

Abstract
Recessive inactivating mutations in human matrix metalloproteinase 2 (MMP2, gelatinase A) are associated with syndromes that include abnormal facial appearance, short stature, and severe bone loss. Mmp2-/- mice have only mild aspects of these abnormalities, suggesting that MMP2 function is redundant during skeletal development in the mouse. Here, we report that Mmp2-/- mice with additional mutations that render type I collagen resistant to collagenase-mediated cleavage to TCA and TCB fragments (Col1a1r/r mice) have severe developmental defects resembling those observed in MMP2-null humans. Composite Mmp2-/-;Col1a1r/r mice were born in expected Mendelian ratios but were half the size of wild-type, Mmp2-/-, and Col1a1r/r mice and failed to thrive. Furthermore, composite Mmp2-/-;Col1a1r/r animals had very abnormal craniofacial features with shorter snouts, bulging skulls, incompletely developed calvarial bones and unclosed cranial sutures. In addition, trabecular bone mass was reduced concomitant with increased numbers of bone-resorbing osteoclasts and osteopenia. In vitro, MMP2 had a unique ability among the collagenolytic MMPs to degrade mutant collagen, offering a possible explanation for the genetic interaction between Mmp2 and Col1a1r. Thus, because mutations in the type I collagen gene alter the phenotype of mice with null mutations in Mmp2, we conclude that type I collagen is an important modifier gene for Mmp2.
Keywords: matrix metalloproteinase, type I collagen, osteopenia
INTRODUCTION
Remodeling of the extracellular matrix (ECM) by matrix metalloproteinases (MMPs) is critical for development of the skeleton, and mice deficient for Mmp9, Mmp13, and Mmp14 (Mt1-mmp) have skeletal defects (Vu et al., 1998; Holmbeck et al., 1999; Zhou et al., 2000; Stickens et al., 2004). Inactivating mutations in human MMP2 are the cause of rare but severe skeletal defect syndromes in patients born from consanguineous marriages (Martignetti et al., 2001; Zankl et al., 2005; Rouzier et al., 2006). The clinical findings in these MMP2-null patients include hyperextension of metacarpophalangeal joints, flexion contractures of large joints, dysmorphic faces, significant growth restrictions and, surprisingly, loss of bone mass with osteolysis (Al Aqeel et al., 2000; Al-Mayouf et al., 2000; Zankl et al., 2005; Rouzier et al., 2006). In contrast to MMP2-null humans, Mmp2-/- mice have mild skeletal defects and no focal osteolysis (Inoue et al., 2006).
Genetic modifiers are genes that interact with a phenotype-associated gene and alter the observed phenotype, for example the severity of the phenotype (Nadeau, 2001). Genetic modifier studies in mice can be powerful tools to identify genetic interactions with relevance for human disease. A recent example comes from cystic fibrosis, one of the most common human autosomal recessive diseases. Mutations in cystic fibrosis transmembrane conductance regulator (CFTR), which encodes a membrane-bound chloride ion channel, cause impaired fluid and salt secretion resulting in mucus accumulation in various tissues. The clinical presentation of cystic fibrosis can vary with age-of-onset and disease severity and this variability has been ascribed both to differences in the specific mutations in the CFTR gene and to genetic modifiers (Nadeau, 2001). Mice deficient for the Cftr gene (Cftr-/- mice) die soon after birth on most genetic backgrounds, but live for many months on others. These observations indicate the existence of a modifier gene that modulates the severity of the phenotype of Cftr-/- mice. Through the intercrossing of mouse strains with different phenotypes and subsequent linkage analysis, a single locus with a semidominant effect on viability was identified (Rozmahel et al., 1996). The identification of the modifier locus in the mouse led to identification of the corresponding human locus, which turned out to be a genetic modifier of the human disease cystic fibrosis (Zielenski et al., 1999).
In the case of MMP2, the difference between the phenotype of Mmp2-/- mice and MMP2-null humans has been attributed to redundancy of Mmp2 gene function in the mouse (Martignetti et al., 2001). Because Mmp14-/- mice have severe skeletal defects with craniofacial abnormalities, growth restrictions, and osteopenia (Holmbeck et al., 1999; Zhou et al., 2000), it has been suggested that MMP14 in the mouse serves a function similar to MMP2 in human skeletal development (Martignetti et al., 2001). However, of the MMP2-null patients with osteolytic syndromes, all but one have been of Arabic descent and all have been born from consanguineous marriages. Thus, it is possible that there are genetic modifiers of MMP2, which have cosegregated with the MMP2 mutations in the affected individuals, and that such genetic modifiers are responsible for the reported different phenotypes of MMP2-null humans and Mmp2-/- mice. We had noticed that a low percentage (∼10%) of Mmp2-/- mice from an inbred line on the C57BL/6 background were severely runted, had abnormally shaped faces, a wide gait, failed to thrive, and rarely lived beyond 4 weeks of age. The runted mice were always Mmp2-/-, never wild-type or Mmp2+/-. The low penetrance of the runted phenotype of Mmp2-/- mice indicated existence of a recessive genetic modifier of Mmp2 in the mouse. Rather than identifying the putative modifier through linkage analysis, we took a candidate approach. The small stature and abnormal craniofacial features of the runted Mmp2-/- mice resembled the appearance of Mmp14-/- mice (Holmbeck et al., 1999; Zhou et al., 2000), but because Mmp2-/-;Mmp14-/- double-deficient mice die immediately after birth (Oh et al., 2004), the spontaneous mutation was unlikely to directly involve Mmp14. MMP14 activates proMMP2 by cleaving off its prodomain (Strongin et al., 1995); thus, we speculated that a genetic modifier of Mmp2 could be another substrate of MMP14, namely type I collagen, which is also abundant in bones (Holmbeck et al., 1999).
Type I collagen is a heterotrimer molecule composed of two α1(I) and one α2(I) polypeptide chains encoded by two separate genes: Col1a1 and Col1a2 (Cutroneo, 2003). Type I collagen has both structural and signaling functions, the latter being mediated by integrins and discoidin domain receptors (Shrivastava et al., 1997; Heino, 2000). Type I collagen is cleaved into characteristic TCA and TCB fragments at approximately one quarter and three quarters of the length of the native molecule by collagenolytic MMPs, including MMP1, MMP2, MMP8, MMP13, and MMP14 (Ohuchi et al., 1997). These cleavage fragments are present in bone (Holmbeck et al., 1999; Stickens et al., 2004) and can initiate signaling that affects cell migration and survival (Montgomery et al., 1994; Petitclerc et al., 1999; Stringa et al., 2000; Fera et al., 2004).
Type I collagen is abundant in the skeleton where it contributes to load-bearing capacity by assembling into ordered fibrils. Naturally occurring mutations in type I collagen genes render type I collagen molecules more fragile and lead to osteogenesis imperfecta, characterized by profound skeletal fragility (Barsh et al., 1982, 1985; Bonadio et al., 1985). Conversely, Col1a1r/r mice with homozygous, engineered mutations (r) in the Col1a1 gene, which render type I collagen resistant to cleavage to TCA and TCB fragments by collagenolytic MMPs (including MMP2, MMP8, MMP13, and MMP14), continually deposit new bone, resulting in progressive thickening of the calvariae and increased trabecular bone density (Liu et al., 1995; Zhao et al., 2000; Chiusaroli et al., 2003). Mice deficient for MMP13 or MMP14, which both have high activity against type I collagen, also display abnormal skeletal development (Holmbeck et al., 1999; Zhou et al., 2000; Stickens et al., 2004).
Here, we test the hypothesis that type I collagen is a modifier of Mmp2 during skeletal development in the mouse. We report that mice that concomitantly are deficient for Mmp2 and carry the mutant Col1a1r gene show skeletal changes resembling those reported in the human skeletal syndromes associated with inactivating mutations in MMP2.
RESULTS
Mmp2-/-;Col1a1r/r Mice Are Severely Growth Retarded
Col1a1r/r mice have mutations in the Col1a1 gene that render type I collagen resistant to cleavage by collagenolytic MMPs (Liu et al., 1995). To determine whether type I collagen was a modifier of Mmp2, we intercrossed Mmp2-/- mice with Col1a1r/r mice. When the resulting Mmp2+/-;Col1a1r/+ mice were further intercrossed, Mendelian ratios of the nine possible genotypes were observed during embryogenesis and immediately after birth (Table 1). However, at weaning, 21 days after birth, the distribution of the genotypes was significantly different from the expected values with fewer than expected Mmp2-/-;Col1a1r/+ and Mmp2-/-;Col1a1r/r mice (Table 1, χ2 = 24.7; P = 0.002). Thus, whereas Mmp2-/- and Col1a1r/r mice had normal survival, Mmp2-/- mice with at least one Col1a1r allele showed reduced postnatal survival.
TABLE 1.
Postnatal Death of Mmp2+/+;Col1a1r/r Micea
| Age |
Mmp2+/+; Col1a1+/+ |
Mmp2+/-; Col1a1+/+ |
Mmp2+/+; Col1a1r/+ |
Mmp2+/-; Col1a1r/+ |
Mmp2+/+; Col1a1r/r |
Mmp2-/-; Col1a1+/+ |
Mmp2+/-; Col1a1r/r |
Mmp2-/-; Col1a1r/+ |
Mmp2-/-; Col1a1r/r |
Total number of mice | χ2 | P value |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E9.5 | 6.10% (5) | 13.41% (11) | 18.29% (15) | 23.17% (19) | 6.10% (5) | 3.66% (3) | 13.41% (11) | 9.76% (8) | 6.10% (5) | 82 | 3.8 | >0.6 |
| E14.5 | 6.67% (4) | 10.00% (6) | 11.67% (7) | 25.00% (15) | 6.67% (4) | 13.33% (8) | 10.00% (6) | 11.67% (7) | 5.00% (3) | 60 | 5.67 | >0.6 |
| p1 | 6.85% (5) | 13.70% (10) | 9.59% (7) | 30.14% (22) | 8.22% (6) | 2.74% (2) | 8.22% (6) | 12.33% (9) | 8.22% (6) | 73 | 4.81 | >0.6 |
| p21 | 6.35% (8) | 15.08% (19) | 12.70% (16) | 25.40% (32) | 11.11% (14) | 4.76% (6) | 19.84% (25) | 3.97% (5) | 0.79% (1) | 126 | 24.7 | 0.002 |
| Expected Mendelian ratios | 6.25% | 12.50% | 12.50% | 25.00% | 6.25% | 6.25% | 12.50% | 12.50% | 6.25% |
Mmp2+/+;Col1a1r/+ mice were intercrossed and the genotypes of their offspring determined during embryogenesis (E9.5 and E14.5), at birth (postnatal day 1, p1), and at weaning (p21). The distributions of genotypes (the number of mice is in parentheses) were compared to the expected Mendelian distributions with χ2 test. The χ2 and P values (8 degrees of freedom) are indicated at each time point.
The Mmp2-/-;Col1a1r/r mice were only approximately half the size of wild-type, Mmp2-/- or Col1a1r/r mice (Fig. 1A). A few Mmp2-/-;Col1a1r/r mice survived for 16 weeks or longer and their body weight was recorded weekly. At birth, the body weight of the Mmp2-/-;Col1a1r/r mice was comparable to wild-type, Mmp2-/-, and Col1a1r/r mice. However, the weight of the Mmp2-/-;Col1a1r/r mice failed to increase from approximately the fourth week of age, coinciding with the growth spurt of the long bones, and remained significantly less than the weights of wild-type, Mmp2-/-, or Col1a1r/r mice for the duration of their lifetime (Fig. 1B). The runted phenotype showed 100% penetrance in Mmp2-/-;Col1a1r/r mice and was also often found in Mmp2-/-;Col1a1r/+ mice, whereas Mmp2+/-;Col1a1r/r animals were not runted (data not shown). None of the Mmp2-/-;Col1a1r/r mice were able to produce offspring.
Fig. 1.
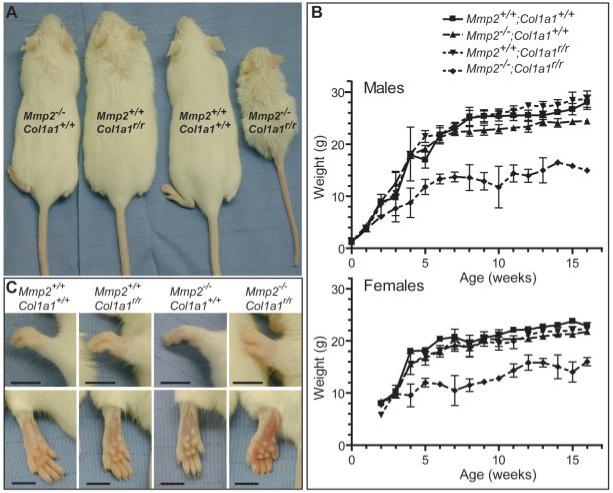
Mmp2-/-;Col1a1r/r mice are severely growth retarded, with swollen and hyperextended metacarpophalangeal joints. A: Adult (age, 8-11 weeks) wild-type (Mmp2+/+;Col1a1+/+), Mmp2-/-;Col1a1+/+, Mmp2+/+; Col1a1r/r, and Mmp2-/-;Col1a1r/r mice (backcrossed to FVB/n). B: Growth curves of male (top) and female (bottom) Mmp2+/+;Col1a1+/+, Mmp2-/-;Col1a1+/+, Mmp2+/+;Col1a1r/r, and Mmp2-/-;Col1a1r/r mice weighed weekly (mean ± SD from repeated measurements), n = 3 for Mmp2+/+;Col1a1+/+, Mmp2-/-;Col1a1+/+, and Mmp2+/+;Col1a1r/r. However, because some Mmp2-/-;Col1a1r/r mice died, six Mmp2-/-;Col1a1r/r mice were followed up for each sex, although not all mice could be weighed at all time points. The curves are significantly different for both males and females using two-way analysis of variance repeated-measurements test. C: Photographs of front (top) and hind (bottom) paws from 7-week-old Mmp2+/+;Col1a1+/+,Mmp2-/-;Col1a1+/+, Mmp2+/+;Col1a1r/r, and Mmp2-/-;Col1a1r/r mice. Scale bar is 5 mm.
Mmp2-/-;Col1a1r/r Mice Have Edematous Paws, Hyperextended Metacarpophalangeal Joints, and Reduced Mobility of Large Joints
Humans with inactivating MMP2 mutations have characteristic swelling of fingers and toes, hyperextension of metacarpophalangeal joints, and either hyperextension or flexion contracture of most other joints (Al Aqeel et al., 2000). While handling Mmp2-/-;Col1a1r/r mice, we noticed that their range of motion, particularly of the knee, hip, shoulder, and elbow joints, was markedly reduced (8 of 8 mice). Strikingly, paws of the Mmp2-/-;Col1a1r/r mice became visibly swollen, and the metacarpophalangeal joints hyperextended around the time of weaning (Fig. 1C). To establish if the increased size of their paws was due to edema, we determined the apparent diffusion coefficient (ADC) using diffusion weighted magnetic resonance imaging (MRI). The ADC is an indirect measure of water content with a higher value reflecting increased water mobility (Le Bihan et al., 1986), which can result from the expanded interstitial space characteristic of edema. The ADC was greater in hind paws of Mmp2-/-;Col1a1r/r mice ([1.30 ± 0.05] × 10-3 mm2/sec, n = 2) than in control mice ([0.98 ± 0.03] × 10-3 mm2/sec, n = 2, one aged-matched wild-type and one Mmp2+/-;Col1a1r/r littermate). These results indicate that the paws of the Mmp2-/-;Col1a1r/r mice were edematous. Tissue involved in edema was most likely soft tissue because the T2-hyperintense regions with the highest ADC values were located at the periphery of the paws (data not shown).
Mmp2-/-;Col1a1r/r Mice Display Abnormal Craniofacial Development With Impaired Calvarial Ossification and Lack of Calvarial Suture Closure
Humans with recessive mutations in MMP2 have flattened faces (Al Aqeel et al., 2000; Al-Mayouf et al., 2000; Zankl et al., 2005). The facial features were also changed in the Mmp2-/-;Col1a1r/r mice, which had shortened snouts and bulging skulls (Fig. 2A,B). Facial changes of the Mmp2-/-;Col1a1r/r mice were accompanied by incomplete intramembranous ossification of calvarial bones (Fig. 2C,D). Even in the single 5-month-old Mmp2-/-;Col1a1r/r mouse that we were able to study, calvarial bones had not completely ossified (data not shown). Instead of bone, a sclerotic membrane was present in adult animals. Mild delays were also evident in Mmp2-/- and Col1a1r/r mice, but the cranial sutures of the single mutants were closed completely by 8 weeks of age (Fig. 2C). The cranial bones that developed in Mmp2-/-;Col1a1r/r mice had a very abnormal histology with a thin and porous appearance (Fig. 2E). The bones from Col1a1r/r mice were slightly osteopetrotic as previously reported (Zhao et al., 2000).
Fig. 2.
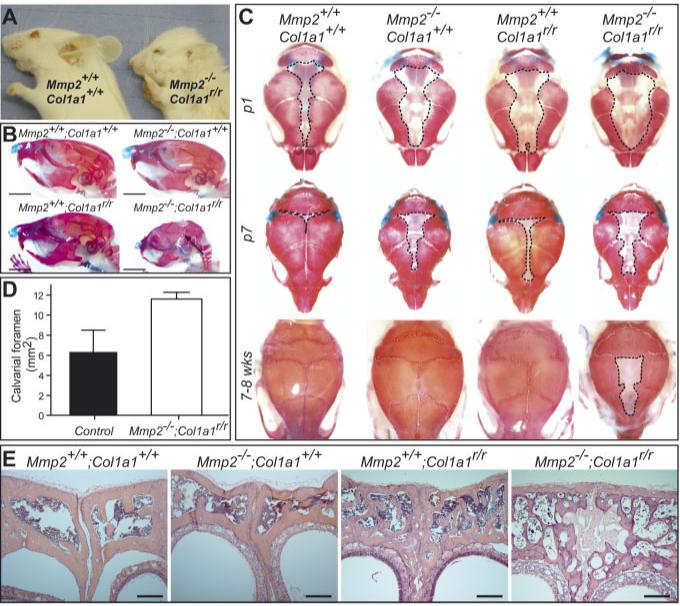
Mmp2-/-;Col1a1r/r mice have abnormal calvarial bone development. A: Side view of heads of 7-week-old Mmp2+/+;Col1a1+/+ and Mmp2-/-;Col1a1r/r mice. B: Side view of skulls of 8-week-old Mmp2+/+;Col1a1+/+, Mmp2-/-; Col1a1+/+, Mmp2+/+;Col1a1r/r, and Mmp2-/-; Col1a1r/r mice stained with Alcian blue (cartilage) and alizarin red (bone). Arrow points to open calvarial sutures in adult Mmp2-/-;Col1a1r/r mice. Scale bar is 5 mm. C: Top-view of skulls from the different genotypes after staining with Alcian blue and Alizarin red at birth (p1), 7 days of age (p7), and 7-8 weeks of age (7-8 wks). D: Quantification of the calvarial foramen (outlined with dashed lines in C) at birth (mean ± SD). The calvarial foramen was significantly larger (P = 0.006, two-sided Student’s t-test) in Mmp2-/-;Col1a1r/r animals (n = 4) than in control littermates with at least one wildtype Mmp2 and Col1a1 allele (n = 3). E: Histology of the frontal bones from 8-week-old mice stained with hematoxylin and eosin. Scale bar = 200 μm.
Long Bones of Mmp2-/-;Col1a1r/r Mice Are Osteopenic
In contrast to the severe defects in cranial development, long bones of Mmp2-/-;Col1a1r/r appeared largely normal, although smaller, when examined by whole skeletal preparation (Fig. 3A). Humans with MMP2-null mutations suffer from osteolytic lesions in their long bones that can be so debilitating that they become wheel-chair-bound (Al-Mayouf et al., 2000). Microcomputed tomography (micro-CT) analysis of Mmp2-/-;Col1a1r/r mice showed reduced trabecular bone density in tibiae, indicating osteopenia (Fig. 3B; due to the reduced survival of the Mmp2-/-;Col1a1r/r mice, only one animal was analyzed at 16 weeks of age). The relative bone volume (BV/TV), which takes into account that Mmp2-/-;Col1a1r/r mice are significantly smaller than their controls, was much lower in tibiae from Mmp2-/-;Col1a1r/r than from wildtype, Mmp2-/-, or Col1a1r/r agedmatched controls (Fig. 3C). Similarly, trabecular thickness (DT-Tb.Th) and cortical thickness at the diaphysis (DT-C.Th-Diaph) were lower in Mmp2-/-;Col1a1r/r mice than in controls. Trabecular separation (TRI-Tb.Sp) was increased in Mmp2-/-;Col1a1r/r mice compared with controls, indicating active bone resorption (Fig. 3C); however, the increase was skewed by the one 16-week-old Mmp2-/-;Col1a1r/r mouse. Interestingly, this mouse had the lowest bone volume and highest trabecular separation of all the mice (Fig. 3C, red triangular symbols). Both type I collagen and MMP2 are expressed by osteoblasts in the trabecular bone (Dacquin et al., 2002; Stickens et al., 2004), and Safranin-O/Fast Green staining demonstrated that there was less trabecular bone in tibiae of Mmp2-/-;Col1a1r/r than in control Mmp2+/-;Col1a1r/r littermates (Fig. 3D). In addition, the number of tartrate-resistant acid phosphatase (TRAP)-positive osteoclasts was increased in both trabecular and cortical bone in Mmp2-/-;Col1a1r/r mice compared with aged-matched controls (Fig. 3E).
Fig. 3.
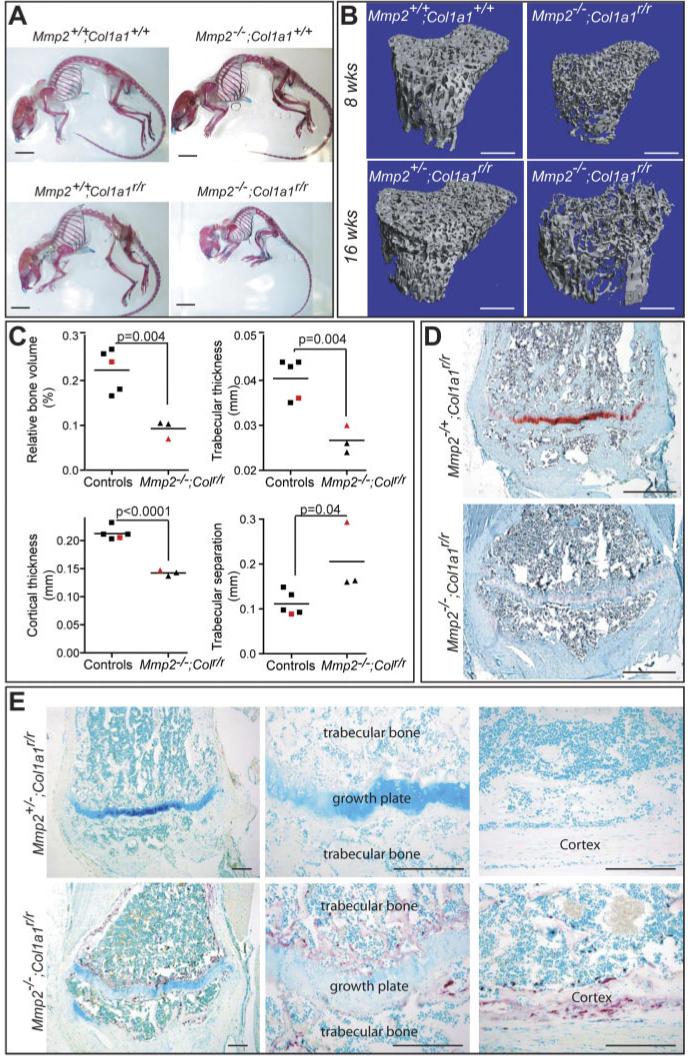
Osteopenia in long bones of Mmp2-/-;Col1a1r/r mice. A: Whole skeletons of 8-week-old animals, stained with Alcian blue (cartilage) and Alizarin red (bone). B: Microcomputed tomography (micro-CT) of tibiae of Mmp2-/-;Col1a1r/r and controls at 8 and 16 weeks of age. C: Relative bone volume (BV/TV), trabecular thickness (DT-Tb.Th), cortical thickness at the diaphysis (DT-C.Th.-Diaph), and trabecular separation (TRI-Tb.Sp.) were determined by micro-CT of tibiae from Mmp2-/-;Col1a1r/r and their aged-matched controls (two Mmp2+/+;Col1a1+/+, one Mmp2-/-;Col1a1+/+, one Mmp2+/+;Col1a1r/r, and one Mmp2+/-;Col1a1r/r). All mice were 7-8 weeks old, except the two mice represented with red symbols that were 16 weeks old. All measurements were significantly different by two-sided Student’s t-test. D: Safranin-O staining of tibia from 16-week-old Mmp2-/-;Col1a1r/r and Mmp2-/+;Col1a1r/r littermates showing reduced trabecular bone in the former. E: Increased tartrate-resistant acid phosphatase (TRAP)-positive (purple) osteoclasts in both trabecular bone and cortex of tibiae from Mmp2-/-;Col1a1r/r counter-stained with fast green. Scale bar = 1 cm in A, 5 mm in B, 500 μm in D, 200 μm in E.
MMP2 Degrades Collagenase-Resistant Type I Collagen at Physiological Temperatures
MMP2 is important for degradation of denatured, cleaved type I collagen, and, although it is often not considered a type I collagenase, it also has activity against native type I collagen (Tournier et al., 1994; Aimes and Quigley, 1995; Tam et al., 2004). The genetic interaction between Mmp2 and Col1a1r/r indicated that type I collagen could be a direct substrate of MMP2. Therefore, we compared the ability of MMP2 to degrade normal and mutant collagen in vitro with two classic collagenolytic MMPs: MMP13 and MMP14. At 25°C, MMP13 and MMP14 degraded wild-type collagen, resulting in generation of the classic TCA fragments, MMP2 had no activity against wild-type collagen, and none of the MMPs degraded the mutant collagen (Fig. 4A), as previously reported (Liu et al., 1995). However, at the physiological temperature of 37°C, MMP2 degraded wild-type and, even more efficiently, mutant collagen, but degradation did not generate TCA fragments. In contrast, MMP13 and MMP14 still had reduced activity against mutant collagen at 37°C (Fig. 4B). Thus, the genetic interaction between Mmp2 and Col1a1r could be caused by a requirement for MMP2 activity to degrade mutant type I collagen.
Fig. 4.
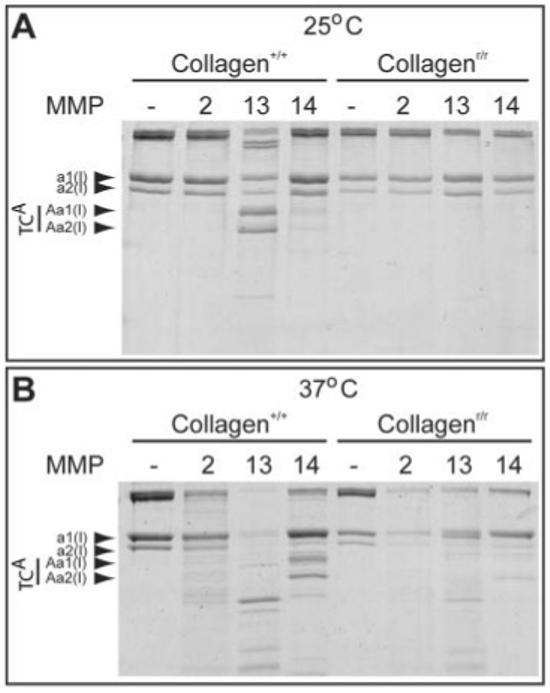
Collagenase-resistant type I collagenr/r is degraded by matrix metalloproteinase 2 (MMP2) at physiological temperature. A,B: Wild-type (collagen+/+) and mutant (collagenr/r) type I collagen was digested in vitro for 1 hr with MMP2, -13, or -14 at 25°C (A) and 37°C (B). Col1α1 (a1(I)) and Col1α2 (a2(I)) chains and their TCA cleavage products (Aa1(I) and Aa2(I)) are indicated. At 37°C, the mutant Col1α1 chain is not cleaved to the normal fragments, but is degraded by MMPs, especially by MMP2. Trace amounts of cleaved TCA (Aa2(I)) from the nonmutant Col1α2 chain were found when digesting mutant type I collagenr/r.
DISCUSSION
Mmp2-/-;Col1a1r/r Mice Have Skeletal Defects Resembling Those of Humans With MMP2 Null Mutations
Humans null for MMP2 have severe osteolytic syndromes, whereas Mmp2-/- mice have only mild skeletal defects (Itoh et al., 1997; Al Aqeel et al., 2000; Al-Mayouf et al., 2000; Martignetti et al., 2001; Zankl et al., 2005; Inoue et al., 2006; Rouzier et al., 2006). The spontaneous occurrence of a low penetrance, runted phenotype with abnormal craniofacial features in our Mmp2-/- mouse colony led us to hypothesize that there were genetic modifiers of Mmp2. Rather than identifying the specific genetic modification in our line, we took a candidate approach: proMMP2 is a substrate of MMP14, and we speculated that type I collagen, another classic MMP14 substrate with established function in bone development, could modify Mmp2. Indeed, severe skeletal developmental defects were observed when Mmp2-/- mice were intercrossed with Col1a1r/r mice, which carry targeted mutations in the Col1a1 gene rendering type I collagen resistant to classic collagenase cleavage. These Mmp2-/-;Col1a1r/r mice had several striking similarities to humans with MMP2-null mutations: in addition to decreased body size, these mice had osteopenic bones, craniofacial defects, failure to close calvarial sutures, decreased range of motion of joints, hyperextended metacarpophalangeal joints, and edematous paws (Table 2). Thus, we conclude that, in mice, type I collagen is a genetic modifier of MMP2, possibly because MMP2 can degrade mutant collagen (r). The Col1a1r mutation is a dominant modifier on the Mmp2-/- background, because the phenotype of Mmp2-/-;Col1a1r/+ resembled the phenotype of Mmp2-/-;Col1a1r/r mice. In contrast, the low penetrance of the spontaneously runted Mmp2-/- phenotype is suggestive of a recessive modifier. Thus, it is unlikely that the spontaneously runted phenotype is due to a mutation that renders type I collagen resistant to MMP-mediated cleavage. It, therefore, remains to be determined if the spontaneously runted Mmp2-/- mice harbor other mutations that also affect collagen metabolism or if additional, collagen-independent, genetic modifiers of Mmp2 exist.
TABLE 2.
Comparison of Phenotypes of Humans With MMP2 Null Mutations and Mmp2-/-;Col1a1r/r Mice
| HumanaMMP2 null | Mouse Mmp2-/-;Col1a1r/r | |
|---|---|---|
| Short stature | + | + |
| Abnormal face | + | + |
| Swollen extremities | + | + |
| Reduced joint mobility | + | + |
| Sclerotic calvarial sutures | + | + |
| Osteopenic long bones | + | + |
| Calvarial bone defects | ND | + |
Phenotypes of human patients with osteolytic syndromes associated with inactivating MMP2 mutations (from references Al Aqeel et al., 2000; Al-Mayouf et al., 2000; Zankl et al., 2005; Rouzier et al., 2006). ND, not determined.
Reduced Extracellular Matrix Degradation Can Result in Reduced Bone Mass
A priori, loss of a matrix-degrading enzyme, such as MMP2, would be expected to result in reduced bone remodeling and an osteopetrotic phenotype. Therefore, it was surprising that MMP2-null mutations were associated with reduced bone mass and osteolysis in human genetic linkage studies (Martignetti et al., 2001). Our study now confirms the human findings: absence of MMP2 activity can result in osteopenia. MMP2 is a substrate of MMP14, and Mmp14-/- mice are another example of an osteopenic phenotype that results from the absence of matrix degrading activity (Holmbeck et al., 1999). Additional similarities between Mmp14-/- and Mmp2-/-;Col1a1r/r mice are the decreased body size, abnormal craniofacial features, and lack of calvarial suture closure. These similarities strongly suggest that proMMP2 and type I collagen are important substrates of MMP14 in bone development. However, MMP14 has other substrates, including proMMP13, which is important for endochondral ossification (Cowell et al., 1998; Stickens et al., 2004). Thus, a detailed comparison of endochondral ossification in Mmp14-/- mice and Mmp2-/-;Col1a1r/r mice is likely to reveal more severe defects in Mmp14-/- than in Mmp2-/-;Col1a1r/r mice. Mmp2-/-;Col1a1r/r mice developed edema of the paws, probably localized to soft tissues. In Mmp14-/- mice, soft tissue changes, arthritis, and joint destruction have been reported (Holmbeck et al., 1999). Thus, it is possible that the edema in the Mmp2-/-;Col1a1r/r paws is secondary to changes in the joints, although a partial analysis of joints did not reveal apparent destruction. As would be expected, Mmp2-/-;Mmp14-/- double null mice also exhibit skeletal defects (Oh et al., 2004); however, due to their perinatal lethality, these defects are not well characterized.
The reduced bone mass in the absence of matrix degradation may be partly explained by a reduced ability to degrade the embryonic cartilage matrix, which is required before osteoblasts can invade into the developing tissue and deposit bone matrix (Holmbeck et al., 1999). However, this finding would not explain the progressive bone loss observed in MMP2-null humans (Al Aqeel et al., 2000; Al-Mayouf et al., 2000; Rouzier et al., 2006). Because of the poor survival of the mice, we were limited to examining bone tissue from relatively young (7-8 weeks old) Mmp2-/-;Col1a1r/r mice and could not perform a proper analysis of whether the bone loss was progressive. Nevertheless, there was a strong trend for progressive bone loss by micro-CT analysis when comparing bones from 7- to 8-week-old mice with that of the one 16-week-old mouse we could analyze. Thus, our data are consistent with the clinical findings that absence of MMP2 activity results in progressive bone loss, suggesting that the balance between bone deposition and bone resorption is skewed.
It may seem particularly surprising that combining Mmp2 null mutations with the Col1a1r mutation results in such a strong osteopenic bone phenotype because Col1a1r/r mice are osteopetrotic and in response to parathyroid hormone they have a severe impairment both in the generation of osteoclasts and in bone resorption (Zhao et al., 1999; Chiusaroli et al., 2003). Intriguingly, the porous calvarial phenotype we observed in the Mmp2-/-;Col1a1r/r mice appeared as if increased bone resorption was taking place on the osteopetrotic background of the Col1a1r/r mice. Osteoclast numbers are increased in the long bones of Col1a1r/r mice even though activity is not (Chiusaroli et al., 2003). Thus, it appears that parathyroid hormone is not the right stimulus for induction of bone resorption in the Col1a1r/r mice, whereas the absence of MMP2 is.
Type I Collagen Metabolism in Bone Development
Type I collagen regulates osteoblast and osteoclast activity in engineered mouse models (Zhao et al., 2000; Kalajzic et al., 2002), but the molecular mechanisms involved are not clear. TCA and TCB fragments of type I collagen are present in developing bone (Holmbeck et al., 1999; Stickens et al., 2004). These collagen fragments can ligate the αvβ3 integrin (Montgomery et al., 1994), and both osteoblasts and osteoclasts express αvβ3 integrins (Cheng et al., 2000; Zhao et al., 2005). Because of the Col1a1r mutation, the TCA and TCB fragments are not generated in the Col1a1r/r mice, but the mutation could also affect the stability and conformation of the collagen triple helix. Changes in stability and conformation could affect both the unfolding of the triple helix that is necessary for collagenase cleavage (Zhao et al., 2000) and the ability of the collagen molecules to interact with other ECM components. Thus, we cannot conclude whether the absence of normal or presence of mutant fibrillar collagen is important for the observed developmental defects.
The Col1a1r mutation affects the ability of all classic collagenolytic MMPs to generate TCA and TCB fragments. In vitro, MMP2 had much higher activity against mutant type I collagen than classic collagenolytic MMPs (MMP8, MMP13, and MMP14) and the strong phenotype of mice with the Col1a1r mutation in the absence of MMP2 activity indicates that other collagenolytic MMPs are unlikely to compensate for MMP2 in vivo. Therefore, the phenotype observed in Mmp2-/-;Col1a1r/r mice is likely attributable not only to a loss of MMP2 activity against type I collagen, but to a near total loss of MMP-mediated degradation of type I collagen.
MMP2 Has Activity Against Type I Collagen
MMP2 is important for degradation of denatured, cleaved, type I collagen but also has direct activity against type I collagen (Tournier et al., 1994; Aimes and Quigley, 1995; Tam et al., 2004). Interestingly, MMP2 had higher activity against mutant type I collagen than normal type I collagen at physiological temperatures, whereas the opposite was true for the classic collagenolytic MMPs. MMP2 unfolds type I collagen by a mechanism distinct from the classic collagenases (Tam et al., 2004), and might, therefore, be more efficient at degrading mutant type I collagen with slight conformational changes. We also observed reduced survival of the Mmp2-/-;Col1a1r/+ mice with one wild-type Col1a1 allele, and these mice were also often runted with stiff joints and abnormal craniofacial features. These phenotypes were not observed in the Mmp2-/+;Col1a1r/r mice with one remaining wild-type Mmp2 allele, suggesting that MMP2 activity is absolutely required for degradation of mutant type I collagen in vivo. In vitro, we demonstrated that MMP2 was able to degrade both wildtype and mutant type I collagen, and the genetic interaction between Mmp2 and Col1a1r might, therefore, be due to direct biochemical interactions between the MMP2 enzyme and one of its substrates, type I collagen.
Conclusions and Future Perspectives
Inactivating mutations in Mmp2 have mild effects on skeletal development in the mouse (Inoue et al., 2006). Here, we show that concomitant mutations in both Mmp2 and Col1a1 result in a severe runted and osteopenic phenotype that resembles the osteolytic syndromes found in humans with MMP2 null mutations. Our results raise the interesting possibility that genetic modifiers of MMP2 may also play a role in the human osteolytic syndromes. MMP2 was much more efficient at degrading mutant type I collagen than were the classic collagenases. Therefore, minor mutations in type I collagen or in enzymes involved in its processing, which would go largely unnoticed on their own, could result in severe disease in the absence of MMP2 activity. Of interest, approximately 35% of the population carries a polymorphism of the COL1A1 gene that increases the ratio of the type I collagen α1(I) chain to α2(I) chain and is associated with a slightly decreased bone mass (Mann et al., 2001; Ralston et al., 2006). The strength of mouse genetics has allowed us to identify a genetic interaction between Mmp2 and type I collagen in skeletal development. Similarly, mouse genetics were instrumental in the identification of a genetic modifier of the CFTR gene (reviewed in Nadeau, 2001). The human syndromes (the Winchester, Torg, and nodulosis-arthropathy-osteolysis [NAO] syndromes) associated with MMP2-null mutations vary in severity and whether or not subcutaneous fibrillar nodules are present (Zankl et al., 2007). Such variation may be accounted for if genetic modifiers of MMP2 also exist in humans. The results of our study indicate that type I collagen and enzymes involved in collagen metabolism are interesting candidate modifiers for these devastating human syndromes. In conclusion, using mouse genetics, we have confirmed the surprising results from the human genetic linkage studies: the lack of a matrix-degrading enzyme, MMP2, can result in increased bone degradation in the context of an abnormal matrix.
EXPERIMENTAL PROCEDURES
Mice
Mmp2-/- mice (Itoh et al., 1997) and Col1a1r/r mice (Liu et al., 1995) were bred into the FVB/n background for four or more generations before generating Mmp2-/-;Col1a1r/r mice. Mice were genotyped by polymerase chain reaction for the wild-type Mmp2 allele (primers GelA2_sense=5′-CAACGATGGAGGCACGAGTG-3′ and GelA_antis=5′-GCCGGGGAACTTGATCATGG-3′), for the Mmp2 null allele (primers GelAmut-1=5′-GACCACCAAGCGAAACAT-3′ and GelAmut-2=5′-CAAGAAGGCGATAGAAGG-3′), for the wild-type Col1a1 allele (primers Col1a1wt-1=5′-TGGACAACGTGGTGTGGTC-3′ and Col1a1-wt-2=5′-TTGAACTCAGGAATTTACCTGC-3′), and for the mutated Col1a1r allele (primers Col1a1mut-1= 5′-TGGACAACGTGGTGCCGCG-3′ and Col1a1wt-2). Mice were housed in a specific-pathogen-free environment, under light-, temperature-, and humidity-controlled conditions with food and water available ad libitum. All mice were maintained and handled according to IACUC procedures.
Embryonic and Neonatal Lethality
Mmp2+/-;Col1a1r/+ mice were interbred to determine whether all genotypes were present in the expected Mendelian ratios. Embryos were collected, and yolk sac DNA was genotyped at embryonic day 9.5 or 14.5, and pups were collected, and tail DNA genotyped at birth (p1) or at weaning (p21).
Weight Curves
Wild-type, Mmp2-/-, Col1a1r/r, and Mmp2-/-;Col1a1r/r male and female mice were weighed weekly. At least three animals of each sex and genotype were followed until 16 weeks of age.
Skull and Skeletal Preparations
Whole skeleton and skull preparations were prepared by removing skin and internal organs, fixing overnight in 4% paraformaldehyde (PFA), washing in increasing concentrations of ethanol, and staining overnight with Alcian blue (Sigma A3157-10G). Skeletons and skulls were then washed in ethanol, digested in 1% trypsin in 30% sodium borate until soft tissue became transparent, rinsed, stained in Alizarin red (Sigma A5533), incubated for 18-48 hr in 0.5% KOH/0.6% H2O2, washed in increasing concentrations of glycerin, and stored in 100% glycerine.
Quantification of Calvarial Bone Formation
Skulls were photographed on a stereo microscope (Leica, MZFL111) with a digital camera (Nikon, DXM1200) using ACT-1 software (Nikon). The area that had not ossified was quantified using NIH Image J 1.34s software.
Histological Staining
Tissues were fixed in 4% PFA and decalcified in 19% ethylenediaminetetraacetic acid for 2 weeks before embedding in paraffin and sectioning at 5 μm. Deparaffinized and rehydrated sections were stained with hematoxylin and eosin using standard methods. For Safranin-O/Fast Green staining, sections were stained in Weigert’s Iron Hematoxylin (Sigma), 0.02% aqueous Fast Green (Sigma), followed by a rinse in 1% acetic acid and 0.1% aqueous Safranin-O (Sigma). Sections were reacted for TRAP activity using a leukocyte acid phosphatase kit and counterstained with 0.02% aqueous Fast Green (Sigma).
MRI
MRI images were acquired on a 1.5T Signa whole body MRI scanner (General Electric Medical Systems). Anesthetized mice were imaged in pairs (an aged-matched wild-type or littermate Mmp2+/-;Col1a1r/r mouse with an Mmp2-/-;Col1a1r/r mouse) using a wrist radiofrequency coil (Medical Advances, Milwaukee, WI) and a customized animal holder. Normal body temperature was maintained during imaging with a heating pad. Axial T2-weighted images were acquired using a two-dimensional multislice fast spin echo sequence (repetition time [TR] = 5.5 sec, echo time [TE] = 85 msec) with an 8-cm field of view (FOV), 256 × 192 matrix, and 3-mm slice thickness to obtain cross-sectional images of the hind paws of the mice. Axial diffusion weighted images were acquired for the same slice locations using a single-shot fast spin echo (TR = 9.5 sec; TE = 85 msec; FOV = 8 cm, thickness = 3 mm, 128 × 128 matrix, and b values of 0 and 600 s/mm2). Water ADC maps demonstrating water mobility in the tissues were calculated as previously described (Partridge et al., 2001), and paw ADC values were determined using regions of interest delineated on T2-weighed images.
Micro-CT
Tibiae were collected at indicated ages and analyzed using a micro-CT system (mCT40, Scanco Medical, Bassersdorf, Switzerland). The trabeculae of the bones were scanned using a Cone-Beam type scan into 240 slices with a voxel of 7 × 7 × 7 μm. Three-dimensional structural parameters were measured directly as described (Jiang et al., 2003).
Preparation of Collagens and Digestion With Collagenases
Collagens were extracted from wild-type or Col1a1r/r mouse tails, digested with APMA-activated recombinant MMPs (Calbiochem, CA) for 1 hr at the indicated temperatures, and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis as described previously (Liu et al., 1995). Each sample contained 1 μg type I collagen.
Statistical Analysis
Statistical analysis was performed using GraphPad Prism 4 and Microsoft Office Excel 2003. The distributions of genotypes after inter-breeding Mmp2-/+;Col1a1r/+ mice were compared with the expected Mendelian distributions with χ2 test (8 degrees of freedom). Growth curves of mice (repeated measurements) were analyzed by two-way analysis of variance repeated measurements test for time points with at least three mice per genotype. For males, curves were significantly influenced by genotype, P < 0.0001 (F = 173.39) and Bonferroni posttests showed that Mmp2-/-;Col1a1r/r at 4 and 6-16 weeks of age were significantly different from the wild-types (P < 0.05). For females, statistical analysis was done only on measurements from 7-16 weeks of age, because there were fewer than three Mmp2-/-;Col1a1r/r mice for weeks 1-6. The curves were significantly influenced by genotype, P < 0.0001 (F = 969.29), and Bonferroni posttests found that Mmp2-/- mice at 15 weeks of age (P < 0.05), Col1a1r/r mice at 11 weeks of age (P < 0.05), and Mmp2-/-;Col1a1r/r mice at 7-16 weeks of age were significantly different (P < 0.001) from wild-type mice. The size of the calvarial foramen, relative bone volume (BV/TV), trabecular thickness (DT-Tb.Th), cortical thickness at the diaphysis (DT-C.Th.-Diaph), and trabecular separation (TRI-Tb.Sp.) were analyzed with two-sided Student’s t-test.
ACKNOWLEDGMENTS
The authors thank Kerstin Dehne and Jake Lee for technical support, Dr. Stephen Krane for Col1a1r/r mice, Dr. Shigeyoshi Itohara for Mmp2-/- mice, and Drs. Sylvain Provot and Andrew J. Ewald for helpful comments on the manuscript. M.E. received a fellowship from The Danish Medical Research Council, and A.E. received a fellowship from The Serono Foundation for the Advancement of Medical Science.
Grant sponsor: National Institutes of Health; Grant number: R01 AR046238; Grant number: CA094168; Grant number: CA098075; Grant sponsor: UCSF Breast Cancer SPORE P50 CA058207 and Core C of P01 CA072006; Grant sponsor: The Sandler Program in Basic Sciences; Grant sponsor: The National Technology Center for Networks and Pathways; Grant number: U54 RR020843; Grant sponsor: The Danish Medical Research Council; Grant sponsor: The Serono Foundation for the Advancement of Medical Science.
REFERENCES
- Aimes RT, Quigley JP. Matrix metalloproteinase-2 is an interstitial collagenase. Inhibitor-free enzyme catalyzes the cleavage of collagen fibrils and soluble native type I collagen generating the specific 3/4- and 1/4-length fragments. J Biol Chem. 1995;270:5872–5876. doi: 10.1074/jbc.270.11.5872. [DOI] [PubMed] [Google Scholar]
- Al Aqeel A, Al Sewairi W, Edress B, Gorlin RJ, Desnick RJ, Martignetti JA. Inherited multicentric osteolysis with arthritis: a variant resembling Torg syndrome in a Saudi family. Am J Med Genet. 2000;93:11–18. doi: 10.1002/1096-8628(20000703)93:1<11::aid-ajmg3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Al-Mayouf SM, Majeed M, Hugosson C, Bahabri S. New form of idiopathic osteolysis: nodulosis, arthropathy and osteolysis (NAO) syndrome. Am J Med Genet. 2000;93:5–10. doi: 10.1002/1096-8628(20000703)93:1<5::aid-ajmg2>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Barsh GS, David KE, Byers PH. Type I osteogenesis imperfecta: a nonfunctional allele for pro alpha 1 (I) chains of type I procollagen. Proc Natl Acad Sci U S A. 1982;79:3838–3842. doi: 10.1073/pnas.79.12.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsh GS, Roush CL, Bonadio J, Byers PH, Gelinas RE. Intron-mediated re-combination may cause a deletion in an alpha 1 type I collagen chain in a lethal form of osteogenesis imperfecta. Proc Natl Acad Sci U S A. 1985;82:2870–2874. doi: 10.1073/pnas.82.9.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonadio J, Holbrook KA, Gelinas RE, Jacob J, Byers PH. Altered triple helical structure of type I procollagen in lethal perinatal osteogenesis imperfecta. J Biol Chem. 1985;260:1734–1742. [PubMed] [Google Scholar]
- Cheng SL, Lai CF, Fausto A, Chellaiah M, Feng X, McHugh KP, Teitelbaum SL, Civitelli R, Hruska KA, Ross FP, Avioli LV. Regulation of alphaVbeta3 and alphaVbeta5 integrins by dexamethasone in normal human osteoblastic cells. J Cell Biochem. 2000;77:265–276. doi: 10.1002/(sici)1097-4644(20000501)77:2<265::aid-jcb9>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Chiusaroli R, Maier A, Knight MC, Byrne M, Calvi LM, Baron R, Krane SM, Schipani E. Collagenase cleavage of type I collagen is essential for both basal and parathyroid hormone (PTH)/PTH-related peptide receptor-induced osteoclast activation and has differential effects on discrete bone compartments. Endocrinology. 2003;144:4106–4116. doi: 10.1210/en.2003-0254. [DOI] [PubMed] [Google Scholar]
- Cowell S, Knauper V, Stewart ML, D’Ortho MP, Stanton H, Hembry RM, Lopez-Otin C, Reynolds JJ, Murphy G. Induction of matrix metalloproteinase activation cascades based on membrane-type 1 matrix metalloproteinase: associated activation of gelatinase A, gelatinase B and collagenase 3. Biochem J. 1998;331(Pt 2):453–458. doi: 10.1042/bj3310453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutroneo KR. How is Type I procollagen synthesis regulated at the gene level during tissue fibrosis. J Cell Biochem. 2003;90:1–5. doi: 10.1002/jcb.10599. [DOI] [PubMed] [Google Scholar]
- Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224:245–251. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- Fera E, O’Neil C, Lee W, Li S, Pickering JG. Fibroblast growth factor-2 and remodeled type I collagen control membrane protrusion in human vascular smooth muscle cells: biphasic activation of Rac1. J Biol Chem. 2004;279:35573–35582. doi: 10.1074/jbc.M400711200. [DOI] [PubMed] [Google Scholar]
- Heino J. The collagen receptor integrins have distinct ligand recognition and signaling functions. Matrix Biol. 2000;19:319–323. doi: 10.1016/s0945-053x(00)00076-7. [DOI] [PubMed] [Google Scholar]
- Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, Ward JM, Birkedal-Hansen H. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99:81–92. doi: 10.1016/s0092-8674(00)80064-1. [DOI] [PubMed] [Google Scholar]
- Inoue K, Mikuni-Takagaki Y, Oikawa K, Itoh T, Inada M, Noguchi T, Park JS, Onodera T, Krane SM, Noda M, Itohara S. A crucial role for MMP-2 in osteocytic canalicular formation and bone metabolism. J Biol Chem. 2006;281:33814–33824. doi: 10.1074/jbc.M607290200. [DOI] [PubMed] [Google Scholar]
- Itoh T, Ikeda T, Gomi H, Nakao S, Suzuki T, Itohara S. Unaltered secretion of beta-amyloid precursor protein in gelatinase A (matrix metalloproteinase 2)-deficient mice. J Biol Chem. 1997;272:22389–22392. doi: 10.1074/jbc.272.36.22389. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Zhao JJ, Mitlak BH, Wang O, Genant HK, Eriksen EF. Recombinant human parathyroid hormone (1-34) [teriparatide] improves both cortical and cancellous bone structure. J Bone Miner Res. 2003;18:1932–1941. doi: 10.1359/jbmr.2003.18.11.1932. [DOI] [PubMed] [Google Scholar]
- Kalajzic I, Terzic J, Rumboldt Z, Mack K, Naprta A, Ledgard F, Gronowicz G, Clark SH, Rowe DW. Osteoblastic response to the defective matrix in the osteogenesis imperfecta murine (oim) mouse. Endocrinology. 2002;143:1594–1601. doi: 10.1210/endo.143.5.8807. [DOI] [PubMed] [Google Scholar]
- Le Bihan D, Breton E, Lallemand D, Grenier P, Cabanis E, Laval-Jeantet M. MR imaging of intravoxel incoherent motions: application to diffusion and perfusion in neurologic disorders. Radiology. 1986;161:401–407. doi: 10.1148/radiology.161.2.3763909. [DOI] [PubMed] [Google Scholar]
- Liu X, Wu H, Byrne M, Jeffrey J, Krane S, Jaenisch R. A targeted mutation at the known collagenase cleavage site in mouse type I collagen impairs tissue remodeling. J Cell Biol. 1995;130:227–237. doi: 10.1083/jcb.130.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann V, Hobson EE, Li B, Stewart TL, Grant SF, Robins SP, Aspden RM, Ralston SH. A COL1A1 Sp1 binding site polymorphism predisposes to osteo-porotic fracture by affecting bone density and quality. J Clin Invest. 2001;107:899–907. doi: 10.1172/JCI10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martignetti JA, Aqeel AA, Sewairi WA, Boumah CE, Kambouris M, Mayouf SA, Sheth KV, Eid WA, Dowling O, Harris J, Glucksman MJ, Bahabri S, Meyer BF, Desnick RJ. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet. 2001;28:261–265. doi: 10.1038/90100. [DOI] [PubMed] [Google Scholar]
- Montgomery AM, Reisfeld RA, Cheresh DA. Integrin alpha v beta 3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen. Proc Natl Acad Sci U S A. 1994;91:8856–8860. doi: 10.1073/pnas.91.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2:165–174. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- Oh J, Takahashi R, Adachi E, Kondo S, Kuratomi S, Noma A, Alexander DB, Motoda H, Okada A, Seiki M, Itoh T, Itohara S, Takahashi C, Noda M. Mutations in two matrix metalloproteinase genes, MMP-2 and MT1-MMP, are synthetic lethal in mice. Oncogene. 2004;23:5041–5048. doi: 10.1038/sj.onc.1207688. [DOI] [PubMed] [Google Scholar]
- Ohuchi E, Imai K, Fujii Y, Sato H, Seiki M, Okada Y. Membrane type 1 matrix metalloproteinase digests interstitial collagens and other extracellular matrix macromolecules. J Biol Chem. 1997;272:2446–2451. doi: 10.1074/jbc.272.4.2446. [DOI] [PubMed] [Google Scholar]
- Partridge SC, McKinnon GC, Henry RG, Hylton NM. Menstrual cycle variation of apparent diffusion coefficients measured in the normal breast using MRI. J Magn Reson Imaging. 2001;14:433–438. doi: 10.1002/jmri.1204. [DOI] [PubMed] [Google Scholar]
- Petitclerc E, Stromblad S, von Schalscha TL, Mitjans F, Piulats J, Montgomery AM, Cheresh DA, Brooks PC. Integrin alpha(v)beta3 promotes M21 melanoma growth in human skin by regulating tumor cell survival. Cancer Res. 1999;59:2724–2730. [PubMed] [Google Scholar]
- Ralston SH, Uitterlinden AG, Brandi ML, Balcells S, Langdahl BL, Lips P, Lorenc R, Obermayer-Pietsch B, Scollen S, Bustamante M, Husted LB, Carey AH, Diez-Perez A, Dunning AM, Falchetti A, Karczmarewicz E, Kruk M, van Leeuwen JP, van Meurs JB, Mangion J, McGuigan FE, Mellibovsky L, del Monte F, Pols HA, Reeve J, Reid DM, Renner W, Rivadeneira F, van Schoor NM, Sherlock RE, Ioannidis JP. Large-scale evidence for the effect of the COLIA1 Sp1 polymorphism on osteoporosis outcomes: the GENOMOS study. PLoS Med. 2006;3:e90. doi: 10.1371/journal.pmed.0030090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzier C, Vanatka R, Bannwarth S, Philip N, Coussement A, Paquis-Flucklinger V, Lambert JC. A novel homozygous MMP2 mutation in a family with Winchester syndrome. Clin Genet. 2006;69:271–276. doi: 10.1111/j.1399-0004.2006.00584.x. [DOI] [PubMed] [Google Scholar]
- Rozmahel R, Wilschanski M, Matin A, Plyte S, Oliver M, Auerbach W, Moore A, Forstner J, Durie P, Nadeau J, Bear C, Tsui LC. Modulation of disease severity in cystic fibrosis transmembrane conductance regulator deficient mice by a secondary genetic factor. Nat Genet. 1996;12:280–287. doi: 10.1038/ng0396-280. [DOI] [PubMed] [Google Scholar]
- Shrivastava A, Radziejewski C, Campbell E, Kovac L, McGlynn M, Ryan TE, Davis S, Goldfarb MP, Glass DJ, Lemke G, Yancopoulos GD. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol Cell. 1997;1:25–34. doi: 10.1016/s1097-2765(00)80004-0. [DOI] [PubMed] [Google Scholar]
- Stickens D, Behonick DJ, Ortega N, Heyer B, Hartenstein B, Yu Y, Fosang AJ, Schorpp-Kistner M, Angel P, Werb Z. Altered endochondral bone development in matrix metalloproteinase 13-deficient mice. Development. 2004;131:5883–5895. doi: 10.1242/dev.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringa E, Knauper V, Murphy G, Gavrilovic J. Collagen degradation and platelet-derived growth factor stimulate the migration of vascular smooth muscle cells. J Cell Sci. 2000;113(Pt 11):2055–2064. doi: 10.1242/jcs.113.11.2055. [DOI] [PubMed] [Google Scholar]
- Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI. Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- Tam EM, Moore TR, Butler GS, Overall CM. Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): the differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J Biol Chem. 2004;279:43336–43344. doi: 10.1074/jbc.M407186200. [DOI] [PubMed] [Google Scholar]
- Tournier JM, Polette M, Hinnrasky J, Beck J, Werb Z, Basbaum C. Expression of gelatinase A, a mediator of extracellular matrix remodeling, by tracheal gland serous cells in culture and in vivo. J Biol Chem. 1994;269:25454–25464. [PubMed] [Google Scholar]
- Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, Shapiro SD, Senior RM, Werb Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zankl A, Bonafe L, Calcaterra V, Di Rocco M, Superti-Furga A. Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin Genet. 2005;67:261–266. doi: 10.1111/j.1399-0004.2004.00402.x. [DOI] [PubMed] [Google Scholar]
- Zankl A, Pachman L, Poznanski A, Bonafe L, Wang F, Shusterman Y, Fishman DA, Superti-Furga A. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester syndrome. J Bone Miner Res. 2007;22:329–333. doi: 10.1359/jbmr.061013. [DOI] [PubMed] [Google Scholar]
- Zhao W, Byrne MH, Boyce BF, Krane SM. Bone resorption induced by parathyroid hormone is strikingly diminished in collagenase-resistant mutant mice. J Clin Invest. 1999;103:517–524. doi: 10.1172/JCI5481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Byrne MH, Wang Y, Krane SM. Osteocyte and osteoblast apoptosis and excessive bone deposition accompany failure of collagenase cleavage of collagen. J Clin Invest. 2000;106:941–949. doi: 10.1172/JCI10158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Ross FP, Teitelbaum SL. Unoccupied alpha(v)beta3 integrin regulates osteoclast apoptosis by transmitting a positive death signal. Mol Endocrinol. 2005;19:771–780. doi: 10.1210/me.2004-0161. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Apte SS, Soininen R, Cao R, Baaklini GY, Rauser RW, Wang J, Cao Y, Tryggvason K. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci U S A. 2000;97:4052–4057. doi: 10.1073/pnas.060037197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielenski J, Corey M, Rozmahel R, Markiewicz D, Aznarez I, Casals T, Larriba S, Mercier B, Cutting GR, Krebsova A, Macek M, Jr, Langfelder-Schwind E, Marshall BC, DeCelie-Germana J, Claustres M, Palacio A, Bal J, Nowakowska A, Ferec C, Estivill X, Durie P, Tsui LC. Detection of a cystic fibrosis modifier locus for meconium ileus on human chromosome 19q13. Nat Genet. 1999;22:128–129. doi: 10.1038/9635. [DOI] [PubMed] [Google Scholar]