

The membrane-targeting domains of the Rac-specific guanine nucleotide-exchange factors Tiam1 and Tiam2 were purified and crystallized.
Keywords: Rac, GTPases, guanine nucleotide-exchange factors, Tiam1, Tiam2
Abstract
T-lymphoma invasion and metastasis 1 and 2 (Tiam1 and Tiam2) are guanine nucleotide-exchange factors that specifically activate Rac GTPase by facilitating the dissociation of GDP. Translocation of Tiam1 and Tiam2 from the cytoplasm to the plasma membrane is an essential step in Rac activation and is mediated by the conserved PH-CC-Ex (pleckstrin-homology, coiled-coil and extra region) region in the N-terminal region. Here, the purification, crystallization and X-ray data collection of the Tiam1 and Tiam2 PH-CC-Ex regions are reported. The regions are shown to exist as a monomer in solution as a folded globular domain. The Tiam2 PH-CC-Ex domain crystallizes in space group P41212 or P43212 with four molecules in the asymmetric unit. An X-ray diffraction data set has been collected to 3.2 Å resolution.
1. Introduction
Rho-family small GTPases, including Rho, Rac and Cdc42, participate in the regulation of cell migration, cell adhesion and cytoskeletal rearrangement by regulating the activity of target proteins (Aelst & D’Souza-Schorey, 1997 ▶; Kaibuchi et al., 1999 ▶). They act as molecular switches that cycle between inactive GDP-bound and active GTP-bound states (Hall, 1990 ▶, 1998 ▶). The GDP/GTP cycle of Rho-family GTPases is controlled by three types of proteins (Takai et al., 1995 ▶; Hakoshima et al., 2003 ▶). Guanine nucleotide-exchange factors (GEFs) activate GTPases by simulating the GDP/GTP-exchange reaction, whereas GTPase-activating proteins (GAPs) and GDP-dissociation inhibitors (GDIs) inactivate GTPases. The balance of activity of these regulatory proteins plays an important role in Rho GTPase signalling, which is dependent on the activation state of the GTPases. Therefore, these regulatory proteins have been the targets of extensive structural studies (Hakoshima et al., 2003 ▶).
GEFs specific for Rho-family GTPases possess the Dbl homology–pleckstrin homology (DH-PH) tandem domain that is responsible for catalyzing the exchange of GDP for GTP (Rossman et al., 2005 ▶). Tiam1 (T-lymphoma invasion and metastasis 1) was identified as a GEF specific for Rac GTPases as determined by proviral tagging in combination with in vitro selection for invasiveness from mouse lymphoma cells (Habets et al., 1994 ▶). SIF (still life) is a Drosophila Tiam1 homologue that is located in synaptic terminals and plays a role in the morphology of axon terminals, which change with differentiation into mature synapses (Sone et al., 1997 ▶). A closely related Tiam1 homologue, Tiam2, also referred to as STEF (SIF and Tiam1-like exchange factor), was isolated by RT-PCR experiments using mRNA extracted from the mouse adult brain (Hoshino et al., 1999 ▶). In addition to the DH-PH tandem domain, these GEFs possess two PEST domains, an N-terminal PH domain and a coiled coil (CC) followed by an adjacent extra conserved sequence (Ex), a Ras-binding domain and a PDZ domain (Mertens et al., 2003 ▶). Translocation of Tiam1-family members to the plasma membrane is crucial for their capacity to induce Rac-mediated membrane ruffles and the activation of c-Jun-activated kinase (Michiels et al., 1997 ▶; Stam et al., 1997 ▶; Matsuo et al., 2002 ▶). Interestingly, deletion of either the N-terminal PH domain or the CC-Ex region abolished translocation to the plasma membrane and association with actin cytoskeletons. Thus, both the N-terminal PH domain and the CC-Ex region, but not the DH-PH tandem domain, were required for membrane translocation.
It has recently been shown that the PH domain followed by the CC-Ex region (the PH-CC-Ex region) mediates Tiam1 binding to integral membrane proteins that play key roles in cell adhesion such as CD44 (Bourguignon et al., 2000 ▶) and ephrin B1 (Tanaka et al., 2004 ▶), the former being a hyaluronic acid (HA) receptor and the latter being a ligand of the Eph receptor that mediates neurite outgrowth. Furthermore, the PH-CC-Ex region interacts directly with scaffolding proteins such as JIP2 (JNK-interaction protein 2), spinophilin and the Par3–Par6–aPKC complex, thus determining the downstream specificity of Rac signalling and regulating neuronal polarity (Buchsbaum et al., 2002 ▶, 2003 ▶; Nishimura et al., 2005 ▶; Chen & Macara, 2005 ▶). However, the precise mechanism by which the PH-CC-Ex region recognizes these target proteins is unknown. Here, we report the purification, crystallization and preliminary X-ray crystallographic characterization of the Tiam1 and Tiam2 PH-CC-Ex regions. These two regions exhibit about 65% sequence identity.
2. Materials and methods
2.1. Protein expression
The nucleotide sequences encoding the PH-CC-Ex regions of mouse Tiam1 (residues 429–702) and Tiam2 (residues 500–757) were amplified by PCR with Tiam1 (gene ID U05245) and Tiam2 (gene ID AB022915) cDNA. Using the BamHI and XhoI restriction-enzyme sites, the coding nucleotides were subcloned into pGEX6P-3 plasmid containing an N-terminal glutathione S-transferase (GST) tag linked by a HRV3C protease site. The constructs produce an additional five residues (GPLGS) at the N-terminus after protease cleavage of the tag. The GST-fusion proteins were expressed in Escherichia coli BL21-CodonPlus-RIL cells.
50 ml of an overnight culture of BL21-CodonPlus-RIL cells transformed with pGEX6P-3-PH-CC-Ex was inoculated into 2 l Super Broth (SB) medium containing 50 µg ml−1 ampicillin. The culture was grown at 310 K until the OD660nm reached 0.6. The expression of recombinant protein was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) to the culture to a final concentration of 1 mM. Recombinant protein was expressed at 298 K for 6 h and cells were then harvested by centrifugation at 5000g for 15 min at 277 K.
The Tiam2 PH-CC-Ex region contains five methionine residues. For multi-wavelength anomalous dispersion (MAD) experiments, selenomethionine-substituted protein was expressed using a technique based on the inhibition of methionine biosynthesis (Van Duyne et al., 1993 ▶; Doublié, 1997 ▶). Cells were grown to mid-log phase prior to the addition of the amino acids required for the inhibition of methionine biosynthesis. Induction using 1.0 mM IPTG was initiated 30 min following the addition of amino acids. Cultures were grown for an additional 12 h following IPTG induction.
2.2. Purification
Cells were disrupted by sonication at 277 K. The supernatant was applied onto a GST affinity column containing glutathione Sepharose 4B (GE Healthcare) and the column was washed with 50 mM Na HEPES buffer pH 7.5 containing 0.3 M NaCl and 1 mM dithiothreitol (DTT). The fusion protein was eluted with 50 mM Na HEPES pH 7.5 buffer containing 0.5 M NaCl, 30 mM glutathione and 1 mM DTT. The fusion protein was cleaved overnight at 277 K using 2 units ml−1 GST-fused HRV3C protease. The cleaved protein was purified by HiTrap SP Sepharose column (GE Healthcare) chromatography using a NaCl concentration gradient (0.2–1.0 M). Fractions containing the PH-CC-Ex region were collected and concentrated. Finally, the PH-CC-Ex region was purified by gel-filtration chromatography on a Superdex75 column (GE Healthcare) equilibrated and eluted with 10 mM Na HEPES buffer pH 7.5 containing 0.3 M NaCl and 1 mM DTT. Similarly, the purified sample was analyzed by gel-filtration chromatography with standard proteins, ribonuclease (13.7 kDa), chymotrypsinogen (25 kDa), ovalbumin (43 kDa) and albumin (67 kDa), to determine the apparent molecular weight in solution.
Protein purity was monitored by 12.5% polyacrylamide gel electrophoresis and subsequent staining with Simply Stain Blue (Invitrogen). Purified samples were verified using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI–TOF MS; JMS ELITE, PerSeptiv Inc) and N-terminal sequence analysis (M492, Applied Biosystems). The N-terminal sequence analysis confirmed the presence of five artificial residues, GPLGS, which remained at the N-terminus of the resultant protein.
2.3. Crystallization and X-ray data collection
For crystallization, the purified PH-CC-Ex region of Tiam2 was concentrated to 30 mg ml−1 in a solution containing 10 mM Na HEPES pH 7.5, 0.3 M NaCl and 1 mM DTT. Initial crystallization conditions were screened by employing the sitting-drop vapour-diffusion method using a Hydra II Plus One crystallization robot (Matrix Technology) with a commercial crystallization-solution kit at 293 K. Crystallization solutions were prepared by mixing 0.2 µl protein solution with 0.2 µl of each reservoir solution. The conditions obtained from the screening were optimized using the hanging-drop vapour-diffusion method at 293 K. The solutions used for the optimized crystallization conditions were prepared by mixing 1.0 µl protein solution with 1.0 µl of each reservoir solution. The crystals obtained were transferred stepwise into a cryoprotective solution containing 30%(v/v) glycerol and flash-cooled at 100 K. Initial diffraction tests were performed using a home-source X-ray generator (Rigaku FR-E) equipped with a Rigaku R-AXIS VII detector at 100 K.
X-ray diffraction data from native crystals of the Tiam1 and Tiam2 PH-CC-Ex regions were collected using an ADSC Quantum 315 CCD detector installed on the BL41XU beamline at SPring-8. Data collection for the native crystal of the Tiam1 PH-CC-Ex region was performed using an angular range of 110°, a step size of 2.0° and an exposure time of 5 s. The camera was fixed at a distance of 250 mm. Data collection for the native crystal of the Tiam2 PH-CC-Ex region was performed using an angular range of 180°, a step size of 1.0° and an exposure time of 1 s. The camera was fixed at a distance of 400 mm. All data were processed and scaled with HKL-2000 (Otwinowski & Minor, 1997 ▶). The set data used for the MAD method was collected on the same beamline at SPring-8.
3. Results and discussion
Gel-filtration chromatography revealed that the purified proteins existed as a monomer in solution with an apparent molecular weight of 31 kDa (the calculated molecular weight was 29 964 Da for Tiam2). This PH-CC-Ex region that exists as a globular monomeric form in solution is referred here to as the PHCCEx domain. Needle-like crystals of the Tiam1 PHCCEx domain were obtained from a solution containing 5 mg ml−1 protein, 33 mM sodium citrate pH 6.0, 1.3%(v/v) Jeffamine M-600, 3.3 mM FeCl3 and 0.5%(v/v) glycerol equilibrated against 100 mM sodium citrate pH 6.0, 4%(v/v) Jeffamine M-600, 10 mM FeCl3 and 1.5%(v/v) glycerol at 293 K. These poorly diffracting crystals belonged to space group P6222 or P6422, with unit-cell parameters a = b = 113.5, c = 113.8 Å. An X-ray data set was collected to a resolution of 4.5 Å (Table 1 ▶); however, efforts to improve the diffraction quality of this Tiam1 PHCCEx domain crystal form by further additive screening and/or redesigning the protein constructs were unsuccessful.
Table 1. X-ray diffraction data of the Tiam1 PHCCEx domain crystal.
Values in parentheses are for the outer resolution shell.
| Space group | P6222 or P6422 |
| Unit-cell parameters (Å, °) | a = b = 113.5, c = 113.8, α = β = 90, γ = 120 |
| Wavelength (Å) | 1.0000 |
| Resolution (Å) | 50–4.5 |
| Reflections measured | 30832 |
| Unique reflections | 2824 |
| Redundancy | 10.9 (8.3) |
| Completeness (%) | 98.7 (96.7) |
| Mean I/σ(I) | 13.8 (2.5) |
| Rmerge† (%) | 15.0 (68.2) |
R
merge = 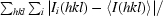
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
The best crystals of the Tiam2 PHCCEx domain were obtained from a solution containing 15 mg ml−1 protein, 50 mM bis-tris pH 6.5, 5%(w/v) polyethylene glycol 3350 (PEG 3350), 100 mM Li2SO4, 150 mM NaCl, 0.5 mM DTT and 5 mM urea equilibrated against 10%(w/v) PEG 3350, 100 mM bis-tris pH 6.5, 200 mM Li2SO4 and 10 mM urea at 293 K. The crystals diffracted to a resolution of 3.0 Å and belonged to space group P41212 or P43212, with unit-cell parameters a = b = 105.6, c = 287.6 Å. A complete data set was collected to a resolution of 3.2 Å using a single crystal (Table 2 ▶). The crystal mosaicity was in the range 0.42–0.53°. MALDI–TOF MS of the Tiam2 sample resulted in a single peak at 29 896 Da (the calculated molecular weight was 29 964 Da). A Matthews coefficient (Matthews, 1968 ▶) of 3.32 Å3 Da−1 was calculated assuming the presence of four PHCCEx domains with an approximate molecular weight of 30 kDa in the asymmetric unit, which corresponds to 62.9% solvent content by volume. Self-rotation functions (Rossmann & Blow, 1962 ▶) were calculated in an effort to search for noncrystallographic symmetry using several Patterson sphere radii, but no significant peaks were found on the maps.
Table 2. X-ray diffraction data of the Tiam2 PHCCEx domain crystal.
Values in parentheses are for the outer resolution shell.
| SeMet Tiam2 | |||
|---|---|---|---|
| Tiam2 | Peak | Edge | |
| Space group | P41212 or P43212 | ||
| Unit-cell parameters (Å) | a = b = 105.6, c = 287.6 | a = b = 105.4, c = 289.7 | a = b = 105.8, c = 288.5 |
| Wavelength (Å) | 1.0000 | 0.9789 | 0.9792 |
| Resolution (Å) | 50–3.2 | 50–3.4 | 50–3.4 |
| Reflections measured | 336843 | 540236 | 503703 |
| Unique reflections | 27986 | 21847 | 21984 |
| Redundancy | 13.1 (9.5) | 24.7 (16.1) | 22.9 (13.7) |
| Completeness (%) | 91.7 (52.1) | 93.6 (61.4) | 94.3 (64.8) |
| Mean I/σ(I) | 38.8 (2.0) | 41.3 (2.9) | 34.6 (2.0) |
| Rmerge† (%) | 7.0 (66.1) | 10.5 (51.3) | 10.7 (55.6) |
R
merge =
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
To facilitate structure determination of the PHCCEx domain of Tiam2 using the MAD method, selenomethionine-substituted PHCCEx domain was prepared and crystallized. SeMet incorporation by metabolic inhibition was successful and a sufficient sample for crystallization was obtained. Validation of the success of the substitution of Met by SeMet was obtained using MALDI–TOF MS, which gave a peak at 30 198 Da (the calculated molecular weight was 30 129 Da). Crystals of the SeMet-substituted PHCCEx domain (Fig. 1 ▶) were obtained using conditions similar to those used for the native crystals and consisting of 15 mg ml−1 protein solution containing 50 mM bis-tris pH 6.1, 3%(w/v) PEG 3350, 100 mM Li2SO4, 150 mM NaCl and 0.5 mM DTT equilibrated against 6%(w/v) PEG 3350, 100 mM bis-tris pH 6.1 and 200 mM Li2SO4 at 293 K. X-ray fluorescence was used to detect and confirm the presence of Se atoms in these crystals in addition to determining the peak and inflection points of the Se K edge for MAD data collection. Table 2 ▶ summarizes the detailed statistics of the MAD data set.
Figure 1.

Crystals of SeMet-derivative Tiam2 PHCCEx domain. The scale bar indicates 0.1 mm.
Automated phasing of the MAD X-ray data using the autoSHARP program (Vonrhein et al., 2007 ▶) allowed the identification of 19 Se atoms in the asymmetric unit. These Se-atom positions were used to obtain initial phases with a figure of merit (FOM) of 0.358 at 3.2 Å resolution. Further density modification using the SOLOMON program (Abrahams & Leslie, 1996 ▶) resulted in phases with an FOM of 0.713 at 3.2 Å. The initial electron-density maps confirmed the presence of four molecules, each consisting of two structural regions (canonical PH domains and additional peptide folds), in the asymmetric unit. Further manual building of the model is in progress.
Acknowledgments
We would like to thank J. Tsukamoto for technical support in performing the MALDI–TOF MS analysis. We gratefully acknowledge K. Kaibuchi for providing mouse STEF cDNA. This work was supported by a Grant-in-Aid for Scientific Research (A) and a Grant-in-Aid for Scientific Research on Priority Areas, Cancer, Macromolecular Assembly, Membrane Interface and Nano-systems in Cells from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. The early stage of the work was supported by Core Research for Evolutional Science and Technology (CREST) from the Japan Science and Technology Corporation (JST). ST was supported by a postdoctoral research fellowship from a Grant-in-Aid for the 21st Century COE Research from MEXT. We acknowledge Drs N. Shimizu, M. Kawamoto and M. Yamamoto at SPring-8 for help with data collection at synchrotron beamline BL41XU.
References
- Abrahams, J. P. & Leslie, A. G. W. (1996). Acta Cryst. D52, 30–42. [DOI] [PubMed]
- Aelst, L. V. & D’Souza-Schorey, C. (1997). Genes Dev.11, 2295–2322. [DOI] [PubMed]
- Bourguignon, L. Y., Zhu, H., Shao, L. & Chen, Y. W. (2000). J. Biol. Chem.275, 1829–1838. [DOI] [PubMed]
- Buchsbaum, R. J., Connolly, B. A. & Feig, L. A. (2002). Mol. Cell. Biol.22, 4073–4085. [DOI] [PMC free article] [PubMed]
- Buchsbaum, R. J., Connolly, B. A. & Feig, L. A. (2003). J. Biol. Chem.278, 18833–18841. [DOI] [PubMed]
- Chen, X. & Macara, I. G. (2005). Nature Cell Biol.7, 262–269. [DOI] [PubMed]
- Doublié, S. (1997). Methods Enzymol.276, 523–530. [PubMed]
- Habets, G. G., Scholtes, E. H., Zuydgeest, D., van der Kammen, R. A., Stam, J. C., Berns, A. & Collard, J. G. (1994). Cell, 77, 537–549. [DOI] [PubMed]
- Hakoshima, T., Maesaki, R. & Shimizu, T. (2003). J. Biochem. (Tokyo), 134, 327–331. [DOI] [PubMed]
- Hall, A. (1990). Science, 249, 635–640. [DOI] [PubMed]
- Hall, A. (1998). Science, 279, 509–514. [DOI] [PubMed]
- Hoshino, M., Sone, M., Fukata, M., Kuroda, S., Kaibuchi, K., Nabeshima, Y. & Hama, C. (1999). J. Biol. Chem.274, 17837–17844. [DOI] [PubMed]
- Kaibuchi, K., Kuroda, S. & Amano, M. (1999). Annu. Rev. Biochem.68, 459–486. [DOI] [PubMed]
- Matsuo, N., Hoshino, M., Yoshizawa, M. & Nabeshima, Y. (2002). J. Biol. Chem.277, 2860–2868. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Mertens, A. E., Roovers, R. C. & Collard, J. G. (2003). FEBS Lett.546, 11–16. [DOI] [PubMed]
- Michiels, F., Stam, J. C., Hordijk, P. L., van der Kammen, R. A., Stalle, L. R., Feltkamp, C. A. & Collard, J. G. (1997). J. Cell Biol.137, 387–398. [DOI] [PMC free article] [PubMed]
- Nishimura, T., Yamaguchi, T., Kato, K., Yoshizawa, M., Nabeshima, Y., Ohno, S., Hoshino, M. & Kaibuchi, K. (2005). Nature Cell Biol.7, 270–277. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol.276, 307–326. [DOI] [PubMed]
- Rossman, K. L., Der, C. J. & Sondek, J. (2005). Nature Rev. Mol. Cell Biol.6, 167–180. [DOI] [PubMed]
- Rossmann, M. G. & Blow, D. M. (1962). Acta Cryst.15, 24–31.
- Sone, M., Hoshino, M., Suzuki, E., Kuroda, S., Kaibuchi, K., Nakagoshi, H., Saigo, K., Nabeshima, Y. & Hama, C. (1997). Science, 275, 543–547. [DOI] [PubMed]
- Stam, J. C., Sander, E. E., Michiels, F., van Leeuwen, F. N., Kain, H. E., van der Kammen, R. A. & Collard, J. G. (1997). J. Biol. Chem.272, 28447–28454. [DOI] [PubMed]
- Takai, Y., Sasaki, T., Tanaka, K. & Nakanishi, H. (1995). Trends Biochem. Sci.20, 227–231. [DOI] [PubMed]
- Tanaka, M., Ohashi, R., Nakamura, R., Shinmura, K., Kamo, T., Sakai, R. & Sugimura, H. (2004). EMBO J.23, 1075–1088. [DOI] [PMC free article] [PubMed]
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol.229, 105–124. [DOI] [PubMed]
- Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. (2007). Methods Mol. Biol.364, 215–230. [DOI] [PubMed]