fig. 1), the major band in lanes 2 and 9 corresponds to the binding of two RPA molecules to the 53-nt flap, with the minor band corresponding to one RPA molecule. This interpretation is consistent with the banding pattern changes seen in an RPA concentration titration on the same substrate, which showed binding of one RPA and then two (data not shown). Dna2 was then titrated into the reaction. As the amount of Dna2 increased, we observed a shift from an RPA-bound band to a Dna2-bound band (Fig. 1, lanes 3–6 and 10–13). Similar results were observed on 37- and 30-nt flap substrates (data not shown). These results are consistent with the idea that Dna2 removes RPA from the flap in a cleavage independent manner, allowing Dna2 access to the flap for cleavage. However, the results do not exclude the possibility that Dna2 cleavage activity aids in further destabilizing RPA binding to the flap.
Interestingly, the addition of ATP did not significantly alter the shift to the Dna2-bound flap (Fig. 1, compare lanes 3–6 and 10–13). However, we observed a minor accumulation of a super-shifted product, in the presence of both Dna2 and RPA (lane 6). This product likely represents an intermediate in which both Dna2 and RPA are bound to the flap substrate. Because ATP is thought to aid Dna2 tracking on the flap (30), the absence of ATP might slow RPA removal because tracking is impaired.
RPA Binds Flaps with a Higher Affinity Than That of Dna2—To assure that Dna2 replacement of RPA (Fig. 1) was derived from an active dissociation process and was not based on a simple difference between binding affinities, we compared the dissociation constants of RPA and Dna2 (Table 2). Increasing amounts of Dna2 or RPA were bound to a fixed amount of the labeled 53-nt flap substrate. Gel shift analysis was then performed to assess binding. The data were then fit by nonlinear least squares regression to determine relative binding affinities (Table 2). The substrate concentration was lowered to assure equilibrium binding (see “Experimental Procedures”). The dissociation constant for RPA binding to a 50-nt segment of ssDNA was previously measured to be ∼0.15 nm (37). Here, we measured the dissociation constant of RPA binding to the 53-nt flap substrate to be 0.35 nm. Both wild type Dna2 and Dna2 E675A were tested and found to have dissociation constants of 5.5 and 3.4 nm, respectively (Table 2). When compared with RPA binding, Dna2 bound with an ∼10-fold lower relative binding affinity. In our binding titration in Fig. 1, the concentration of Dna2 never exceeded 5 times the concentration of RPA. Nevertheless, Dna2 displaced virtually all of the RPA. These observations suggest that, based on binding affinities, Dna2 would not have out-competed RPA for binding. Instead, it must have interacted with RPA in a specific manner to dissociate the higher affinity RPA.
Removal of RPA Is Species-specific—Previously, Kim et al. (9, 31) demonstrated that stimulation of Dna2 by RPA is species-specific; for example, human RPA (hRPA) inhibits instead of stimulates S. cerevisiae Dna2 cleavage activity. Using the 53-nt flap, we also saw inhibition of yeast Dna2 cleavage activity by hRPA (Fig. 2A). Because Dna2 dissociates RPA (Fig. 1), we tested whether dissociation is also species-specific (Fig. 2B). Yeast RPA (lanes 2–4) and hRPA (lanes 6–8) were pre-bound to the flap. When progressively greater amounts of yeast Dna2 were then incubated with the hRPA-bound flaps the quantity of bound hRPA was unaltered (lanes 6–8). The inability of yeast Dna2 to remove hRPA from the flap might explain why single-stranded binding proteins from other species inhibit Dna2 cleavage. If the binding proteins block either the motion or catalytic activity of Dna2 then cleavage would be prevented. These data further suggest a specific protein-protein interaction between Dna2 and RPA, which is required for RPA removal followed by Dna2 cleavage.
FIGURE 2.

Yeast Dna2 cannot dissociate human RPA. A, Dna2 (50 fmol) cleavage activity was measured by denaturing PAGE with increasing amounts of hRPA (10, 50, 100, 200, and 500 fmol) and 5 fmol of the 53-nt flap substrate (D1:U1:T1). B, gel shift analysis was performed with 100 fmol of either RPA (lanes 2–4) or hRPA (lanes 6–8), 5 fmol of the 53-nt flap substrate (D1:U1:T1), and increasing amounts of Dna2 E675A (100 and 200 fmol). RPA was pre-bound to the substrate as indicated under “Experimental Procedures.” Dna2 was then added into the reaction (lanes 3–4 and 7–8). Lanes 1 and 5 are the substrate alone and substrate with Dna2 E675A (500 fmol) alone, respectively. The substrate, used is both A and B, is depicted above the gel in A, and the asterisk indicates the site of the 5′-32P radiolabel.
Dna2 Tracking Is Not Required for RPA Removal—Dna2 employs a tracking mechanism to cleave on flap substrates. If the 5′-end of the flap is blocked by a double-stranded region or a streptavidin-biotin conjugate then cleavage is inhibited (30). Significantly, a block to tracking inhibits cleavage by Dna2, but not Dna2 binding. Previously, we showed that Dna2 bound a flap substrate blocked at the 5′-end by a streptavidin-biotin conjugate (34). Because Dna2 can bind the flap without tracking, we asked whether it could still remove RPA when the 5′-end of the flap was blocked (Fig. 3). Streptavidin was preincubated with a flap containing a biotin attached to the 5′-end. RPA was added into the reaction and allowed to bind the blocked substrate. Increasing amounts of Dna2 were then added into the reaction. Surprisingly, Dna2 was still able to remove the flap-bound RPA even when tracking was blocked (Fig. 3, lanes 4–6). The ability of Dna2 to remove RPA on substrates blocked at the 5′-end is an additional confirmation that cleavage is not required for the dissociation of RPA.
RPA/Dna2 Interaction Differs on ssDNA—To further analyze the mechanism of RPA removal, we evaluated the effect of Dna2 on RPA bound to a single-stranded segment of DNA (Fig. 4). Use of ssDNA allowed us to determine whether structural features of the flap are required for RPA dissociation. A biotin was attached to the 3′-end of the ssDNA, because blocking the end may prevent Dna2 from tracking off the DNA. The 50-nt sequence used was the same sequence as the 53-nt flap minus three nucleotides at the 5′-end. (These nucleotides were omitted because of a length requirement for 3′-biotin modification.) The ssDNA was prepared with and without pre-bound streptavidin. RPA was then bound to the substrate followed by the addition of Dna2 and the products were then analyzed by gel shift (Fig. 4).
Dna2 binding to the ssDNA produced two bands, which likely correspond to one and two molecules of Dna2 bound to DNA (Fig. 4, lane 7). The presence of streptavidin slowed the migration of both bands corresponding to bound Dna2, as anticipated (lane 14). The shift observed when RPA was bound to the ssDNA (compare lanes 2 and 9) was similar with and without streptavidin. By titrating progressively more RPA with the ssDNA, we found that the similar shift resulted from the binding of only two RPA molecules when streptavidin was present versus three in its absence (data not shown). When Dna2 was titrated into the reaction with the bound RPA in Fig. 4, we were surprised to find that the Dna2 did not shift the labeled ssDNA to a position that would indicate Dna2 displacement of the RPA (lanes 3–6, 10–13). Instead, the initial RPA band was super-shifted to a higher mobility complex. This shift likely corresponds to the binding of both Dna2 and RPA. The shifting pattern was not significantly altered by the presence or absence of streptavidin (compare lanes 1–7 and 8–14). In addition, the conversion from an RPA band to the super-shifted band was incomplete at the highest Dna2 concentration with ssDNA, whereas the same concentration of Dna2 achieved almost complete RPA dissociation with the flap substrate (compare Fig. 4, lanes 6 and 13, to Fig. 1, lanes 6 and 13). From these results it is difficult to assess how Dna2 and RPA might interact and bind the ssDNA. It is readily apparent, however, that the interactions of RPA and Dna2 on ssDNA are different from those on a flap substrate (compare Figs. 4 and 1).
RPA Stimulates Dna2 to Cleave More Efficiently and Past the Flap Base—We expected that detailed examination of the effects of RPA interaction on the catalytic activities of Dna2 would reveal more about the movements of both Dna2 and RPA on flaps. Previous studies have shown RPA stimulation of both Dna2 helicase and cleavage activities (9, 10). To further examine the manner in which RPA stimulates Dna2 cleavage, we analyzed the cleavage patterns of Dna2 with and without RPA. Because Dna2 cleaves multiple times on the DNA and must enter at the 5′-end for tracking and cleavage (17, 30, 38), we could visualize both the first and last cleavage sites of Dna2 by radiolabeling the flap substrates at either the 5′- or 3′-end of the flap-primer, respectively. Denaturing PAGE was used to determine the location of the first cleavage site (5′ radiolabel) and the last cleavage site (3′ radiolabel) (Fig. 5). When comparing the cleavage patterns with and without RPA present, we saw a shift in cleavage with respect to both the first and last sites. It is interesting to note that the stimulation of Dna2 cleavage activity by RPA is lower on the 30-nt flap substrate that we employed than has been previously observed with other substrates (9, 10, 38). The stimulation by RPA is about 2-fold or less, a value that will be addressed later under “Results” (Fig. 7). In Fig. 5A, the first cleavages by Dna2 were distributed along the length of the flap. The distribution was shifted upon the addition of RPA (Fig. 5A, lanes 4–7), with the first cleavages occurring closer to the 5′-end of the flap. This result suggests that Dna2 becomes more efficient at cleavage once it begins tracking onto the 5′-end of the flap substrate, because the bound RPA stimulates Dna2. Alternatively, the Dna2 cleavage pattern could simply be shifted because RPA binding to the flap decreases the amount of exposed DNA accessible to Dna2 for cleavage.
FIGURE 5.

Dna2 cleavage patterns in the presence of RPA. Reactions were performed with 5 fmol of the 30-nt flap substrate (D4:U1:T1). A, cleavage by wild type Dna2 (10 fmol) was measured on a 5′-radiolabeled flap substrate with increasing amounts of RPA (10, 50, 100, and 500 fmol). B, cleavage reactions were performed as in A, except a 3′-radiolabeled flap substrate was used. C, Dna2 K1080E (5 fmol), a helicase-deficient mutant, was used to measure cleavage activity with increasing amounts of RPA (10, 50, 100, and 200 fmol), in the absence of ATP, using the 3′-radiolabeled flap substrate. In all gels, lanes 1–3 are the substrate alone, substrate with RPA (500 fmol) alone, and substrate with Dna2 alone, respectively. Percent cleavage is defined as (cleaved/(cleaved + uncleaved)) × 100. The substrates used are depicted above the gel with the asterisk indicating the site of the 32P radiolabel.
FIGURE 7.

Dna2 cleavage activity in the presence of RPA on unstructured and structured flap substrates. Dna2 (10 fmol) cleavage activity was measured by denaturing PAGE with increasing amounts of RPA (10, 50, 100, and 200 fmol) and 5 fmol of substrate. Lanes 1, 8, and 15 are substrate alone. Lanes 2, 9, and 16 are substrate with RPA (200 fmol) alone and lanes 3, 10, and 17 are substrate with Dna2 (10 fmol) alone. Lanes 1–7 contain a poly(dT) sequence in the flap region of the substrate (D6:U2:T3). Lanes 8–14 contain a 6-nt duplex in the flap region (D7:U2:T3) with lengths a = 6 nt; b = 6 nt; c = 4 nt; and d = 10 nt. Lanes 15–21 contain an 18-nt duplex in the flap region of the substrate (D8:U2:T3) with lengths a = 12 nt; b = 18 nt; c = 6 nt; and d = 33 nt. Percent cleavage is defined as (cleaved/(cleaved + uncleaved)) × 100. The substrates are depicted above the gel with the asterisk indicating the site of the 3′-32P radiolabel.
Next we examined the final cleavage site positions (Fig. 5B). When Dna2 alone was incubated with the flap substrate the furthest cleavage site occurred near the base of the flap (lane 3). Upon addition of RPA, the cleavage distribution was shifted further down the flap. Not only were the sites of cleavage shifted closer to the base of the flap but cleavage was also increased past the flap base into the originally annealed portion of the primer (Fig. 5B, lanes 4–7).
Previously, Dna2 was shown to cleave past the flap base in high ATP concentrations and RPA had also been shown to stimulate Dna2 helicase function (10, 21, 32, 38). Based on these data, we presumed that the shift further down the flap and into the originally annealed region was caused by RPA stimulation of Dna2 helicase activity. To test this expectation, Dna2 and RPA were incubated in the absence of ATP on the 30-nt flap substrate (data not shown). We were surprised to see that the cleavage patterns were the same as those in Fig. 5B. To assure that no residual ATP was purified with Dna2, we performed the same experiment with the helicase-deficient mutant Dna2 K1080E (Fig. 5C) and still found cleavage further down the flap and into the annealed region in the presence of RPA. This suggests that cleavage into the originally annealed portion of the primer derives from a property of RPA and not Dna2 helicase function.
RPA Strand Melting Facilitates Dna2 Cleavage—Because RPA is known to have strand melting activity (39), we wondered whether cleavage into the originally annealed region occurred because the RPA melted the double strands near the flap base. To test this idea, increasing amounts of RPA were incubated with the 30-nt flap substrate, used in Fig. 5. The reaction products were then resolved by non-denaturing PAGE (Fig. 6A). RPA was able to fully displace the radiolabeled flap-primer in a protein concentration-dependent manner (Fig. 6A). The annealed portion of this flap had a GC-content of 52% (see Table 1). AT-rich regions have previously been shown to promote RPA strand melting activity (40). Accordingly, we tested several other flap substrates with a >75% GC content in the annealed region. These substrates exhibited virtually no RPA-induced unwinding nor Dna2 cleavage past the flap base in the presence of RPA (data not shown and Fig. 7A).
FIGURE 6.

RPA strand melting activity allows Dna2 cleavage past the base of the flap. Non-denaturing PAGE was performed to measure the strand melting activity of RPA. A, increasing amounts of RPA (50, 100, 200, and 500 fmol) and 5 fmol of flap substrate were incubated in the absence of MgCl2 and ATP (lanes 2–5). The 30-nt substrate (D4:U1:T1) used was the same as in Fig. 5B. B, an unwinding assay was performed as in A, except 2 mm MgCl2 and 1 mm ATP were included in the reaction buffer. C, an unwinding assay was performed as in A, except the GC content of the 30-nt substrate was lowered from 52 to 33% (D5:U1:T2) in the annealed region of the primer (see Table 1). D, an unwinding assay was performed as in C, except 2 mm MgCl2 and 1 mm ATP were included in the reaction buffer. In all cases, lane 1 represents the substrate alone. Percent unwound is defined as (unwound/(unwound + annealed)) × 100. Substrate and product bands are identified. The substrate is depicted above the gel in A with the asterisk indicating the site of the 3′-32P radiolabel.
RPA strand melting is suppressed by increased magnesium concentration (41). Because the experiments in Fig. 6A were done in the absence of MgCl2 and ATP, we tested whether unwinding would still occur under more physiological conditions (2 mm MgCl2 and 1 mm ATP). Consistent with previous studies, RPA strand melting was inhibited on the 30-nt flap substrate, used in Fig. 6A, when MgCl2 and ATP were added to the reaction (Fig. 6B). Although RPA strand melting was inhibited, the assay only measured the complete removal of the primer and so did not account for partial melting. Partial melting of the primer would account for Dna2 cleavage at sites past the original base of the flap as we observed (see Fig. 5, B and C), even though the primer was not completely removed when MgCl2 and ATP were present (Fig. 6B).
Because RPA strand melting was more active on AT-rich regions of DNA (40), we altered the nucleotide composition to increase the amount of RPA strand melting with MgCl2 and ATP. We modified the 30-nt flap substrate, used in Fig. 6A, to create an AT-rich region in the annealed portion of the labeled primer (see Table 1). The GC content in the annealed region was lowered from 52 to 33%. This AT-rich substrate was then used to measure RPA strand melting in Fig. 6, C and D. We first tested RPA strand melting without MgCl2 and ATP present (Fig. 6C), for comparison with the previous flap substrate used in Fig. 6A. We saw a significant increase in strand unwinding (compare Fig. 6, A and C). RPA unwinding activity was then tested in the presence of 2 mm MgCl2 and 1 mm ATP (Fig. 6D). Although RPA strand melting was lowered, we still saw a significant amount of substrate melting on the AT-rich substrate. Based on these findings, we propose that RPA strand melting activity can facilitate flap elongation under physiological conditions.
RPA Removal of DNA Secondary Structure Promotes Dna2 Cleavage—RPA strand melting was previously shown to aid Dna2 cleavage activity (21). In the study, a flap substrate able to form a 10-bp duplex in the central region of the flap was used to show that RPA improved Dna2 cleavage past the duplex. Because these results were not compared with results of an unstructured flap, we tested a 30-nt poly(dT) flap substrate to determine whether stimulation was solely due to RPA strand melting activity. The unstructured flap was compared with two structured flaps, one predicted to form a 6-bp duplex (ΔG = –3.3) and another with a potential 18-bp duplex (ΔG = –21) (see Table 1). These latter substrates also contain a GC content of >75% in the annealed portion of the labeled flap-primer, which inhibited strand melting of the annealed region by RPA (data not shown).
Dna2 and increasing amounts of RPA were incubated with the flaps and the cleavage products were analyzed by denaturing PAGE (Fig. 7). The addition of RPA produced little increase in the amount of Dna2 cleavage of both the unstructured flap and the 18-bp duplex, whereas the 6-bp duplex showed a significant increase in cleavage activity (compare Fig. 7, lanes 4–7, 11–14, and 18–21). Consideration of the expected melting properties of RPA provides a plausible explanation for these results. The unstructured flap has no secondary structure to hinder movement or cleavage by Dna2, and so RPA would not stimulate Dna2. The 18-bp duplex structure may be too stable for RPA to fully melt, preventing RPA from stimulating Dna2. The 6-bp duplex structure may be just the right stability for RPA melting such that it promotes Dna2 cleavage activity.
The RPA-induced shift in cleavage pattern, apparent with the 18-bp duplex substrate, can be explained by partial melting of the duplex by RPA followed by Dna2 cleavage (Fig. 7, lanes 17–21). Despite the lack of increased cleavage product on the unstructured flap, the addition of RPA did shift the final Dna2 cleavage site further down the flap (Fig. 7, lanes 4–7) and decreased the size of the first product (data not shown), similar to the results in Fig. 5. Based on these findings, we can conclude that the removal of structure is a major, but not complete determinant of RPA effects on Dna2 cleavage activity. This idea is consistent with other reports of increased Dna2 cleavage activity by RPA, in which the flaps used were predicted to contain DNA secondary structure (9, 10, 42, 43). Additionally, the poor RPA stimulation of Dna2 in Fig. 5 is likely explained by the weak DNA secondary structure of the 30-nt flap substrate (ΔG = –1.2). However, RPA stimulation of Dna2 does appear to be limited to the strand melting capacity of RPA, because stimulation was not observed on the more stable 18-nt duplex (Fig. 7A, lanes 17–21).
DISCUSSION
In one proposed pathway of eukaryotic Okazaki fragment processing, the 5′-end region of the fragment is displaced into a flap long enough to bind RPA (9, 42, 43). The RPA stimulates the tracking and cleavage activity of the Dna2 helicase/nuclease, which cleaves the flap to a length that does not support RPA binding, allowing FEN1 access. In this study, we explored the mechanism by which Dna2 enters and cleaves an RPA-coated flap. Using a nuclease-defective mutant of Dna2 (E675A), we showed that Dna2 removes RPA before the flap is reduced in length. This result is striking because RPA binds DNA with such high affinity and, in fact, binds more tightly than Dna2 (Table 2). In addition, we have examined the mechanism of RPA stimulation by Dna2 and found that RPA strand melting activity increases Dna2 cleavage through the removal of the DNA secondary structure.
Dna2 and RPA have been shown to interact physically and RPA stimulates Dna2 cleavage activity (9, 22). Based on these observations, our initial hypothesis was that Dna2 binds to flaps coated with RPA, allowing the Dna2 and RPA to bind each other. Because Dna2 requires tracking to cleave the flap, we envisioned that Dna2 would enter at the 5′-end and track down the flap associating with successive RPA molecules. The interactions would stimulate Dna2 to cleave the flap, releasing a mixture of RPA-bound flap segments, free RPA, and free DNA. Following cleavage by Dna2, the remaining short flap would be free of RPA for subsequent tracking and cleavage by FEN1. Instead, by using the Dna2 cleavage mutant, we found that Dna2 dissociates RPA independent of the cleavage process (Fig. 1). We believe that this is indicative of an RPA remodeling event caused by Dna2, which lowers the binding affinity of RPA. Because our reactions were done with a nuclease-defective Dna2 mutant, we cannot exclude the possibility that concurrent Dna2 cleavage activity aids in further destabilization of RPA binding.
A major function of RPA within the cell is to coat ssDNA, reducing its melting temperature, thus preventing the formation of secondary structure and reannealing (23–25). RPA must then be removed or displaced to permit other proteins access to the DNA. Fanning and colleagues (27, 44) have proposed that displacement occurs through protein-protein interactions, which remodel the conformation of RPA from an extended (high affinity) to a more compact (low affinity) form. The weakly bound form of RPA is then readily displaced by other proteins, allowing enzyme access to the DNA. We believe that Dna2 interacts with RPA in a similar manner to elicit its dissociation. Additionally, we have shown that RPA stimulates Dna2 by the removal of DNA secondary structure (Fig. 7). For this to occur, RPA must be displaced coordinately with the nearby binding of Dna2. This is the only way that we can visualize for RPA to relinquish the DNA to Dna2 before reformation of stable secondary structures.
Moreover, the ability of Dna2 to quantitatively dissociate RPA may require a polymer structure unique to a flap substrate, because Dna2 does not remove RPA from ssDNA but instead forms an RPA-Dna2 complex (Fig. 4). Possibly, Dna2 must also be conformationally altered by initial interaction with the base of the flap, before it can accomplish efficient removal of RPA. Additional studies are required to elucidate why binding and dissociation of RPA by Dna2 did not occur on ssDNA but these results indicate the importance of the flap structure.
Consistent with this interpretation, we discovered that tracking of Dna2 on the flap is not required for RPA removal (Fig. 3). We previously reported that Dna2 binds to a flap substrate even when tracking is blocked by a biotin-streptavidin at the flap 5′-end (34). When Dna2 binds in this non-tracking mode cleavage cannot occur but the Dna2 can still clear the flap of RPA, thus allowing cleavage by either another Dna2 molecule or FEN1. This would allow more rapid progression toward the final product, even if Dna2 binds the flap in the non-tracking mode. We have also considered that both Dna2 and RPA are proposed to be involved in overlapping cellular pathways, such as DNA repair, recombination, and telomere maintenance (20, 27–29, 45–48). Dissociation of RPA by Dna2 could extend to these other pathways in which RPA removal is necessary for further processing of the DNA.
To better understand how RPA alters the movements and nuclease functions of Dna2 on the flap, we conducted a detailed analysis of the Dna2 cleavage pattern in the presence of RPA. As long flaps arise during lagging strand replication, they have an increasing propensity to form DNA secondary structure. When this structure becomes stable enough to interfere with tracking, it can inhibit the nuclease activity of both Dna2 and FEN1. Dna2 helicase activity has previously been shown to provide little aid to tracking on structured flap substrates. In one study, Dna2 helicase function was shown to enhance Dna2 cleavage past a predicted 10-nt duplex in the flap region, which blocked cleavage by Dna2 (21). Although cleavage past the duplex was increased, the predominant products still occurred before the duplex region. In another study by our group, Dna2 helicase activity provided only minor stimulation of Dna2 cleavage past the predicted duplex regions in the flaps, as the amount of structure increased (38). Furthermore, we have shown here that cleavage by Dna2 alone is significantly reduced on a structured flap, containing a predicted 6-nt duplex, compared with cleavage on an unstructured flap (Fig. 7). These results indicate that Dna2 helicase activity alone is not sufficient to remove stable DNA secondary structure.
In our current work, the presence of RPA stimulated Dna2 cleavage on the predicted 6-nt duplex flap but not an unstructured flap (Fig. 7). RPA not only increased Dna2 cleavage but shifted cleavage further down the flap past the duplex. These results allow us to attribute Dna2 stimulation to relief of DNA secondary structure by RPA. Although, when a flap with a predicted 18-nt duplex was used, Dna2 cleavage was not increased, suggesting that RPA strand melting capacity is limited.
Although RPA strand melting may not fully unwind very stable structures, one could imagine that in vivo RPA would hinder such stable structures from forming. Reconstitution studies of Okazaki fragment processing have shown that a small fraction of flaps formed dynamically during lagging strand synthesis escape FEN1 cleavage and reach a length to which RPA can bind (14). Because RPA is an abundant protein within the nucleus and RPA binds ssDNA very rapidly (27–29), flaps not readily cleaved by FEN1 would be stably bound by RPA once they reach an adequate length. In this sense, RPA binding prepares the flap for Dna2 cleavage and stimulates cleavage activity by the removal of secondary structure. The cooperative action of RPA strand melting and the additional helicase activity of Dna2 may also be required if flaps have already formed stable secondary structure prior to RPA binding.
From these findings, we have developed a model of how RPA and Dna2 interact during Okazaki fragment processing (Fig. 8). When long flaps arise, they have a greater tendency to form DNA secondary structure. In the absence of RPA, Dna2 cleavage would be inhibited on these structured flaps. Binding by RPA would melt out the structure in the flap. Our current results show that Dna2 requires a flap structure for facile dissociation of RPA. Based on these observations, we propose that Dna2 recognizes the flap and begins the tracking process. It then dissociates RPA, moving through the ssDNA before the reformation of DNA secondary structure. During this process it cuts the flap to successively shorter lengths. Following cleavage by Dna2, the remaining short flap, which RPA cannot bind, would then be subject to cleavage by FEN1, creating a nick for ligation.
FIGURE 8.

A model of the proposed RPA/Dna2 interaction during Okazaki fragment processing. Strand displacement synthesis by pol δ creates flap intermediates that can become long and form DNA secondary structure. This structure inhibits cleavage activity by Dna2 or FEN1. RPA then uses its strand melting capacity to remove the structure and bind the flap. Dna2 tracking is then employed to remove RPA from the flap and cleave. Finally, the remaining RPA-free flap is cleaved by FEN1 to create a nick for ligation by DNA Ligase I.
We must also reconcile this model with the observation that Dna2 can dissociate RPA without tracking. Dna2 must track to cleave, and must acquire the unfolded DNA at the moment of RPA dissociation. In view of these facts, it is reasonable to conclude that RPA dissociation is accomplished during tracking. Blocked tracking is an unnatural condition during Okazaki fragment processing. The fact that RPA dissociation can still be accomplished reveals a feature of the dissociation mechanism, but may not represent the coordinated interactions that occur when tracking is allowed.
An additional value of the facile dissociation of RPA by Dna2 is the likelihood that Dna2 slows RPA-promoted flap elongation. We and the Seo group (9, 14) have proposed that the two-nuclease pathway for flap removal has evolved to cleave flaps that become too long for processing by FEN1 alone. Although RPA strand melting activity would aid Dna2 in flap cleavage, an additional consequence of strand melting is the potential for further flap elongation. RPA strand melting capacity was shown to bind to AT-rich regions of the SV40 genome and catalyze unwinding of the DNA (40). Additionally, we have demonstrated that RPA stimulates Dna2 cleavage into the annealed portion of the primer, as a consequence of RPA strand melting activity (Fig. 5). RPA-promoted strand melting, as assessed by RPA unwinding, was increased when AT-rich downstream double-stranded regions were examined and diminished when AT content was minimized. We suggest that Dna2-directed dissociation of RPA would help to slow flap displacement, minimizing unnecessary degradation of the DNA portion of Okazaki fragments during processing.
Finally, the interaction between RPA and Dna2 appears to be specific to proteins from the same species. Previous analysis of Dna2 cleavage reactions with RPA revealed that RPA of the same species stimulated Dna2 cleavage, but when homologues of RPA and Dna2 from different species were tested together Dna2 cleavage was inhibited (31). We were able to reproduce this phenomenon using the 53-nt flap substrate with hRPA and yeast Dna2 (Fig. 2A). We extended this analysis to show that dissociation of bound hRPA by yeast Dna2 is inhibited suggesting that dissociation of the flap-bound RPA is required for cleavage. Even hRPA, which shares ∼30% identical and 45% similar sequence homology with S. cerevisiae RPA (28), is unable to be dissociated by Dna2 (Fig. 2B). This contrasts with the facile dissociation of RPA seen when yeast RPA and Dna2 were tested (see Fig. 1). Importantly, this result is also further confirmation that Dna2-promoted dissociation of RPA is a very specific phenomenon, and not a consequence of a simple binding competition. Specificity is additionally supported by results with the E. coli homolog of RPA, single-stranded binding protein, which was found to inhibit yeast Dna2 cleavage (9). Taken together, we can conclude that Dna2 specifically interacts with RPA to facilitate dissociation followed by Dna2 cleavage.
In conclusion, we have more closely defined the interactions between RPA and Dna2. Our findings show that RPA strand melting capacity can relieve the DNA secondary structure in the flap to facilitate rapid and efficient tracking and cleavage by Dna2. Concurrently, Dna2 dissociates the flap-bound RPA to access the flap for cleavage, leaving a short RPA-free flap. FEN1 cleavage then creates a nicked product for subsequent ligation and fragment joining. In this fashion, Dna2 and RPA have evolved to coordinate their functions to ensure proper primer removal during Okazaki fragment processing.
Acknowledgments
We thank Drs. Sara Binz and Marc Wold for the purified yeast and human RPA proteins. In addition, we thank members of the Bambara and Campbell laboratories for valuable discussion and critical review of the manuscript.
This work was supported by National Institutes of Health Grant GM024441 (to R. A. B.), with additional support from Grant GM087666 (to J. L. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: pol, polymerase; FEN1, flap endonuclease 1; RPA, replication protein A; hRPA, human RPA; nt, nucleotide; ss, single-stranded.
References
- 1.Kornberg, A., and Baker, T. A. (1992) DNA Replication, 2nd Ed., pp. 140–144, W. H. Freeman, New York
- 2.Rossi, M. L., Purohit, V., Brandt, P. D., and Bambara, R. A. (2006) Chem. Rev. 106 453–473 [DOI] [PubMed] [Google Scholar]
- 3.Shevelev, I. V., and Hubscher, U. (2002) Nat. Rev. Mol. Cell. Biol. 3 364–376 [DOI] [PubMed] [Google Scholar]
- 4.Ayyagari, R., Gomes, X. V., Gordenin, D. A., and Burgers, P. M. (2003) J. Biol. Chem. 278 1618–1625 [DOI] [PubMed] [Google Scholar]
- 5.Maga, G., Frouin, I., Spadari, S., and Hubscher, U. (2001) J. Biol. Chem. 276 18235–18242 [DOI] [PubMed] [Google Scholar]
- 6.Podust, V. N., Podust, L. M., Goubin, F., Ducommun, B., and Hubscher, U. (1995) Biochemistry 34 8869–8875 [DOI] [PubMed] [Google Scholar]
- 7.Jin, Y. H., Ayyagari, R., Resnick, M. A., Gordenin, D. A., and Burgers, P. M. (2003) J. Biol. Chem. 278 1626–1633 [DOI] [PubMed] [Google Scholar]
- 8.Liu, Y., Kao, H. I., and Bambara, R. A. (2004) Annu. Rev. Biochem. 73 589–615 [DOI] [PubMed] [Google Scholar]
- 9.Bae, S. H., Bae, K. H., Kim, J. A., and Seo, Y. S. (2001) Nature 412 456–461 [DOI] [PubMed] [Google Scholar]
- 10.Bae, S. H., and Seo, Y. S. (2000) J. Biol. Chem. 275 38022–38031 [DOI] [PubMed] [Google Scholar]
- 11.Arunkumar, A. I., Stauffer, M. E., Bochkareva, E., Bochkarev, A., and Chazin, W. J. (2003) J. Biol. Chem. 278 41077–41082 [DOI] [PubMed] [Google Scholar]
- 12.Bochkareva, E., Korolev, S., Lees-Miller, S. P., and Bochkarev, A. (2002) EMBO J. 21 1855–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henricksen, L. A., Umbricht, C. B., and Wold, M. S. (1994) J. Biol. Chem. 269 11121–11132 [PubMed] [Google Scholar]
- 14.Rossi, M. L., and Bambara, R. A. (2006) J. Biol. Chem. 281 26051–26061 [DOI] [PubMed] [Google Scholar]
- 15.Budd, M. E., and Campbell, J. L. (1995) Proc. Natl. Acad. Sci. U. S. A 92 7642–7646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuo, C., Nuang, H., and Campbell, J. L. (1983) Proc. Natl. Acad. Sci. U. S. A 80 6465–6469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bae, S. H., Choi, E., Lee, K. H., Park, J. S., Lee, S. H., and Seo, Y. S. (1998) J. Biol. Chem. 273 26880–26890 [DOI] [PubMed] [Google Scholar]
- 18.Budd, M. E., and Campbell, J. L. (1997) Mol. Cell. Biol. 17 2136–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masuda-Sasa, T., Polaczek, P., and Campbell, J. L. (2006) J. Biol. Chem. 281 38555–38564 [DOI] [PubMed] [Google Scholar]
- 20.Budd, M. E., Tong, A. H., Polaczek, P., Peng, X., Boone, C., and Campbell, J. L. (2005) PLoS Genet 1 e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bae, S. H., Kim, D. W., Kim, J., Kim, J. H., Kim, D. H., Kim, H. D., Kang, H. Y., and Seo, Y. S. (2002) J. Biol. Chem. 277 26632–26641 [DOI] [PubMed] [Google Scholar]
- 22.Bae, K. H., Kim, H. S., Bae, S. H., Kang, H. Y., Brill, S., and Seo, Y. S. (2003) Nucleic Acids Res. 31 3006–3015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brill, S. J., and Stillman, B. (1991) Genes Dev. 5 1589–1600 [DOI] [PubMed] [Google Scholar]
- 24.Heyer, W. D., Rao, M. R., Erdile, L. F., Kelly, T. J., and Kolodner, R. D. (1990) EMBO J. 9 2321–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haring, S. J., Mason, A. C., Binz, S. K., and Wold, M. S. (2008) J. Biol. Chem. 283 19095–19111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wold, M. S., and Kelly, T. (1988) Proc. Natl. Acad. Sci. U. S. A 85 2523–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fanning, E., Klimovich, V., and Nager, A. R. (2006) Nucleic Acids Res. 34 4126–4137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wold, M. S. (1997) Annu. Rev. Biochem. 66 61–92 [DOI] [PubMed] [Google Scholar]
- 29.Iftode, C., Daniely, Y., and Borowiec, J. A. (1999) Crit. Rev. Biochem. Mol. Biol. 34 141–180 [DOI] [PubMed] [Google Scholar]
- 30.Kao, H. I., Campbell, J. L., and Bambara, R. A. (2004) J. Biol. Chem. 279 50840–50849 [DOI] [PubMed] [Google Scholar]
- 31.Kim, D. H., Lee, K. H., Kim, J. H., Ryu, G. H., Bae, S. H., Lee, B. C., Moon, K. Y., Byun, S. M., Koo, H. S., and Seo, Y. S. (2005) Nucleic Acids Res. 33 1372–1383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Budd, M. E., Choe, W., and Campbell, J. L. (2000) J. Biol. Chem. 275 16518–16529 [DOI] [PubMed] [Google Scholar]
- 33.Sibenaller, Z. A., Sorensen, B. R., and Wold, M. S. (1998) Biochemistry 37 12496–12506 [DOI] [PubMed] [Google Scholar]
- 34.Stewart, J. A., Campbell, J. L., and Bambara, R. A. (2006) J. Biol. Chem. 281 38565–38572 [DOI] [PubMed] [Google Scholar]
- 35.Mathews, D. H., Sabina, J., Zuker, M., and Turner, D. H. (1999) J. Mol. Biol. 288 911–940 [DOI] [PubMed] [Google Scholar]
- 36.Zuker, M. (2003) Nucleic Acids Res. 31 3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim, C., Paulus, B. F., and Wold, M. S. (1994) Biochemistry 33 14197–14206 [DOI] [PubMed] [Google Scholar]
- 38.Kao, H. I., Veeraraghavan, J., Polaczek, P., Campbell, J. L., and Bambara, R. A. (2004) J. Biol. Chem. 279 15014–15024 [DOI] [PubMed] [Google Scholar]
- 39.Georgaki, A., Strack, B., Podust, V., and Hubscher, U. (1992) FEBS Lett. 308 240–244 [DOI] [PubMed] [Google Scholar]
- 40.Treuner, K., Ramsperger, U., and Knippers, R. (1996) J. Mol. Biol. 259 104–112 [DOI] [PubMed] [Google Scholar]
- 41.Georgaki, A., and Hubscher, U. (1993) Nucleic Acids Res. 21 3659–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kao, H. I., Henricksen, L. A., Liu, Y., and Bambara, R. A. (2002) J. Biol. Chem. 277 14379–14389 [DOI] [PubMed] [Google Scholar]
- 43.Masuda-Sasa, T., Imamura, O., and Campbell, J. L. (2006) Nucleic Acids Res. 34 1865–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arunkumar, A. I., Klimovich, V., Jiang, X., Ott, R. D., Mizoue, L., Fanning, E., and Chazin, W. J. (2005) Nat. Struct. Mol. Biol. 12 332–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith, J., Zou, H., and Rothstein, R. (2000) Biochimie (Paris) 82 71–78 [DOI] [PubMed] [Google Scholar]
- 46.Schramke, V., Luciano, P., Brevet, V., Guillot, S., Corda, Y., Longhese, M. P., Gilson, E., and Geli, V. (2004) Nat. Genet. 36 46–54 [DOI] [PubMed] [Google Scholar]
- 47.Salas, T. R., Petruseva, I., Lavrik, O., Bourdoncle, A., Mergny, J. L., Favre, A., and Saintome, C. (2006) Nucleic Acids Res. 34 4857–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choe, W., Budd, M., Imamura, O., Hoopes, L., and Campbell, J. L. (2002) Mol. Cell. Biol. 22 4202–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]