

Abstract
Astrocytes and oligodendrocytes are characterized by a very negative resting potential and a high resting permeability for K+ ions. Early pharmacological and biophysical studies suggested that the resting potential is established by the activity of inwardly rectifying, Ba2+ sensitive, weakly rectifying Kir channels. Molecular cloning has identified 16 Kir channels genes of which several mRNA transcripts and protein products have been identified in glial cells. However, genetic deletion and siRNA knock-down studies suggest that the resting conductance of astrocytes and oligodendrocytes is largely due to Kir4.1. Loss of Kir4.1 causes membrane depolarization, and a break-down of K+ and glutamate homeostasis which results in seizures and wide-spread white matter pathology. Kir channels have also been shown to act as critical regulators of cell division whereby Kir function is correlated with an exit from the cell cycle. Conversely, loss of functional Kir channels is associated with re-entry of cells into the cell cycle and gliosis. A loss of functional Kir channels has been shown in a number of neurological diseases including temporal lobe epilepsy, amyotrophic lateral sclerosis, retinal degeneration and malignant gliomas. In the latter, expression of Kir4.1 is sufficient to arrest the aberrant growth of these glial derived tumor cells. Kir4.1 therefore represents a potential therapeutic target in a wide variety of neurological conditions.
Keywords: astrocyte, brain tumor, cell proliferation, glioma, ion channel, K+ buffering
In the early 1960s, Kuffler and colleagues (Kuffler et al. 1966) examined the membrane properties of glial cells in the salamander. They found that the membrane of glial cells in the optic nerve was almost exclusively permeable to K+. Even small changes in extracellular [K+], such as those arising from the electrical activity induced by a single flash of light to the salamander’s eye, readily resulted in a small depolarization of the glial membrane potential (Orkand et al. 1966). Many studies have since demonstrated a similar K+ permeability in glial cells from many preparations across several species and tissue types. It is now commonly accepted that the membrane of most glial cells, and astrocytes in particular, is highly K+ selective. These early experiments preceded the discovery of ion channels by almost two decades. Yet these investigators formulate an attractive hypothesis concerning the role of the glial K+ conductance that remains compelling to date. Kuffler and his colleagues reasoned that an unequal distribution of K+ conductances (i.e. channels) along the membrane of a single cell, or a small network of cells connected by gap-junctions, would allow glial cells to engage in a spatial redistribution of K+ from a brain region with elevated K+ to a region where K+ is low. This idea came to be known as the spatial potassium buffering hypothesis (Orkand 1980).
While early studies such as these relied largely on recordings with sharp voltage electrodes, later patch-clamp recordings from glial cells showed a remarkable complement of voltage-activated ion channels in many glial cell types. Glial progenitor cells and immature astrocytes in addition to voltage-activated K+ channels even express voltage-gated Na+ and Ca2+ channels previously thought to be reserved for excitable cells (Sontheimer 1994). Much work went into a detailed cataloging and characterization of these channels and particularly the many K+ channels expressed (Olsen and Sontheimer 2004b).
As it pertains to Kuffler’s initial discovery, the search for the K+ channel(s) responsible for the large resting K+ conductance in astrocytes and hence the channel underlying spatial buffering has recently come to closure. As discussed in more detail below, most researchers now believe that Kir4.1 (KCNJ10), an inwardly rectifying K+ channel, is largely responsible for establishing and maintaining the glial resting membrane potential, and indeed appears, at least partially, responsible for transmembrane K+ fluxes in spatial buffering. Kir4.1 and its putative role in glial biology including some unexpected roles will be discussed in this article. Where appropriate we will refer the reader to complete reviews on specific topics.
Macroscopic properties of inwardly rectifying K+ channels
In the nervous system, K+ channels are typically engaged in the repolarization of membranes following the influx of Na+ or Ca2+ in the context of action potentials. Since the equilibrium potential for K+ (EK) is very negative in most cells, i.e. around −85 mV, the opening of any K+ channel leads to a repolarization of the membrane. At potentials positive of EK, this will be accompanied by an efflux of K+ down its electrochemical gradient. The vast majority of K+ channels are outwardly rectifying, i.e. show higher open probability, hence permitting greater conduction at more depolarized potentials. These channels are therefore ideally suited to truncate an action potential as a depolarized potential will enhance their activity thereby accelerating membrane repolarization. Essentially all excitable cells utilize outwardly rectifying K+ channels for the repolarization of the resting potential.
In 1949, Katz described a K+ current that behaves opposite to those described above (Katz 1949). A depolarized membrane actually reduces current flow, while a more negative potential enhances it. The resulting currents were termed ‘anomalous rectifiers’ now more commonly referred to as inwardly rectifying K+ currents (Kir). Such currents have since been described in many excitable and inexcitable cells. The first of the Kir channels was cloned in 1993 (Kubo et al. 1993) and since then molecular cloning identified 16 Kir channel subunits that can each give rise to inwardly rectifying currents (Doupnik et al. 1995). These channels all share a similar membrane topology with two membrane spanning domains, and cytoplasmic N and C terminals. Each inward rectifier channel is composed of four alpha subunits, assembled as homomultimers or heteromeric channels. The 16 subunits can be grouped into seven subfamilies by homology which are designated Kir1.x-Kir7.x (Nichols and Lopatin 1997). The resulting channels differ with regards to the degree of rectification, single channel conductance and relative sensitivity to certain pharmacological blockers. Several members are regulated by G-proteins, ATP or H+.
Despite originating from 16 different genes, all Kir channels share some common signature features. For example, in all cases inward current increases with increasing [K+]o concentrations, whereby currents are proportional to the square root of [K+]o (Hagiwara and Takahashi 1974; Sakmann and Trube 1984; Newman 1993) a feature discussed further with regards to Kir channels in glia being involved in K+ homeostasis. Furthermore, nearly all of the known Kir channel subunits are highly sensitive to micromolar concentrations of extracellular Ba2+ (Coetzee et al. 1999) and, as the name implies they each preferentially conduct inward current, although some subunits pass outward current as well. Indeed, it should be noted that Kir channels, while rectifying, are not voltage-dependent in the strictest sense. Instead intracellular Mg2+ or polyamine ions enter the channel pore, thereby blocking ion permeation (Oliver et al. 2000). This effect increases with more positive potentials resulting in a reduction of current flow at positive potentials. A typical recording along with a corresponding current-voltage (IV) curve is shown in Fig. 1 illustrating inward rectification, seen as larger currents at potentials negative of the cells resting potential than at positive potentials (Fig. 1b).
Fig. 1.
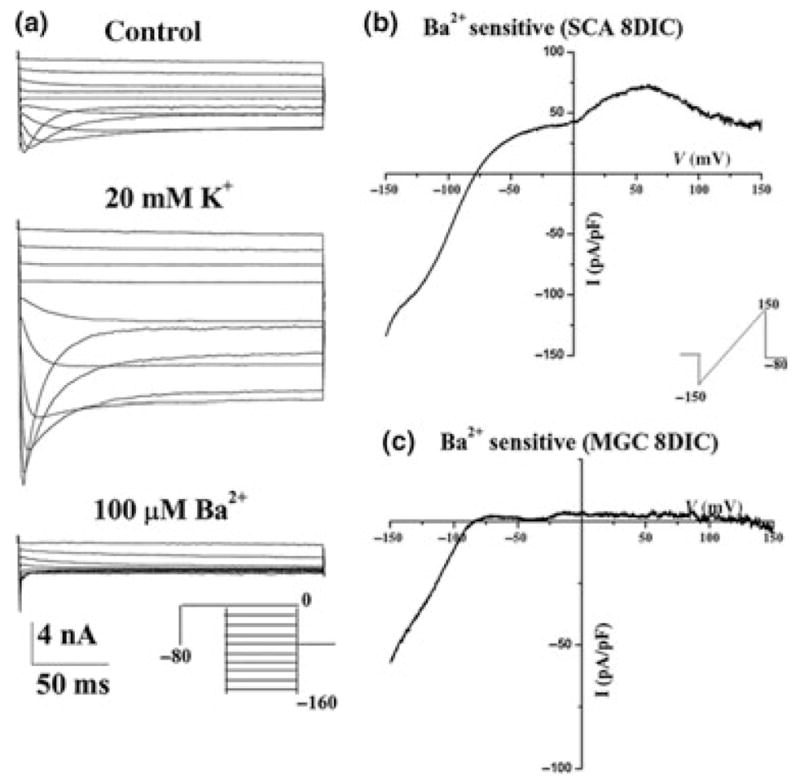
Signature features of astrocytic inward rectifiers: (a) Representative recordings from spinal cord astrocytes demonstrating currents that activate with negative voltage steps. Current amplitude increases markedly with elevated extracellular K+ (control 5 mM). At the most negative voltages currents show time-dependent inactivation because of a block by extracellular Mg2+. Inward currents are completely inhibited by 100 μM Ba2+. (b) A continuous ramp-like change in voltage ranging from −150 mM to 150 mV was used before and after application of Ba2+, and the subtracted Ba2+ sensitive current was plotted in (b) and (c). The resulting current-voltage curve was weakly rectifying in (b) recorded from a spinal cord astrocytes after 8 days in culture, showing smaller outward currents than inward currents. The same approach was taken in (c) in a recording from a microglial cells. The resulting current-voltage curve showed strong rectification with essentially no outward current and only inward currents at potentials negative of the Ek. [With permission from Olsen et al. 2006].
While all Kir channels rectify and hence pass K+ ions into the cell more readily then they efflux K+, the degree of rectification varies between channel subtypes. The Kir1.x, Kir4.x, Kir5.x and Kir7.x subfamilies encode weak inward rectifiers, i.e. they also permit considerable outward K+ currents, as the example in Fig. 1(b), recorded from a spinal cord astrocyte demonstrates, while the Kir2.x and Kir3.x subfamilies rectify strongly, permitting little if any K+ efflux as illustrated in a recording from a microglial cell that expresses Kir2.1 in Fig. 1(c). Generally speaking most inwardly rectifying currents recorded from glial cells to date showed weak rectification, suggesting an absence of homomeric Kir2.x and Kir3.x channels despite the apparent presence of transcripts and protein for some of these channels.
Another feature of vertebrate astrocytic Kir currents is a slow time-dependent inactivation or relaxation of currents that increases at very negative potentials (Fig. 1a) and actually creates the negative-bend in the IV-curve when one plots the steady-state current (not shown). While this could be interpreted as a voltage-dependence of gating, it actually reflects a block of the channel by extracellular Na+ ions. Glial Kir channels are particularly sensitive to even small changes in [Na+]o and a reduction by just 15 mM results in a loss of this relaxation and a significantly enhanced current at negative potentials (Ransom et al. 1996). The most defining features shared among Kir channels is the reversible block by in low micro-millimolar extracellularly applied Ba2+ (Fig. 1a bottom). Not surprisingly therefore, Ba2+ is frequently used as a relatively selective inhibitor for Kir channels since higher concentrations of Ba2+ are required to block other K+ channels. At millimolar concentrations, Cs+ is also a useful blocker, yet it lacks the specificity of Ba2+ for inwardly rectifying channels.
Molecular identification of glial Kir channels
Early studies on astrocytes in culture and slices revealed Kir channel expression in nearly every preparation examined, however, it was not until the cloning of these channels that molecular identification became possible. Many investigators have examined Kir channels in glial cells by PCR, Western blot and immunohistochemistry. Transcripts and protein products for many Kir channel subunits have been shown repeatedly in astrocytes, oligodendrocytes and retinal Müller glial cells. For example, single cell PCR studies in hippocampal astrocytes identified transcripts for four Kir genes namely Kir2.1, 2.2, 2.3, and 4.1 (Schroder et al. 2002). Our own work has demonstrated transcripts for 15 of 16 Kir channel transcripts in cultured spinal cord astrocytes (Olsen et al. 2006). Kir2.1 and 2.2 were detected by immunohistochemistry using antibodies directed against these proteins in hind brain and retinal Müller cells. These subunits were found in astrocyte membranes opposed to neuronal cell bodies, where it is thought that they are responsible for K+ uptake (Stonehouse et al. 1999). Kir6.1 protein has been identified in fine distal processes in hippocampus, cortical cerebellar astrocytes and Bergmann glial cells (Thomzig et al. 2001). However, despite the large number of Kir channel subunits found in glial cells throughout the CNS, multiple lines of evidence suggest Kir4.1 is the principle pore forming subunit in glial cells.
Initial evidence supporting a role for Kir4.1 as the predominant Kir channel subunit came from a mouse line with a targeted disruption in Kir4.1. Müller cells in these animals demonstrated a greater than 10-fold increase in input resistance, indicative of decreased potassium permeability, and a depolarized resting membrane potential (−13.2 ± 2.9 mV vs. −85.2 ± 0.7 mV in wild-type littermates). Similar findings were reported in oligodendrocytes in the spinal cord (Neusch et al. 2001), astrocytes in the spinal cord (Olsen et al. 2006), astrocytes in the ventral respiratory group (Neusch et al. 2006) and astrocytes from the hippocampus (Djukic et al. 2007). These observations were extended to rats using siRNA knockdown in spinal cord (Olsen et al. 2006) and cortical astrocytes (Kucheryavykh et al. 2007). The results from these studies demonstrate a complete lack of Ba2+ sensitive currents attributable to Kir4.1 channels (Fig. 2a and b). These cells show a 5-fold increase in input resistance i.e. a 5-fold decrease in resting ion permeability and are significantly depolarized (Fig. 2c and d). Finally, inducible, astrocyte specific elimination of Kir4.1 produces glia that lack Ba2+ sensitive inwardly rectifying channels (Fig. 2e, Djukic et al. 2007). Hence in vitro, and in situ, the genetic elimination of Kir4.1 is sufficient to remove any trace of K+ permeability attributable to Kir channels. This supports the conclusion that Kir4.1 is an essential component of the glial Kir channel.
Fig. 2.
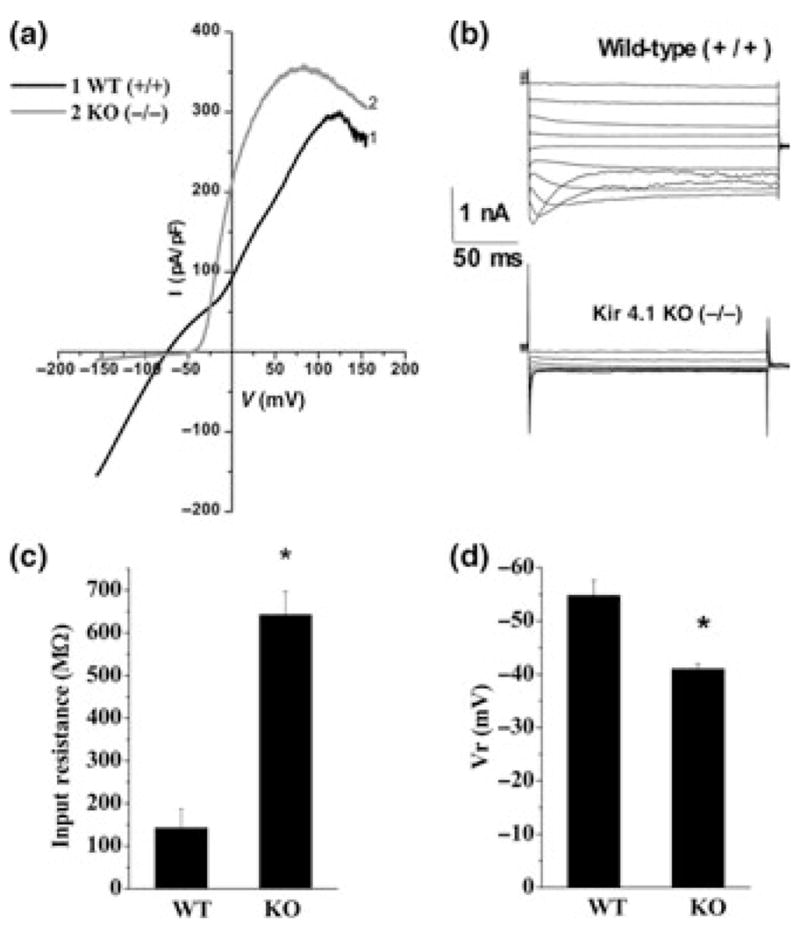
Inwardly rectifying K+ currents in astrocytes are mediated by Kir4.1: (a) Voltage-ramps were used to elicit currents in astrocytes isolated from wildtype (WT,1) and Kir4.1 knockout (KO,2) animals. Only wildtype recordings showed any significant inward currents. (b) Using voltage step protocols, wildtype astrocytes showed pronounced inward currents with characteristic inactivation at negative potentials. Inward currents were completely absent in astrocytes from Kir4.1 knockout animals. (c) Astrocytes cultured from knock-out animals demonstrated a near 5-fold increase in input resistance. (d) Resting membrane potential is significantly depolarized in astrocytes cultured from knock out animals (*p < 0.05). [With permission from Olsen et al. 2006].
It is possible that astrocytic Kir channels are formed by the heteromeric association of multiple Kir genes, and hence the disruption of Kir4.1 may be sufficient to eliminate functional channels. A heteromeric assembly from Kir4.1 and 5.1 has been shown for astrocytes in neocortex and in the glomeruli of the olfactory bulb (Hibino et al. 2004). For a more extensive discussion of heteromeric Kir channels in glia the reader is referred to a recent review (Butt and Kalsi 2006). However, most electrophysiological data published to date does not suggest the presence of functional heteromeric channels in astrocytes or oligodendrocytes.
Roles for Kir channels in normal glia
Without question, the expression of Kir channels confers in large part the very deep resting potential and high K+ permeability that characterizes glial cells (Ransom and Goldring 1973). These two functions of Kir channels have been inferred from pharmacological studies that showed a marked depolarization upon application of Ba2+. For example, in spinal cord astrocytes Ba2+ depolarizes the membrane of astrocytes up to 35 mV (Ransom and Sontheimer 1994). Pharmacological studies are now complemented by studies targeted to specifically disrupt Kir4.1. Collectively they demonstrate similar depolarizing shifts in resting potential and massive changes in input resistance. These changes in electrophysiological properties indicate a high open probability of Kir4.1 at resting membrane potentials. The high open probability in turn contributes to the negative resting membrane potential and a large portion of K+ permeability. The high K+ permeability is required for potassium clearance or uptake while the negative resting potential of glial cells has been postulated to be essential for a number of glial functions including K+ buffering, neurotransmitter uptake, and pH regulation. It may additionally serve to induce or maintain a differentiated cell phenotype, as further discussed below.
Kir4.1 and K+ buffering
One of the most discussed functions of glial cells pertains to their role in extracellular K+ homeostasis [for review see, (Kofuji and Newman 2004)]. The narrowness of the extracellular space requires effective mechanisms to regulate K+ as a single action potential can raise extracellular [K+] by close to 1 mM and high frequency neuronal discharge can raise K+ by several millimolar (Ransom et al. 1995). Since any increase in extracellular K+ could compromise normal neuronal firing, it is attractive to consider the possibility that glial cells may buffer these changes by uptake. This can occur in several ways. The most popular of the hypothesized ways involves diffusional uptake of K+ in areas of elevated K+ and release at distant sites where K+ is low. This spatial redistribution, termed ‘spatial buffering’ (Orkand 1980) requires no additional energy, is rapid, and can effectively shuttle K+ through intracellular routes over long distances. Indeed, K+ could spread to neighboring cells via gap-junctions. This mechanism of potassium clearance has been postulated to be more effective than pure diffusion in several preparations (Gardner-Medwin 1983). A second proposed K+ buffer mechanism is analogous to that suggested by Boyle and Conway (Boyle and Conway 1941) in muscle cells. Here K+ uptake is balanced by the uptake of Cl−, essentially importing KCl, which is uncharged together with water, which would swell the cell. Glial cell swelling following neuronal activity has been demonstrated following electrical stimulation (MacVicar and Hochman 1991; MacVicar et al. 2002). Evidence suggesting that Kir channels are involved in these processes was provided in acute slices from guinea pig olfactory cortex. Using double barreled ion–sensitive microelectrodes glial Cl− and K+ levels were shown to increase following stimulation and the intracellular rise in K+ was inhibited by Ba2+ application (Ballanyi et al. 1987). Similarly, in hippocampal slices, extracellular K+ increases were augmented when Kir channels were blocked with Ba2+ (Heinemann et al. 2000). For both mechanisms, K+ uptake has been proposed to be accomplished through inwardly rectifying K+ channels with recent evidence pointing to Kir4.1 as the principle channel employed. The most likely Cl− uptake pathway is via ClC-2 Cl− channels (Sik et al. 2000).
In support of Kir4.1 being the channel that supports spatial buffering and homeostasis, knockdown (Olsen et al. 2006; Kucheryavykh et al. 2007) and knockout studies (Neusch et al. 2006) show impairment in K+ uptake. The examples in Fig. 3(a) and (b) compare side-by-side K+ currents in response to a 50 mM K+ challenge in wildtype and knockout astrocytes from the ventral respiratory group; the latter lacking any measurable K+ uptake current (Neusch et al. 2006). Astrocytes from Kir4.1 conditional knockout mice demonstrate a near complete lack of Ba2+ sensitive uptake currents following Schaffer collateral stimulation (Fig. 3c and d). Consequently the larger and prolonged extracellular [K+] increases following neuronal activity make Kir4.1 knockout mice more prone to neuronal hyperexcitability (Djukic et al. 2007). In retinal Müller cells channel mediated K+ redistribution from synaptic layers of the retina to the vitreous humor has been elegantly demonstrated (Newman 1984). This directed K+ clearance termed K+ siphoning in the retina has also been conclusively linked to Kir4.1 (Kofuji et al. 2000). However, while these studies show a requirement for Kir4.1 channels, they can not rule out that K+ is taken up together with Cl− and H2O in a Boyle-Conway type scenario. In support of such a scenario Kir4.1 and Aqp4 (Nagelhus et al. 1999) colocalize to astrocytic endfeet at the immuno-electronmicroscopic level and co-immunoprecipitation studies suggest a protein-protein interaction through a multi-protein dystrophin-glycoprotein complex (Nagelhus et al. 2004). Another study suggests that ClC-2 Cl− channels also localize to the astrocytic endfeet (Sik et al. 2000).
Fig. 3.

Loss of Kir4.1 inhibits K+ and glutamate uptake. (a,b) Patch-clamp recordings were obtained in astrocytes in slices from the ventral respiratory group in wildtype and Kir4.1 knockout animals. Application of 50 mM K+ induced an inward current in wildtype astrocytes but not in astrocytes from Kir4.1 knockout mice (*p < 0.05). [(a,b) With permission from Neusch et al. 2006; (c,d) With permission from Djukic et al. 2007].
It should be noted that the weakly-rectifying nature of Kir4.1 permits bidirectional movement of K+ into and out of the cell that is dependent on the transmembrane potassium gradient. This feature allows for K+ efflux at the vitreous humor (or K+ sink) during K+ siphoning in retinal Müller cells. This aspect of Kir4.1 channels may be important with regards to glial mediated K+ release back into the extracellular space following neuronal activity. During neuronal activity, an increase in neuronal intracellular sodium activates the Na+/K+-ATPase. Sustained pump activation because of elevated levels of intracellular Na+ after cessation of neuronal activity leads to an undershoot or a reduction in baseline potassium levels in the extracellular space. Consistent with the idea of astrocyte Kir mediated K+ release providing a return pathway for pathway for K+, Ba2+ (200 μM) caused an increase in the pump mediated undershoot (D’Ambrosio et al. 2002). Importantly, this K+ release was shown to be directly mediated by Kir4.1 in astrocytes from the ventral respiratory group using Kir4.1 knockout animals (Neusch et al. 2006).
Although the role of glial Kir channels and specifically Kir4.1 seems to be well established in K+ homeostasis and buffering, it remains an excellent hypothesis and other mechanisms of K+ clearance are likely utilized. For instance, one study in the hippocampus using Ba2+ and oubain, a Na+/K+-ATPase inhibitor, to differentiate between glial Kir channel and pump mediated K+ clearance demonstrated Kir channels regulate baseline K+ accumulation but are not involved in K+ clearance, even during high frequency stimulation (D’Ambrosio et al. 2002). Further it was recently shown that Ba2+ application following neuronal activity in hippocampal slices had little impact on baseline [K+]o levels or stimulus induced rises in [K+]o (Meeks and Mennerick 2007). Similarly, in the optic nerve, K+ clearance under physiological conditions does not involve Ba2+ -sensitive Kir channels but is instead accomplished predominately by axonal reuptake via the Na+/K+-ATPase (Ransom et al. 2000). And data from Kir4.1 knockout studies demonstrate, surprisingly normal basal synaptic transmission in the hippocampus (Djukic et al. 2007) as well as normal rhythmic bursting activity in ventral respiratory group (Neusch et al. 2006), suggesting that at least in these tissues other mechanisms contribute to K+ homeostasis. In each instance, however, when channels are involved, Kir4.1 is the most likely candidate to permit the diffusional entry of K+ along a favorable electrochemical gradient.
Kir4.1 and glutamate transport
One of the most important astrocytic functions is the clearance of neuronally released glutamate (Anderson and Swanson 2000). To accomplish this, astrocytic processes are positioned in the close vicinity of glutamatergic synapses and are richly endowed with Na+-dependent glutamate transporters (Ueda et al. 2001). These transporters are electrogenic and hence take up glutamate much more effectively at negative resting potentials. One would therefore postulate that expression of Kir4.1, which sets the resting membrane potential close to the EK, typically around −85 mV, would ensure maximal transport rates via either of the two glial glutamate transporters Glt-1 or GLAST. This question was examined in a recent study in which Kir4.1 channels were suppressed using a siRNA approach. This paper indeed showed a reduction in glutamate uptake via Glt-1 by ~30% following Kir4.1 channels knockdown or its functional block by Ba2+ (Kucheryavykh et al. 2007). Importantly, reduced glutamate uptake is also is seen following astrocyte specific knockout of Kir4.1 in vivo (Fig. 3c and d, Djukic et al. 2007). These findings may in part explain why these animals were more prone to seizures. A similar seizure phenotype was observed in animals in which glutamate uptake was compromised by knockdown of Glt-1 transporters (Rothstein et al. 1996). Taken together these data suggest that the negative resting potential imparted by Kir4.1 is essential to maintain a sufficient electrochemical gradient for astrocytes to engage in effective glutamate homeostasis.
Association of glial Kir channels with aquaporins
A fascinating and somewhat controversial story emerged from studies that examined the localization of Kir4.1 channels to astrocytic endfeet and their relationship to blood vessels (for review see: Nagelhus et al. 2004). As alluded to above, the hypothesis that K+ buffering may be associated with Cl− and water fluxes led some investigators to examine a possible co-localization of these proteins. Using immunogold-EM, Kir4.1 channels were shown to co-localize with Aqp4 (Nagelhus et al. 1999), the principle water channel expressed in astrocytes on astrocytic membranes facing endothelial cells (Tait et al. 2008). Furthermore, immunoprecipitation experiments using Kir4.1 antibodies isolated a protein complex consisting of Kir4.1, Aqp4 and several members of the dystrophin-glycoprotein family (Connors et al. 2004). Supporting a functional interaction between these proteins, alpha syntrophinh−/− mice (alpha-syntrophin is a component of the dystrophin-glycoprotein complex), display marked mislocalization of Aqp4, and demonstrate delayed K+ clearance, although Kir4.1 localization remains unchanged (Amiry-Moghaddam et al. 2003). In the retina it is hypothesized that this association with members of the dystrophin associated complex tethers Kir4.1 and Aqp4 together (Connors et al. 2004) and anchors them to the cell membrane (Puwarawuttipanit et al. 2006). It appears that even during early postnatal development these proteins may traffic together as both proteins are found in horizontal neurons in retina from young animals and after P15 are found to localize together in Müller cells (Bosco et al. 2005). When a member of the dystrophin associated proteins, dystrophin 71, is knocked-out, Aqp4 and Kir4.1 expression were shown to decrease around blood vessels (Dalloz et al. 2003). An examination of protein revealed a general decrease in Aqp4 expression and diffuse labeling of Kir4.1 which led to a redistribution of Müller cell K+ conductance (Fort et al. 2008) typically highly enriched in Müller cell endfeet (Newman 1986). In disagreement with these studies, Aqp4 knockout mice display no changes in the electrophysiological properties of Müller cells or Kir4.1 localization but show a marked decrease in water permeability (Ruiz-Ederra et al. 2007). Further, this group went on to demonstrate that functional inhibition of Kir4.1 by RNAi or inhibition with Ba2+ in primary cultured brain cells did not significantly alter water permeability and molecular interactions between AQ4 and Kir4.1 could not be shown in hippocampal astrocytes (Zhang and Verkman 2008). It is possible that the co-localization may have been the result of a clustering of the proteins in specialized lipid raft domains which bring important molecules to a common site on the membrane without them necessarily interacting at the protein level (Hibino and Kurachi 2007).
Developmental expression of Kir channels
A consistent observation in glia cells in culture and in situ is the developmental up-regulation of Kir channel activity. For instance, early in culture A2B5+, O4+ cells of oligodendrocytes lineage demonstrate little if any Kir currents. However, as these cells differentiate into mature oligodendrocytes Kir channel activity appears (Sontheimer et al. 1989). Similar results were observed in cultured spinal cord astrocytes (Ransom and Sontheimer 1994) and Schwann cells (Wilson and Chiu 1990). In acutely dissociated Müller cells, Kir channel activity appears after postnatal day 7 (P7) (Bringmann et al. 1999). And in hippocampal slices, Kir currents increase from P5 to P20 in complex astrocytes (Bordey and Sontheimer 1997). The increase in channel activity is associated with a more hyperpolarized resting membrane potential which is now largely attributed to Kir4.1. Using immunohistochemistry Kir4.1 immunoreactivity in fine processes of astrocytes and oligodendrocytes cell bodies in the optic nerve was evident at P10 and peaked at P15 (Kalsi et al. 2004). Labeling on processes was also developmentally up-regulated and detected in astrocytes from the ventral respiratory group (Neusch et al. 2006). In spinal cord, Kir4.1 immunoreactivity labels astrocytes in the gray matter and co-localizes with Glt-1 (Fig. 4a–c) and oligodendrocytes cell bodies but not in neurons (Olsen et al. 2007, Neusch et al. 2001). When examining Kir4.1 expression in spinal cord in vivo, we found a profound developmental up-regulation of Kir4.1 in whole spinal cord lysates probed by Western blot (Fig. 4d). Kir4.1 appears to be weakly expressed at birth (P0), prominent at P15 and strongly expressed in the adult animal (P30) (Olsen et al. 2006). The developmental expression pattern of Kir4.1 channels, which are believed to underlie the [K+] homeostasis absolutely correlates with the developmental regulation of extracellular K+ dynamics in vivo (Connors et al. 1982) where K+ fluctuates widely at birth and becomes increasingly regulated by about P30 when Kir4.1 expression saturates (Fig. 4e).
Fig. 4.

Kir4.1 is developmentally regulated in glia in the spinal cord. (a) Images from the ventral horn demonstrating Kir 4.1 largely overlapped with GLT-1 as demonstrated by the yellow color in the merged image. (b) Kir4.1 and Neu-N labeling appear distinct in the ventral horn. (c) High magnification images from the ventral horn demonstrate Kir4.1 staining is most intense surrounding neuronal cell bodies. [Scale bars (a) and (b), 100 μm, (c) 50μm]. (d) Whole-cell lysates from wild-type rat spinal cord were probed with antibodies to Kir4.1 and two bands at 55 kD and ~200 kD characteristic of monomeric and tetrameric Kir4.1 alpha subunits. Expression was very weak at P0, increased notably at P15 and was high at P30 suggesting a developmental gain in expression of Kir4.1 in spinal cord. (e) Over the same developmental time period, recordings with extracellular K+ electrodes in optic nerve in situ suggest a tightening of extracellular K+ fluctuations with developmental age from P2 to adulthood. [(a–c) With permission from Olsen et al., 2007; (d) With permission from Olsen et al. 2006; (e) with permission from Connors et al. 1982].
Kir4.1 and cell differentiation
For the past two decades, numerous studies have examined ion channels in inexcitable cells and their possible role in cell proliferation and differentiation. From essentially all cell systems examined to date a common finding that dividing cells have relatively positive resting membrane potentials, typically between −30 mV to −50 mV when compared to terminally differentiated cells (Sontheimer 1995). This is clearly the case when comparing glial progenitor cells to differentiated oligodendrocytes (Sontheimer et al. 1989) or astrocytes (Bordey and Sontheimer 1997) and when comparing immature neurons to differentiated neurons (Ribera and Spitzer 1992). The mechanistic link between membrane potential and cell division is poorly understood. However, studies dating back over 40 years demonstrated that cells with a negative resting membrane potential rarely enter mitosis (Cone 1970). The transition from immature astrocyte to differentiated astrocyte has been well characterized, and these studies suggests that the exit of the cell cycle is associated with a negative shift in resting potential that correlates with the up-regulation of Kir channel. Indeed, when Kir channels are pharmacologically inhibited, cell differentiation and exit from the cell cycle is delayed (MacFarlane and Sontheimer 2000). Consistent with this oligodendrocytes cultured from Kir4.1 null mice display a depolarized resting membrane potential and immature morphology (Neusch et al. 2001) and cultured cortical astrocytes demonstrate significantly lower Kir4.1 mRNA levels compared to confluent cells (Li et al. 2001).
Changes in Kir expression in malignancy
Similar to proliferating glial progenitors cells, glial-derived tumors (gliomas) maintain a relatively depolarized membrane potential around −20 mV to −40 mV (Ullrich et al. 1998). Patch-clamp recordings showed no evidence for Kir currents in gliomas but instead revealed the expression of Ca2+-activated K+ channels (Ransom and Sontheimer 2001) along with ClC-2 and ClC-3 Cl− channels (Olsen et al. 2003). As the former require changes in [Ca2+] or a very large depolarization to be activated, the resting conductance for K+ ions is very low in gliomas. However, PCR and Western blots show prominent expression of several Kir channel genes at the transcript and protein level. These included Kir2.1; 2.3; 3.1, and 4.1 (Olsen and Sontheimer 2004a). Closer examination showed Kir4.1 channels to be absent from the plasma membrane but prominently expressed in the nucleus (Fig. 5a) whereas it was expressed throughout the plasma membrane in wild-type spinal cord astrocytes (Fig. 5b). Upon cloning and sequencing, no mutations were found in the Kir4.1 gene that could explain this mislocalization (H. Higashimori, University of Alabama at Birmingham, unpublished). This prompted the question of whether Kir4.1 mislocalization to this region may necessary to allow cells to divide. Another way of phrasing the question would be to ask whether an absence of Kir from the membrane is required for malignant glioma cell division and whether insertion of these channels would inhibit proliferation and induce differentiation. This indeed appears to be the case (Higashimori and Sontheimer 2007). When recombinant Kir4.1 was either transiently or stably transfected into human glioma cells, functional Kir4.1 channels were expressed that gave rise to Ba2+sensitive inwardly rectifying channels with properties identical to those observed in astrocytes (Fig. 5c). Importantly, the cell membrane became as hyperpolarized as that of non-malignant cells (Fig. 5d). This alone was sufficient to arrest cell growth (Fig. 5e) and shifted cells into the G0/G1 phase of the cell cycle. And the same cells, when chronically treated with Ba2+ so as to block Kir4.1 channels, caused a membrane depolarization and cells returned to proliferation rates comparable to that of wild type glioma cells. Furthermore, simply depolarizing cells that express functional Kir4.1 channels with 20 mM [K+]o was sufficient to induce cell division suggesting that the role of Kir4.1 regarding cell proliferation ultimately comes down to changes in resting membrane potential. More specifically, Kir4.1, by hyperpolarizing the resting membrane potential causes cell cycle arrest. Understanding what causes the mislocalization of Kir4.1 in glioma to the nucleus rather than the plasma membrane may provide insight as to how to interfere with the aberrant growth of these cancers. Similarly, regulation of Kir4.1 function in non-malignant astrocytes may hold the promise to influence the differentiation/dedifferentiation of astrocytes in normal brain or following injury.
Fig. 5.

Role of Kir4.1 in glial growth control: (a) Immunostaining with antibodies to Kir4.1 and the cytoskeleton with Phalloiden (Kir4.1: green, Phalloiden:red) show Kir4.1 labels the cell nucleous. (b) In contrast in astrocytes Kir4.1 (green) labels the cell membrane (the cytoskeleton is labeled with GFAP, red). (c) This difference is also reflected in patch-clamp recordings from another glioma cells line (D54) which lack inward currents compared to SC astrocytes that express prominent inward currents. However, upon transfection of D54MG cells with a GFP containing plasmid encoding Kir4.1, these glioma cells express inward currents that are indistinguishable from astrocytic Kir currents. (d) Expression of Kir4.1 in glioma caused their resting potential to shift ~30 mV negative making it similar to that of differentiated astrocytes. (e) The presence or absence of functional Kir4.1 channels determined the rate of growth of D54 glioma cells, in which growth was retarded when Kir4.1 channels were functional. However, depolarizing the cell with 20 mM K+ was sufficient to rescue growth even when Kir4.1 was over-expressed [(a) With permission from Olsen and Sontheimer 2004a; (b) With permission Olsen et al. 2006; (c–e) With permission from Higashimori and Sontheimer 2007].
Glial Kir4.1 channels following injury and implications for neurological disease
A large body of work demonstrates that the glial Kir4.1 expression and function decreases following acute injury and is often associated with glial fibrillary acidic protein (GFAP) up-regulation and gliotic cell proliferation. Our work has demonstrated that when proliferation is experimentally induced by a mechanical scar type injury in spinal cord astrocytes Kir channel activity decreases (MacFarlane and Sontheimer 1997). Gliotic proliferation near the scar was assessed by BRDU labeling. BRDU+ cells showed a 3-fold decrease in Kir currents, while these same cells up-regulated outwardly rectifying fast inactivating (Ka) and delayed rectifying (Kd) currents). In BRDU− cells, the primary conductance was IKir. In the retina following ischemic injury, Müller cell Kir currents were reduced to 20% that of control cells within three days of injury (Pannicke et al. 2005). Injury appeared to cause dedifferentiation of glial cells (Bringmann et al. 2000). Proliferative vitreoretinopathy in the retina is associated with proliferative gliosis of Müller cells. In proliferative vitreoretinopathy retinas from humans, rabbits and rodents, K+ conductance mediated by Kir channels is nearly abolished (Ulbricht et al. 2008). This lack of channel activity is not associated with an overall decrease in Kir4.1 but a mislocalization and possible inactivation of the channel (Ulbricht et al. 2008). Abnormal K+ accumulation was seen in hippocampal slices following traumatic brain injury which was attributed to a decrease in Kir currents as blockade of Kir channels with Ba2+ in control slices produced similar results (D’Ambrosio et al. 1999). In addition to altered ion homeostasis, the decreased channel activity would lead to membrane depolarizations and thus less efficient glutamate clearance. Reduced glutamate uptake has been demonstrated in astrocytes in Kir4.1 conditional knockout mice (Djukic et al. 2007) and in rat astrocytes in which Kir4.1 levels were decreased with siRNA (Kucheryavykh et al. 2007). Logically, these changes would impact the excitability of neurons. A recent study targeted Kir4.1 using siRNA in satellite glial cells surrounding primary sensory neurons in the rat trigeminal ganglion; the region responsible for pain sensation in the face (Vit et al. 2008). The decrease in Kir4.1 channel activity led to spontaneous and evoked pain like behavior in free moving rats. Further, when these authors constricted the infraorbital nerve, Kir4.1 levels decreased ~40%. Although not directly assessed, these results suggest that decreases in Kir4.1 caused changes in neuronal excitability, which led to abnormal sensory perception. Similar results may occur in the spinal cord as we observed an ~80% decrease in Kir4.1 immunoreactivity as assessed by Western blot following a severe crush injury to the spinal cord 7 days post-injury (M. L. Olsen, University of Alabama at Birmingham, unpublished).
Neurological disorders lead to cellular changes that take place on a slower time scale than that of acute injury. Glial hypertrophy and GFAP up-regulation are also associated with diseases including epilepsy, amyotrophic lateral sclerosis, and Alzheimer’s. Astrocytes from several animals models of neurological disease and disorders show marked decreases in Kir channel activity or function. For example, a progressive decrease in Kir4.1 immunoreactivity was observed in the ventral horn of the SOD1G93A mutant, a common model for amyotrophic lateral sclerosis. This loss was associated with a greater susceptibility of motor neuron cell death induced by high K+ suggesting that the loss of glial K+ buffering via Kir4.1 contributes to motor neuron cell death (Kaiser et al. 2006). Epilepsy is unique in that although several animal models are available, human tissue is readily obtainable from patients with intractable epilepsies. Studies examining glial membrane properties in such tissue demonstrate a near complete lack of inwardly rectifying currents (Bordey and Sontheimer 1998) immature current patterns (Hinterkeuser et al. 2000) and an impaired ability to clear K+ (Gabriel et al. 1998; Jauch et al. 2002). And both Kir4.1 null mice and conditional knock-out mice demonstrate severe seizures prior to death. Finally, KCNJ10, the gene that codes for Kir4.1 is a putative seizure susceptibility gene in mice (Ferraro et al. 2004) and humans (Buono et al. 2004). It must be noted that in all of the above studies it is difficult to separate cause and effects. It is conceivable that any insult to the nervous system that causes gliosis and potentially glial proliferation is associated with the loss of Kir4.1, simply to permit cell division. This in turn may lead to a loss in K+ homeostasis, which may worsen the underlying condition. Kir4.1 certainly must be considered an important player in a number of neurological conditions and could be the target of some therapeutic intervention through influencing channel regulation. It is important to note that enhanced Kir4.1 expression, which would be desirable under such circumstances, has been shown to result from a 48 h exposure of astrocytes to guanosine (Benfenati et al. 2006). Furthermore glial Kir currents are acutely modulated by beta-adrenergic agonists (Roy and Sontheimer 1995).
Lessons learned from Kir4.1 knockout animals
Knockout animals were essential in defining Kir4.1 as the astrocytic Kir channel involved in setting the resting membrane potential and as the likely diffusional pathway for K+ in spatial buffering. However, these animals also harbored several surprises. The first generation of Kir4.1 knockout (KO) animals died within 8–20 days and presented with pronounced CNS pathology. Unexpectedly, the largest pathology was observed in white matter in the spinal cord and hippocampus (Neusch et al. 2001). While this could have suggested that the loss of Kir4.1 in astrocytes somehow negatively affects the development or maintenance of white matter, i.e. axons and myelin, it also made plausible the idea that oligodendrocytes may rely on Kir4.1 expression. Instead, what emerged, and is now supported by conditional knockout studies that eliminated Kir4.1 only in GFAP expressing cells (Djukic et al. 2007) is the notion that Kir4.1 is required for normal differentiation of glial progenitor cells that also give rise to myelinating oligodendrocytes. While it has been clear for some time that oligodendrocytes also express Kir channels (Girsch and Peracchia 1985), the role of Kir4.1 in supporting myelination only became apparent through studies of knockout animals. Moreover what became apparent is that glial progenitor cells, possibly O2A cells that are destined to become oligodendrocytes express the GFAP gene, and hence are targeted by this deletion. These animals also died at 20–25 days of age, suggesting that the early death was a direct consequence of a CNS loss of Kir4.1 channel function. Yet another surprise from the conditional KO studies was the finding that a subpopulation of astrocytes termed ‘complex glia’ appears to be decimated in these animals. This suggest that perhaps complex glial cells are the equivalent of a ‘type 2 astrocyte’ and derive from the same progenitor cell (O2A) that gives rise to oligodendrocytes. The loss of Kir4.1 may prevent the differentiation of these cells into either astrocytes or olidogodendrocyte. Finally, a possibly more direct role for Kir4.1 in myelination remains to be explored. Taken together, these findings certainly raise the stature of Kir4.1 with regards to it role in normal brain development and the channels potential implication in neurological disease.
Acknowledgments
The author is grateful for the continued supported by grants from the National Institutes of Health RO1 NS-31234, RO1 NS-52634 and RO1 NS-36692.
Abbreviations used
- GFAP
glial fibrillary acidic protein
- KO
knockout
- siRNA
small interfering RNA
References
- Amiry-Moghaddam M, Williamson A, Palomba M, Eid T, de Lanerolle NC, Nagelhus EA, Adams ME, Froehner SC, Agre P, Ottersen OP. Delayed K+ clearance associated with aquaporin-4 mislocalization: phenotypic defects in brains of alpha-syntrophin-null mice. Proc Natl Acad Sci USA. 2003;100:13615–13620. doi: 10.1073/pnas.2336064100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CM, Swanson RA. Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia. 2000;32:1–14. [PubMed] [Google Scholar]
- Ballanyi K, Grafe P, TenBruggencate G. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol (London) 1987;382:159–174. doi: 10.1113/jphysiol.1987.sp016361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Nobile M, Rapisarda C, Ferroni S. Guanosine promotes the up-regulation of inward rectifier potassium current mediated by Kir4.1 in cultured rat cortical astrocytes. J Neurochem. 2006;98:430–445. doi: 10.1111/j.1471-4159.2006.03877.x. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Postnatal development of ionic currents in rat hippocampal astrocytes in situ. J Neurophysiol. 1997;78:461–477. doi: 10.1152/jn.1997.78.1.461. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Properties of human glial cells associated with epileptic seizure foci. Epilepsy Res. 1998;32:286–303. doi: 10.1016/s0920-1211(98)00059-x. [DOI] [PubMed] [Google Scholar]
- Bosco A, Cusato K, Nicchia GP, Frigeri A, Spray DC. A developmental switch in the expression of aquaporin-4 and Kir4.1 from horizontal to Muller cells in mouse retina. Invest Ophthalmol Vis Sci. 2005;46:3869–3875. doi: 10.1167/iovs.05-0385. [DOI] [PubMed] [Google Scholar]
- Boyle PJ, Conway EJ. Potassium accumulation in muscle and associated changes. J Physiol (London) 1941;100:1–63. doi: 10.1113/jphysiol.1941.sp003922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Biedermann B, Reichenbach A. Expression of potassium channels during postnatal differentiation of rabbit Muller glial cells. Eur J Neurosci. 1999;11:2883–2896. doi: 10.1046/j.1460-9568.1999.00706.x. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Francke M, Pannicke T, Biedermann B, Kodal H, Faude F, Reichelt W, Reichenbach A. Role of glial K+ channels in ontogeny and gliosis: a hypothesis based upon studies on Muller cells. Glia. 2000;29:35–44. doi: 10.1002/(sici)1098-1136(20000101)29:1<35::aid-glia4>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Buono RJ, Lohoff FW, Sander T, et al. Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res. 2004;58:175–183. doi: 10.1016/j.eplepsyres.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Butt AM, Kalsi A. Inwardly rectifying potassium channels (Kir) in central nervous system glia: a special role for Kir4.1 in glial functions. J Cell Mol Med. 2006;10:33–44. doi: 10.1111/j.1582-4934.2006.tb00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee WA, Amarillo Y, Chiu J, et al. Molecular diversity of K+ channels. Ann N Y Acad Sci. 1999;868:233–285. doi: 10.1111/j.1749-6632.1999.tb11293.x. [DOI] [PubMed] [Google Scholar]
- Cone CD., Jr Variation of the transmembrane potential level as a basic mechanism of mitosis control. Oncology. 1970;24:438–470. doi: 10.1159/000224545. [DOI] [PubMed] [Google Scholar]
- Connors BW, Ransom BR, Kunis DM, Gutnick MJ. Activity-dependent K+ accumulation in the developing rat optic nerve. Science. 1982;216:1341–1343. doi: 10.1126/science.7079771. [DOI] [PubMed] [Google Scholar]
- Connors NC, Adams ME, Froehner SC, Kofuji P. The potassium channel Kir4.1 associates with the dystrophin-glycoprotein complex via alpha-syntrophin in glia. J Biol Chem. 2004;279:28387–28392. doi: 10.1074/jbc.M402604200. [DOI] [PubMed] [Google Scholar]
- D’Ambrosio R, Maris DO, Grady MS, Winn HR, Janigro D. Impaired K+ homeostasis and altered electrophysiological properties of post-traumatic hippocampal glia. J Neurosci. 1999;19:8152–8162. doi: 10.1523/JNEUROSCI.19-18-08152.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- Dalloz C, Sarig R, Fort P, et al. Targeted inactivation of dystrophin gene product Dp71: phenotypic impact in mouse retina. Hum Mol Genet. 2003;12:1543–1554. doi: 10.1093/hmg/ddg170. [DOI] [PubMed] [Google Scholar]
- Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik CA, Davidson N, Lester HA. The inward rectifier potassium channel family. Curr Opin Neurobiol. 1995;5:268–277. doi: 10.1016/0959-4388(95)80038-7. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, et al. Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm Genome. 2004;15:239–251. doi: 10.1007/s00335-003-2270-3. [DOI] [PubMed] [Google Scholar]
- Fort PE, Sene A, Pannicke T, et al. Kir4.1 and AQP4 associate with Dp71- and utrophin-DAPs complexes in specific and defined microdomains of Muller retinal glial cell membrane. Glia. 2008;56:597–610. doi: 10.1002/glia.20633. [DOI] [PubMed] [Google Scholar]
- Gabriel S, Kivi A, Kovacs R, Lehmann TN, Lanksch WR, Meencke HJ, Heinemann U. Effects of barium on stimulus-induced changes in [K+]o and field potentials in dentate gyrus and area CA1 of human epileptic hippocampus. Neurosci Lett. 1998;249:91–94. doi: 10.1016/s0304-3940(98)00420-0. [DOI] [PubMed] [Google Scholar]
- Gardner-Medwin AR. Analysis of potassium dynamics in mammalian brain tissue. J Physiol (London) 1983;335:393–426. doi: 10.1113/jphysiol.1983.sp014541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girsch SJ, Peracchia C. Lens cell-to-cell channel protein: II. Conformational change in the presence of calmodulin. J Mem Biol. 1985;83:227–233. doi: 10.1007/BF01868697. [DOI] [PubMed] [Google Scholar]
- Hagiwara S, Takahashi K. The anomalous rectification and cation selectivity of the membrane of a starfish egg cell. J Mem Biol. 1974;18:61–80. doi: 10.1007/BF01870103. [DOI] [PubMed] [Google Scholar]
- Heinemann U, Gabriel S, Jauch R, Schulze K, Kivi A, Eilers A, Kovacs R, Lehmann TN. Alterations of glial cell function in temporal lobe epilepsy. Epilepsia. 2000;41:S185–S189. doi: 10.1111/j.1528-1157.2000.tb01579.x. [DOI] [PubMed] [Google Scholar]
- Hibino H, Kurachi Y. Distinct detergent-resistant membrane microdomains (lipid rafts) respectively harvest K(+) and water transport systems in brain astroglia. Eur J Neurosci. 2007;26:2539–2555. doi: 10.1111/j.1460-9568.2007.05876.x. [DOI] [PubMed] [Google Scholar]
- Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem. 2004;279:44065–44073. doi: 10.1074/jbc.M405985200. [DOI] [PubMed] [Google Scholar]
- Higashimori H, Sontheimer H. Role of Kir4.1 channels in growth control of glia. Glia. 2007;55:1668–1679. doi: 10.1002/glia.20574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinterkeuser S, Schröder W, Hager G, Seifert G, Blümcke I, Elger CE, Schramm J, Steinhäuser C. Astrocytes in the hippocampus of patients with temporal lobe epilepsy display changes in potassium conductances. Eur J Neurosci. 2000;12:2087–2096. doi: 10.1046/j.1460-9568.2000.00104.x. [DOI] [PubMed] [Google Scholar]
- Jauch R, Windmuller O, Lehmann TN, Heinemann U, Gabriel S. Effects of barium, furosemide, ouabaine and 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) on ionophoretically-induced changes in extracellular potassium concentration in hippocampal slices from rats and from patients with epilepsy. Brain Res. 2002;925:18–27. doi: 10.1016/s0006-8993(01)03254-1. [DOI] [PubMed] [Google Scholar]
- Kaiser M, Maletzki I, Hulsmann S, Holtmann B, Schulz-Schaeffer W, Kirchhoff F, Bahr M, Neusch C. Progressive loss of a glial potassium channel (KCNJ10) in the spinal cord of the SOD1 (G93A) transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;99:900–912. doi: 10.1111/j.1471-4159.2006.04131.x. [DOI] [PubMed] [Google Scholar]
- Kalsi AS, Greenwood K, Wilkin G, Butt AM. Kir4.1 expression by astrocytes and oligodendrocytes in CNS white matter: a developmental study in the rat optic nerve. J Anat. 2004;204:475–485. doi: 10.1111/j.0021-8782.2004.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B. Les Constantes electriques de la membrane du muscle. Arch Sci Physiol. 1949;3:285–299. [Google Scholar]
- Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1043–1054. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo Y, Baldwin TJ, Jan YN, Jan LY. Primary structure and functional expression of a mouse inward rectifier potassium channel [see comments] Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–281. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- Kuffler SW, Nicholls JG, Orkand RK. Physiological properties of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:768–787. doi: 10.1152/jn.1966.29.4.768. [DOI] [PubMed] [Google Scholar]
- Li L, Head V, Timpe LC. Identification of an inward rectifier potassium channel gene expressed in mouse cortical astrocytes. Glia. 2001;33:57–71. doi: 10.1002/1098-1136(20010101)33:1<57::aid-glia1006>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Electrophysiological changes that accompany reactive gliosis in vitro. J Neurosci. 1997;17:7316–7329. doi: 10.1523/JNEUROSCI.17-19-07316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacFarlane SN, Sontheimer H. Changes in ion channel expression accompany cell cycle progression of spinal cord astrocytes. Glia. 2000;30:39–48. doi: 10.1002/(sici)1098-1136(200003)30:1<39::aid-glia5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- MacVicar BA, Hochman D. Imaging of synaptically evoked intrinsic optical signals in hippocampal slices. J Neurosci. 1991;11:1458–1469. doi: 10.1523/JNEUROSCI.11-05-01458.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia. 2002;37:114–123. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Astrocyte membrane responses and potassium accumulation during neuronal activity. Hippocampus. 2007;17:1100–1108. doi: 10.1002/hipo.20344. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Horio Y, Inanobe A, Fujita A, Haug FM, Nielsen S, Kurachi Y, Ottersen OP. Immunogold evidence suggests that coupling of K+ siphoning and water transport in rat retinal Muller cells is mediated by a coenrichment of Kir4.1 and AQP4 in specific membrane domains. Glia. 1999;26:47–54. doi: 10.1002/(sici)1098-1136(199903)26:1<47::aid-glia5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Nagelhus EA, Mathiisen TM, Ottersen OP. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience. 2004;129:905–913. doi: 10.1016/j.neuroscience.2004.08.053. [DOI] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, et al. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J Neurophysiol. 2006;95:1843–1852. doi: 10.1152/jn.00996.2005. [DOI] [PubMed] [Google Scholar]
- Newman EA. Regional specialisation of retinal glial cell membrane. Nature. 1984;309:155–157. doi: 10.1038/309155a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. High potassium conductance in astrocyte endfeet. Science. 1986;233:453–454. doi: 10.1126/science.3726539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. Inward-rectifying potassium channels in retinal glial (Müller) cells. J Neurosci. 1993;13:3333–3345. doi: 10.1523/JNEUROSCI.13-08-03333.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels. Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Oliver D, Baukrowitz T, Fakler B. Polyamines as gating molecules of inward-rectifier K+ channels. Eur J Biochem. 2000;267:5824–5829. doi: 10.1046/j.1432-1327.2000.01669.x. [DOI] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. Mislocalization of Kir channels in malignant glia. Glia. 2004a;46:63–73. doi: 10.1002/glia.10346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Sontheimer H. In: Voltage-activated ion channels in glial cells, in Neuroglia. 2. Ransom BR, Kettenmann H, editors. Oxford University Press; New York: 2004b. pp. 112–130. [Google Scholar]
- Olsen ML, Schade S, Lyons SA, Amaral MD, Sontheimer H. Expresssion of voltage-gated chloride channels in human glioma cells. J Neurosci. 2003;23:5572–5582. doi: 10.1523/JNEUROSCI.23-13-05572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Higashimori H, Campbell SL, Hablitz JJ, Sontheimer H. Functional expression of K(ir)4.1 channels in spinal cord astrocytes. Glia. 2006;53:516–528. doi: 10.1002/glia.20312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen ML, Campbell SL, Sontheimer H. Differential distribution Kir4.1 in spinal cord astrocytes suggests regional differences in K+ homeostasis. J Neurophysiol. 2007;98:786–793. doi: 10.1152/jn.00340.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkand RK. Extracellular potassium accumulation in the nervous system. Fed Proc. 1980;39:1515–1518. [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Uckermann O, Iandiev I, Biedermann B, Wiedemann P, Perlman I, Reichenbach A, Bringmann A. Altered membrane physiology in Muller glial cells after transient ischemia of the rat retina. Glia. 2005;50:1–11. doi: 10.1002/glia.20151. [DOI] [PubMed] [Google Scholar]
- Puwarawuttipanit W, Bragg AD, Frydenlund DS, et al. Differential effect of alpha-syntrophin knockout on aquaporin-4 and Kir4.1 expression in retinal macroglial cells in mice. Neuroscience. 2006;137:165–175. doi: 10.1016/j.neuroscience.2005.08.051. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Goldring S. Ionic determinants of membrane potential of cells presumed to be glia in cerebral cortex of cat. J Neurophysiol. 1973;36:855–868. doi: 10.1152/jn.1973.36.5.855. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. Soc Neurosci Abs. 1994;20(Suppl PT 1 2) doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. BK channels in human glioma cells. J Neurophysiol. 2001;85:790–803. doi: 10.1152/jn.2001.85.2.790. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Ransom BR, Sontheimer H. Activity-dependent K+ accumulation in the rat optic nerve is cleared by temperature-sensitive and temperature-insensitive mechanisms. Soc Neurosci Abs. 1995;21(Part 1) [Google Scholar]
- Ransom CB, Sontheimer H, Janigro D. Astrocytic inwardly rectifying potassium currents are dependent on external sodium ions. J Neurophysiol. 1996;76:626–630. doi: 10.1152/jn.1996.76.1.626. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol. 2000;522:427–442. doi: 10.1111/j.1469-7793.2000.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribera AB, Spitzer NC. Developmental regulation of potassium channels and the impact on neuronal differentiation. Ion Channels. 1992;3:1–38. doi: 10.1007/978-1-4615-3328-3_1. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Roy M-L, Sontheimer H. b-Adrenergic modulation of glial inwardly rectifying potassium channels. J Neurochem. 1995;64:1576–1584. doi: 10.1046/j.1471-4159.1995.64041576.x. [DOI] [PubMed] [Google Scholar]
- Ruiz-Ederra J, Zhang H, Verkman AS. Evidence against functional interaction between aquaporin-4 water channels and Kir4.1 K+ channels in retinal muller cells. J Biol Chem. 2007;282:21866–21872. doi: 10.1074/jbc.M703236200. [DOI] [PubMed] [Google Scholar]
- Sakmann B, Trube G. Conductance properties of single inwardly rectifying potassium channels in ventricular cells from guinea-pig heart. J Physiol (London) 1984;347:641–657. doi: 10.1113/jphysiol.1984.sp015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder W, Seifert G, Huttmann K, Hinterkeuser S, Steinhauser C. AMPA receptor-mediated modulation of inward rectifier k(+) channels in astrocytes of mouse hippocampus. Mol Cell Neurosci. 2002;19:447–458. doi: 10.1006/mcne.2001.1080. [DOI] [PubMed] [Google Scholar]
- Sik A, Smith RL, Freund TF. Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience. 2000;101:51–65. doi: 10.1016/s0306-4522(00)00360-2. [DOI] [PubMed] [Google Scholar]
- Sontheimer H. Voltage-dependent ion channels in glial cells. Glia. 1994;11:156–172. doi: 10.1002/glia.440110210. [DOI] [PubMed] [Google Scholar]
- Sontheimer H. Ion channels in inexcitable cells. The Neuroscientist. 1995;1:64–67. [Google Scholar]
- Sontheimer H, Trotter J, Schachner M, Kettenmann H. Channel expression correlates with differentiation stage during development of oligodendrocytes from their precursor cells in culture. Neuron. 1989;2:1135–1145. doi: 10.1016/0896-6273(89)90180-3. [DOI] [PubMed] [Google Scholar]
- Stonehouse AH, Pringle JH, Norman RI, Stanfield PR, Conley EC, Brammar WJ. Characterisation of Kir2.0 proteins in the rat cerebellum and hippocampus by polyclonal antibodies. Histochem Cell Biol. 1999;112:457–465. doi: 10.1007/s004180050429. [DOI] [PubMed] [Google Scholar]
- Tait MJ, Saadoun S, Bell BA, Papadopoulos MC. Water movements in the brain: role of aquaporins. Trends Neurosci. 2008;31:37–43. doi: 10.1016/j.tins.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Thomzig A, Wenzel M, Karschin C, Eaton MJ, Skatchkov SN, Karschin A, Veh RW. Kir6.1 is the principal pore-forming subunit of astrocyte but not neuronal plasma membrane K-ATP channels. Mol Cell Neurosci. 2001;18:671–690. doi: 10.1006/mcne.2001.1048. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Doi T, Tokumaru J, Yokoyama H, Nakajima A, Mitsuyama Y, Ohya-Nishiguchi H, Kamada H, Willmore LJ. Collapse of extracellular glutamate regulation during epileptogenesis: down-regulation and functional failure of glutamate transporter function in rats with chronic seizures induced by kainic acid. J Neurochem. 2001;76:892–900. doi: 10.1046/j.1471-4159.2001.00087.x. [DOI] [PubMed] [Google Scholar]
- Ulbricht E, Pannicke T, Hollborn M, et al. Proliferative gliosis causes mislocation and inactivation of inwardly rectifying K(+) (Kir) channels in rabbit retinal glial cells. Exp Eye Res. 2008;86:305–313. doi: 10.1016/j.exer.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Ullrich N, Bordey A, Gillespie GY, Sontheimer H. Expression of voltage-activated chloride currents in acute slices of human gliomas. Neuroscience. 1998;83:1161–1173. doi: 10.1016/s0306-4522(97)00456-9. [DOI] [PubMed] [Google Scholar]
- Vit JP, Ohara PT, Bhargava A, Kelley K, Jasmin L. Silencing the Kir4.1 potassium channel subunit in satellite glial cells of the rat trigeminal ganglion results in pain-like behavior in the absence of nerve injury. J Neurosci. 2008;28:4161–4171. doi: 10.1523/JNEUROSCI.5053-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GF, Chiu SY. Potassium channel regulation in Schwann cells during early developmental myelinogenesis. J Neurosci. 1990;10:1615–1625. doi: 10.1523/JNEUROSCI.10-05-01615.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Verkman AS. Aquaporin-4 independent Kir4.1 K+ channel function in brain glial cells. Mol Cell Neurosci. 2008;37:1–10. doi: 10.1016/j.mcn.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]