

Abstract
C-terminal-truncated (ΔC) microdystrophin is being developed for Duchenne muscular dystrophy gene therapy. Encouraging results have been achieved in the mdx mouse model. Unfortunately, mdx mice do not display the same phenotype as human patients. Evaluating ΔC microdystrophin in a symptomatic model will be of significant relevance to human trials. Utrophin/dystrophin double-knockout (u-dko) mice were developed to model severe dystrophic changes in human patients. In this study we evaluated the therapeutic effect of the ΔR4-R23/ΔC microdystrophin gene (ΔR4/ΔC) after serotype-6 adeno-associated virus-mediated gene transfer in neonatal u-dko muscle. At 2 months after gene transfer, the percentage of centrally nucleated myofiber was reduced from 89.2 to 3.4% and muscle weight was normalized. Furthermore, we have demonstrated for the first time that ΔC microdystrophin can eliminate interstitial fibrosis and macrophage infiltration and restore dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex. Interestingly neuronal nitric oxide synthase was not restored. The most impressive results were achieved in muscle force measurement. Neonatal gene therapy increased twitch- and tetanic-specific force. It also brought the response to eccentric contraction-induced injury to the normal range. In summary, our results suggest that the ΔR4/ΔC microgene holds great promise in preventing muscular dystrophy.
Keywords: Duchenne muscular dystrophy, adeno-associated virus, microdystrophin, muscle contraction, utrophin/dystrophin double-knockout mice
Introduction
The molecular basis of Duchenne and Becker muscular dystrophies (DMD, BMD) is the dystrophin gene mutation [1]. In DMD patients, mutations in the dystrophin gene completely abolish protein expression and result in severe muscle pathology. Eventually, patients die from respiratory and/or cardiac failure before age 30 [1]. BMD patients display a much milder clinical profile. Some BMD patients are ambulant beyond age 60 [2]. The benign manifestation of BMD is due mainly to residual expression of a truncated but partially functional dystrophin protein [3]. This suggests that gene therapy with the mini- or microdystrophin gene may convert deadly DMD to mild BMD and improve life quality.
The 427-kDa full-length dystrophin protein has four distinctive functional domains including the N-terminal, the rod, the cysteine-rich (CR), and the C-terminal domains. The functional significance of these domains has been elucidated by a series of carefully designed transgenic studies (reviewed in [4]). The domains most critical to DMD pathology are those participating directly in the mechanical linkage between the extracellular matrix and the cytoskeleton, such as the CR domain and the N-terminal domain. Recombinant dystrophins lacking the majority of the rod domain and/or the C-terminal domain are relatively functional in transgenic mdx mice. Based on these results, the C-terminal-deleted microdystrophin genes were developed recently (reviewed in [4]). Among these microgenes, a 3.8-kb ΔR4/ΔC microgene is especially promising. Despite deletion of the majority of the rod domain (R4-R23) and the entire C-terminus, adeno-associated virus (AAV)-mediated expression of the ΔR4/ΔC microgene has resulted in encouraging improvement in skeletal muscle and the heart in mdx mice [5–9].
The mdx mice share the same genetic defect as DMD patients [10]. However, they display an extremely mild clinical phenotype. Numerous therapeutic studies have been performed in mdx mice. However, given the benign phenotype in mdx mice, it is conceivable that a more reliable therapeutic predication should have been achieved with disease models that recapitulate the clinical picture seen in human patients.
Utrophin is an autosomal paralogue of dystrophin. It is ubiquitously expressed in a wide range of tissues and is especially enriched at the neuromuscular and myotendinous junctions in adult skeletal muscle (reviewed in [11]). In adult mdx mice, utrophin is up-regulated and it can be detected along the entire myofiber. This up-regulation has been thought to be at least partially responsible for the mild phenotype in mdx mice. In support of this notion, mice deficient in both dystrophin and utrophin (u-dko mice) are weak and small at the time of weaning [12,13]. They present rapidly aggressive muscle wasting and many other classic DMD phenotypes such as growth retardation, weight loss, scoliosis, contractures, and heart diseases. Most u-dko mice die prematurely between 8 and 10 weeks. Since the catastrophic progression in u-dko mice closely reproduces the clinical manifestation of DMD patients, these mice are considered an ideal model to predict more accurately the outcome of experimental gene therapy in human patients.
In this study, we evaluated the therapeutic efficacy of the ΔR4/ΔC microgene in the extensor digitorum longus (EDL) muscle of newborn u-dko mice. Consistent with previous findings in mdx mice, AAV-mediated ΔR4/ΔC microdystrophin also reduced the pathological degeneration/regeneration process and prevented myofiber necrosis in u-dko mice [5,8,9]. We also observed a dramatic reduction of interstitial fibrosis and macrophage infiltration. Furthermore, we demonstrated restoration of dystrobrevin and syntrophin, but not neuronal nitric oxide synthase (nNOS) in the sarcolemma. AAV-mediated ΔR4/ΔC expression has been shown to protect adult mdx muscle from contraction-induced injury, but muscle specific force was not improved [8,9]. Interestingly, neonatal AV.ΔR4/ΔC transduction increased muscle specific force. Taken together, our results have, for the first time, demonstrated therapeutic potential of a microdystrophin gene in a severe animal model for DMD.
Results
AV.ΔR4/ΔC Expression Restores the Entire Dystrophin-Associated Glycoprotein Complex (DGC), but Not nNOS
The u-dko model was developed in 1997 by two independent research groups [12,13]. In this study we used the mouse line reported by Grady et al. [13]. The u-dko mice have been considered an ideal preclinical model system to test gene therapy strategies for DMD [12,13]. The mdx mice are in the C57BL/10 (BL10) background [10]. However, the originally reported u-dko mice are not in a pure genetic background. To facilitate functional comparison with BL10 mice, we performed an additional three rounds of backcrossing with mdx mice. The experimental male u-dko mice were obtained from crossing the N3 generation of heterozygous mice (mdx/utr+/−). Backcrossing to a purer genetic background did not alter the severe phenotype of muscular dystrophy in u-dko mice (data not shown).
To target the EDL muscle in newborn mice, we delivered AAV-6 AV.ΔR4/ΔC to the entire anterior muscle compartment in the hind limb of 3-day-old u-dko pups. At 9 weeks after gene transfer, we harvested the EDL muscle for analysis. Despite a blind gene delivery approach, efficient transduction was achieved in the EDL muscle (76.62 ± 6.20%; range, 59.48–92.31%) (Figs. 1A and 3A). Furthermore, ΔR4/ΔC microdystrophin seemed to be enriched at the neuromuscular junction (Fig. 1B). We confirmed microgene expression further by western blot (Supplementary Fig. 1). Efficient long-term gene transfer has been obtained with AAV-6 in mdx muscle [7,19]. To determine whether AAV-6 infection in u-dko muscle can elicit immune response, we examined neonatally infected u-dko muscle for CD4+ T cells, CD8+ T cells, and B cells. Disease-associated CD4+ and CD8+ T cells, as well as a small number of B cells, were evident in 2-month-old untreated u-dko muscle, but were reduced or eliminated in AV.ΔR4/ΔC-infected muscle (Supplementary Figs. 2–4).
FIG. 1.
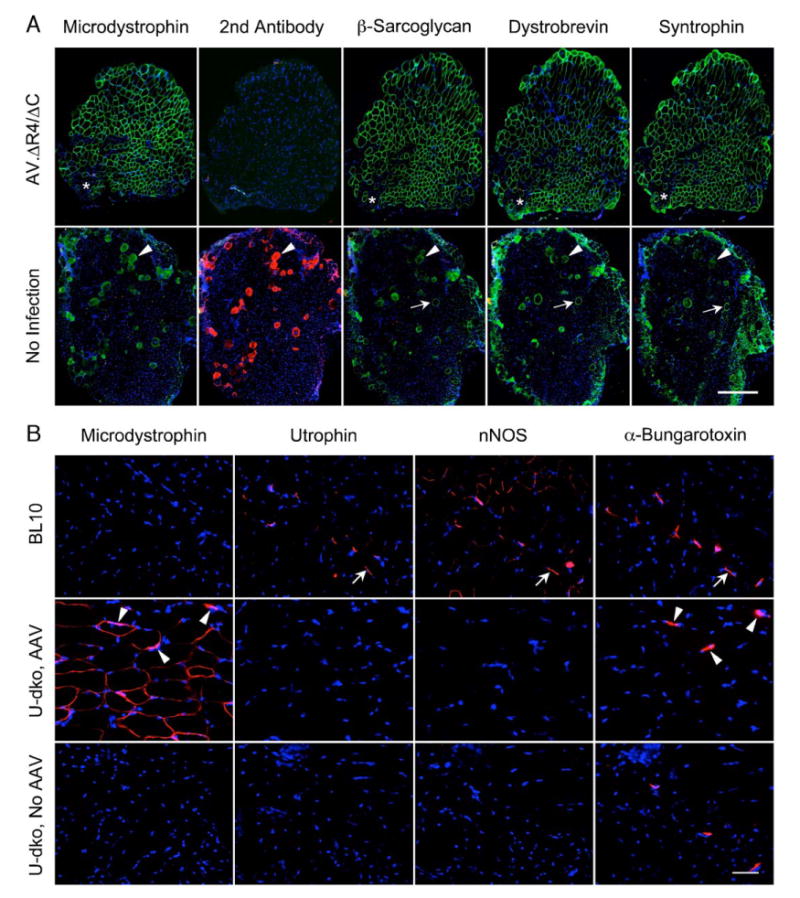
C-terminal-truncated microdystrophin restores the DGC, but not nNOS, in the sarcolemma of u-dko mice. 6 × 1010 vg particles of AAV-6 AV.ΔR4/ΔC were delivered to the anterior compartment of the left hind limb in 3-day-old neonatal u-dko mice. The contralateral limb was injected with an equal volume of saline buffer. The DGC components were examined by immunostaining at 9 weeks after gene transfer. (A) Representative photomicrographs of u-dko muscle serial sections immunostained with monoclonal antibodies for the dystrophin N-terminal domain, β-sarcoglycan, dystrobrevin, and syntrophin, respectively. Secondary antibody alone (exactly the same antibody as used for other immunostainings except it is conjugated to Alexa 594) is included as a negative control for nonspecific staining. Secondary antibody alone also reveals immunoglobulin uptake in injured myofibers. Nuclei are revealed by DAPI staining. Asterisk, a region not transduced by AAV; arrow, a revertant myofiber; arrowhead, an injured myofiber with sarcolemma leakage. Scale bar, 300 μm. (B) Representative photomicrographs of serial sections immunostained with monoclonal antibodies for the dystrophin N-terminal domain (specific for AV.ΔR4/ΔC) and utrophin and with polyclonal antibody for nNOS. Neuromuscular junctions are revealed with Alexa 594-conjugated α-bungarotoxin. Nuclei are revealed by DAPI staining. Arrow, expression of utrophin and nNOS at the neuromuscular junctions in BL10 muscle; arrowhead, microdystrophin expression is enhanced at the neuromuscular junctions in AV.ΔR4/ΔC-infected u-dko muscle. Scale bar, 50 μm.
FIG. 3.
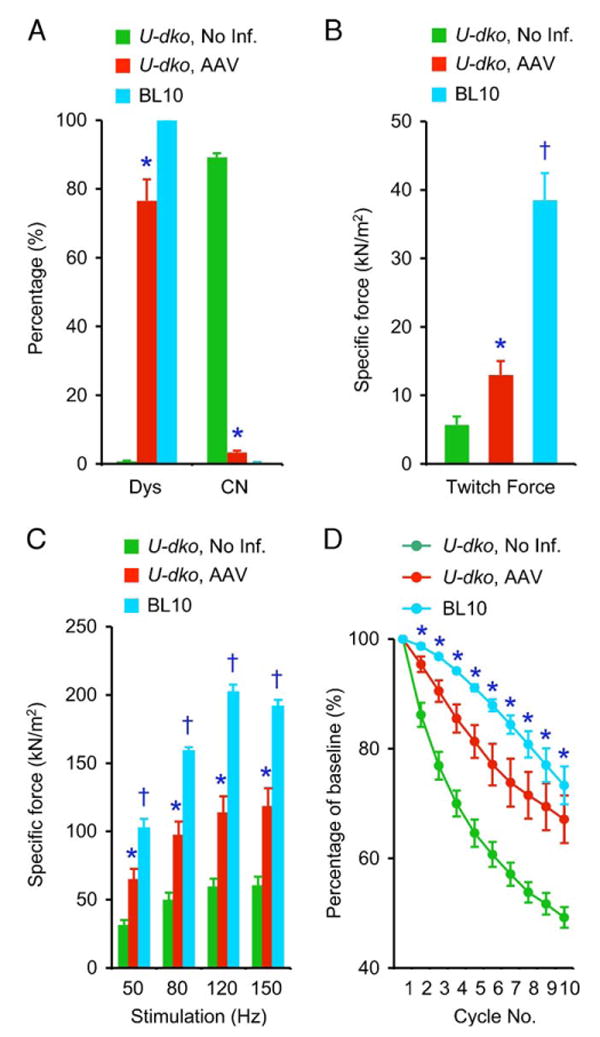
Neonatal AV.ΔR4/ΔC treatment reduces muscle degeneration and improves muscle force in u-dko mice. The analyses were performed in the EDL muscle at 9 weeks after neonatal AV.ΔR4/ΔC infection in u-dko mice (red). Untreated contralateral muscle served as negative control (green). Results from age- and sex-matched BL10 mice are also included (blue). (A) Quantitative analysis of transduction efficiency and central nucleation. Dys, percentage of dystrophin-positive myofiber. In uninfected u-dko and BL10 mice (N = 3 each), the number of dystrophin-positive cells was determined with an anti-dystrophin C-terminus-specific monoclonal antibody. The dystrophin-positive myofibers seen in uninfected u-dko muscle are revertant myofibers. In AV.ΔR4/ΔC-infected muscle (N = 5), the number of dystrophin-positive cells was determined with an anti-human dystrophin N-terminus-specific monoclonal antibody. CN, percentage of centrally nucleated myofibers. In uninfected u-dko muscle (N = 4) and BL10 muscle (N = 3), quantification was performed on all myofibers in a cryosection. In AV.ΔR4/ΔC-infected muscle (N = 7), quantification was performed only in microdystrophin-positive myofibers. *The value in AV.ΔR4/ΔC-infected u-dko muscle is significantly different from that in uninfected u-dko muscle or in BL10 muscle. (B) Specific twitch force in uninfected u-dko muscle (N = 7), AAV-infected u-dko muscle (N = 7), and BL10 muscle (N = 5). *The result in AV.ΔR4/ΔC-treated muscle is significantly higher than that of untreated muscle. †The result in BL10 muscle is significantly different from that of u-dko muscle (both infected and uninfected). (C) The force–frequency relationship in uninfected u-dko muscle (N = 7), AAV-infected u-dko muscle (N = 7), and BL10 muscle (N = 5). *The results in AV.ΔR4/ΔC-treated muscle are significantly higher than those in untreated muscle. †The results in BL10 muscle are significantly higher than those in u-dko muscle (both infected and uninfected). (D) Relative force decline following eccentric contraction-induced injury in uninfected u-dko muscle (N = 7), AAV-infected u-dko muscle (N = 7), and BL10 muscle (N = 5). *The results from AAV-infected u-dko muscle and BL10-muscle are significantly different from those of uninfected u-dko muscle. However, there is no significant difference between AAV-infected u-dko muscle and BL10 muscle. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
We next examined whether ΔR4/ΔC microdystrophin could recruit other partners in the DGC. We immunostained serial sections from AAV-infected and uninfected muscles with monoclonal antibodies specific for representative components of the dystroglycan, sarcoglycan, and cytoplasmic subcomplexes of the DGC. Previous studies suggest that C-terminal-truncated microdystrophin can restore dystroglycan and sarcoglycan subcomplexes in mdx muscle [6,14]. We achieved similar results in u-dko muscle (Fig. 1A, data not shown for the dystroglycan subcomplex).
The cytoplasmic subcomplex of the DGC includes dystrobrevin and syntrophin. In the context of a full-length dystrophin protein, the absence of the C-terminal domain seems to have no detrimental effect on the cytoplasmic subcomplex [15]. In the context of microdystrophin, only one study has examined the cytoplasmic subcomplex [16]. Using a transgenic mdx mouse model, Harper et al. revealed the restoration of dystrobrevin and syntrophin in mdx muscle by a C-terminal-inclusive microdystrophin [16]. It is not clear whether a C-terminal-truncated microdystrophin can achieve the same effect. To address this issue, we examined dystrobrevin and syntrophin (Fig. 1A) expression in AV.ΔR4/ΔC-infected u-dko muscle. Interestingly, both dystrobrevin and syntrophin were detected in microdystrophin- positive myofibers. The PDZ domain of syntrophin has been shown to interact with nNOS [17,18]. We next examined nNOS expression. In normal muscle, nNOS can be detected in the sarcolemma, especially at the neuromuscular junction (Fig. 1B). In u-dko muscle, sarcolemmal nNOS expression was lost. Surprisingly, syntrophin expression in AV.ΔR4/ΔC-transduced myofiber failed to restore sarcolemmal nNOS expression (Fig. 1).
C-Terminal-Truncated Microdystrophin Ameliorates Dystrophic Pathology in u-dko Mice
Overall growth retardation and severe muscle pathology are striking clinical features seen in both DMD patients and u-dko mice. As expected, local gene therapy in one limb muscle did not halt disease progression. These mice were significantly smaller than age-matched BL10 mice (Table 1). However, muscle mass and cross-sectional area were restored to the normal range in AV.ΔR4/ΔC treated muscle (Table 1).
TABLE 1.
Characteristics of mice and the EDL muscle
| EDL characteristics at harvest | ||||||||
|---|---|---|---|---|---|---|---|---|
| Strain | AV.ΔR4/ΔC | N | Age at infection (days) | Age at harvest (days) | Body wt at harvest (g) | Wt (mg) | Lo(mm) | CSA (mm2) |
| BL10 | No | 5 | N/A | 67.2 ± 1.3 | 25.1 ± 0.3* | 8.10 ± 0.20 | 10.74 ± 0.22 | 1.62 ± 0.02 |
| u-dko | No | 7 | N/A | 67.1 ± 2.1 | 19.2 ± 0.9 | 10.11 ± 0.26* | 9.07 ± 0.10 | 2.39 ± 0.07* |
| u-dko | Yes | 7 | 3 | 67.1 ± 2.1 | 19.2 ± 0.9 | 7.33 ± 0.74 | 9.11 ± 0.13 | 1.74 ± 0.19 |
Significantly different from other groups in the same category (P < 0.05).
Muscles from both mdx and u-dko mice are characterized by increased sarcolemmal permeability to macromolecules such as Evans blue dye and immunoglobulin [12,13,19]. Similar to what has been shown in mdx muscle [6,14], C-terminal-truncated microdystrophin enhanced sarcolemma integrity and reduced immunoglobulin leakage (Fig. 1A).
On histology examination, u-dko muscle was dominated by centrally nucleated regenerative myofibers (Figs. 2 and 3A). Clusters of small-size, basophilic early regenerating myofibers were also seen frequently, indicating an active regenerating process (Fig. 2). Furthermore, there were extensive interstitial fibrosis and macrophage infiltration (Fig. 2). All these pathological changes were effectively arrested by ΔR4/ΔC microdystrophin (Figs. 2 and 3A). In particular, the percentage of centrally nucleated fiber dropped from 89.2% in untreated muscle to 3.4% in AV.ΔR4/ΔC-positive myofibers (Fig. 3A). Impressively, interstitial fibrosis and macrophage infiltration were completely eliminated in regions transduced by AV.ΔR4/ΔC (Fig. 2).
FIG. 2.
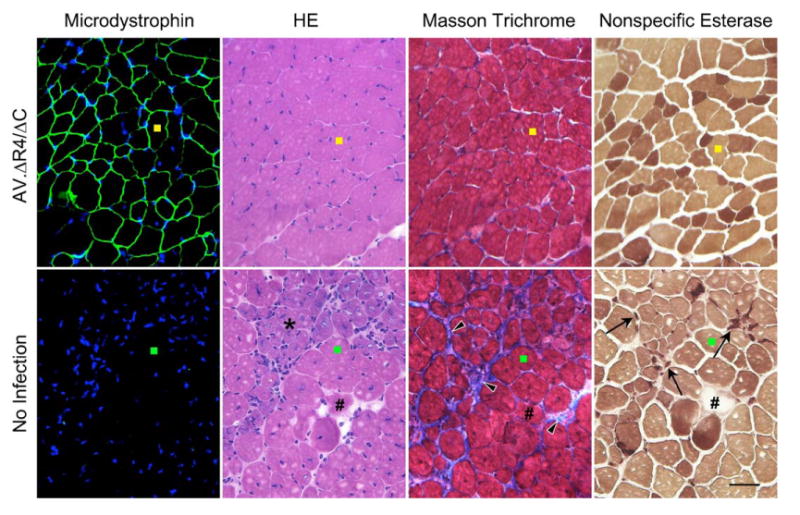
AAV-mediated microdystrophin expression reduces muscle pathology in u-dko mice. C-terminal-truncated microdystrophin was delivered to neonatal u-dko muscle by AAV-6 vector. Representative photomicrographs of serial sections in AAV-infected muscle (top) and uninfected contralateral muscle (bottom) show transgene expression and muscle pathology at 9 weeks later. Colored square marks (yellow in AAV-infected muscle; green in uninfected muscle) identify the same myofiber in serial sections. Microdystrophin expression was revealed by immunostaining with a monoclonal antibody specific for the human dystrophin N-terminal domain. DAPI staining reveals nuclei in immunostaining photomicrographs. HE staining illustrates general muscle morphology and the presence of centrally nucleated myofibers. Masson trichrome staining reveals interstitial fibrosis. Nonspecific esterase staining identifies macrophages. Asterisk, early regenerating myofibers; pound, a necrotic myofiber; arrowhead, extensive fibrosis in uninfected muscle; arrow, macrophage infiltration in uninfected muscle. Scale bar, 50 μm.
ΔR4/ΔC Microdystrophin Improves Contractile Performance in u-dko Muscle
To evaluate thoroughly the therapeutic effect on phenotypic u-dko mice, we measured muscle force (Fig. 3). Compared with the contralateral uninfected muscle, specific twitch and tetanic forces were doubled in AV.ΔR4/ΔC-treated muscle. Although this represented a significant improvement, it did not reach the BL10 level (Figs. 3B and 3C).
We next determined the relative force production after applying eccentric contraction-induced injury. In untreated u-dko muscle, we observed a precipitous tetanic force drop following eccentric contraction. The damage is especially remarkable in the first three rounds of eccentric contraction. In contrast to u-dko muscle, force production in BL10 muscle was better preserved (Fig. 3D). In a paired t test, the AV.ΔR4/ΔC-treated muscle was significantly better than the untreated muscle. In Fig. 3D, the average percentage of force drop in AV.ΔR4/ΔC-treated muscle fell in the range between that of untreated u-dko muscle and BL10 muscle in each eccentric contraction cycle. Encouragingly, a Bonferroni post hoc multiple comparison analysis revealed no statistically significant difference between AV.ΔR4/ΔC-treated muscle and BL10 muscle. Our results provide the first evidence that application of C-terminal-truncated microdystrophin prior to disease onset may significantly strengthen muscle force in a severe DMD animal model.
Discussion
AAV has been considered one of the most promising vectors to treat inherited muscle diseases such as DMD (reviewed in [4,20]). Unfortunately, AAV has a very small packaging capacity. This size limitation has excluded the possibility of delivering a fully functional gene (such as the 11-kb full-length dystrophin cDNA and the 6-kb minidystrophin gene) in a single AAV virion. To overcome this hurdle, a series of C-terminal-truncated microdystrophin genes were developed (reviewed in [4]). These microgenes are about 3.8 to 4.1 kb. The therapeutic expression cassettes carrying these microgenes can be efficiently packaged in a single AAV virion. In this study, we focused on the 3.8-kb ΔR4/ΔC microgene. This microgene has been demonstrated to ameliorate muscle pathology in mdx mice, a mild mouse model for DMD [5–9].
The ultimate goal of DMD gene therapy is to treat patients who are suffering from severe muscle disease. So far, none of the published microgene studies has evaluated gene therapy effect in a symptomatic model. In an attempt to address this issue, Liu et al. and Abmayr et al. delivered AV.ΔR4/ΔC to 9- to 13-month-old mdx mice [8,9]. Aged mdx mice have been shown to display a moderate DMD-like phenotype [21]. Despite clear evidence of muscle protection in young mice, a less favorable response was observed in aged mdx mice [8]. It is not clear whether the diminished protection is due to inefficient gene transfer in aged muscle or intrinsic functional incompetence of the massively truncated microgene. To validate further the therapeutic efficacy of the ΔR4/ΔC microgene, we delivered AV.ΔR4/ΔC to the EDL muscle of newborn u-dko mice.
Similar to DMD patients, u-dko mice are severely disabled. Consistent with previous reports [12,13], we noted a significant weight loss in our experimental u-dko colony (Table 1). We also observed active degeneration and regeneration, widespread inflammation, sarcolemmal leakage, and fibrosis in u-dko muscle (Figs. 1A and 2). The degenerative/necrotic process in u-dko muscle starts at approximately 2 to 2½ weeks and it continues throughout the entire life span [12,13,22]. To express microdystrophin right on or before the onset of muscle degeneration in u-dko muscle, we delivered AAV-6 AV.ΔR4/ΔC to 3-day-old u-dko mice. AAV-6 is one of the most potent AAV serotypes for muscle gene transfer [23,24]. Efficient transduction can be achieved within a week in neonatal u-dko muscle (Supplementary Fig. 5). In this study, we examined muscle pathology and physiology at 9 weeks after neonatal AAV therapy.
In normal muscle, dystrophin is concentrated at the neuromuscular junction [25]. Interestingly, we observed a similar distribution pattern for ΔR4/ΔC microdystrophin. In AV.ΔR4/ΔC-positive fibers, a condensed expression was seen at the neuromuscular junctions that were highlighted by α-bungarotoxin in serial sections (Fig. 1B). This result provides additional localization evidence for the functional competence of the ΔR4/ΔC microgene. As expected, ΔR4/ΔC microdystrophin successfully stabilized the dystroglycan and sarcoglycan subcomplexes to the sarcolemma. Interestingly, immunolocalization of the cytoplasmic subcomplex revealed restoration of dystrobrevin and syntrophin, but not nNOS (Fig. 1).
The dystrobrevins and syntrophins are multimember protein families. They are regular members in the sarcolemmal DGC and they play an important signaling role [26,27]. It is thought that the binding motifs at the dystrophin C-terminal domain are responsible for recruiting dystrobrevin and syntrophin to the DGC [28–30]. However, a previous study suggests that the assembly of syntrophin and dystrobrevin does not require the dystrophin C-terminal domain in the context of the full-length dystrophin gene [15]. The expression pattern of dystrobrevin and syntrophin has never been investigated in any published studies on the C-terminal-truncated microdystrophin genes. It will be of great therapeutic relevance to see whether these components are recovered in AV.ΔR4/ΔC-transduced myofibers. To our surprise, the lack of the binding motifs in microdystrophin did not abolish sarcolemmal expression of dystrobrevin and syntrophin (Fig. 1A). It is currently not clear how dystrobrevin and syntrophin are preserved in the DGC when their binding motifs are eliminated. Additional studies are needed to clarify these intriguing results further.
Among all the partners of the DGC, nNOS is unique. nNOS-null mice display no muscle pathology [31,32]. Removing nNOS from mdx mice does not aggravate the dystrophic phenotype [32,33]. However, low nNOS expression may interfere with blood flow in exercising muscle and lead to functional ischemia in mdx mice and DMD patients [34–36]. More recently, overexpression of nNOS has been shown to ameliorate muscle disease in mdx mice [37,38]. These results suggest that restoring nNOS expression on the sarcolemma may help to reduce dystrophic pathology in DMD. nNOS is recruited to the DGC through its interaction with the syntrophin PDZ domain [17]. Since syntrophin expression was reestablished in ΔR4/ΔC microdystrophin-positive myofibers (Fig. 1A), we initially hypothesized that nNOS should be restored too. Surprisingly, we could not detect nNOS expression in AV.ΔR4/ΔC-infected u-dko muscle (Fig. 1B). Interestingly, several other studies revealed similar results. These studies show that the presence of syntrophin by itself, or both syntrophin and dystrobrevin, may not be sufficient to restore nNOS expression on the sarcolemma [15,19,39]. The exact molecular mechanisms underlying these findings are not clear yet. There are several possibilities. First, the affinity of the PDZ nNOS binding domain may be modulated by other unknown factors. For example, both β1- and β2-syntrophin share the same PDZ domain as α-syntrophin. However, neither β1- nor β2-syntrophin can recruit nNOS to the sarcolemma [40]. Even α-syntrophin itself may sometimes fail to interact with nNOS [41]. Second, each syntrophin has only one PDZ domain. It may need the coordinated action of two perfectly paired PDZ domains to recruit nNOS successfully to the sarcolemma. The syntrophin dimerization process may have been compromised under these situations.
The most exciting finding from our study is the physiological improvement in muscle contractile properties (Fig. 3). We have previously shown that delivering AV.ΔR4/ΔC to adult mdx mice reduced eccentric contraction-induced injury but specific force was only marginally enhanced [8]. By delivering AV.ΔR4/ΔC to newborn u-dko mice, we have now achieved an exceptionally impressive recovery in muscle function. In particular, both specific twitch force and tetanic forces were strengthened (Figs. 3B and 3C). The response to eccentric contraction even reached the level of BL10 mice, although it was still in the low end of the normal range (Fig. 3D). These results agree with previously published transgenic data [4,5]. Taken together, results from the transgenic study and our current study emphasize the importance of early intervention. A much better therapeutic effect can be achieved from preventing muscle disease rather than treating the existing muscle pathology.
In this study, we have demonstrated for the first time that a C-terminal-truncated microdystrophin could improve muscle function in a symptomatic DMD model. Despite the exciting findings, one should be cautious in extrapolating it to human gene therapy. It is very likely that AAV-mediated microdystrophin gene therapy may at most convert a severe DMD to a mild BMD. However, by taking advantage of some recent developments such as a dual AAV vector approach to express a more competent minidystrophin gene [19,42] and coadministration of several therapeutic AAV vectors [9], we may further enhance AAV-mediated DMD gene therapy.
Materials and Methods
Animals
All animal experiments were approved by the Animal Care and Use Committee at the University of Missouri and were in accordance with NIH guidelines. All experimental mice were housed in a specific pathogen-free facility. BL10 mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The u-dko mice were originally generated by crossing utrophin-deficient mice and mdx mice [13]. Utrophin-deficient mice were derived from targeted mutagenesis in the R1 embryonic stem cells (from 129X1/SvJ × 129S1/Sv-p) [43] and these mice were on a hybrid genetic background of C57B5 (black)/129J (chinchilla/agouti). The heterozygous breeders (mdx/utr+/−) were kindly provided by Dr. Mark Grady at Washington University [13]. To ensure genetic purity, we performed additional three generations of backcrossing to the mdx background. The u-dko mice are infertile [13]. Experimental u-dko mice were obtained by crossing heterozygous male and female mice. The utrophin genotype was determined according to a previously published three-primer PCR protocol [44]. The primers include one common reverse primer (DL242, 5′-CTGAGTCAAACAGCTTGGAAGCCTCC) and two distinctive forward primers for wild-type utrophin (DL241, 5′-TGCAGTGTCTCCCAATAAGGTATGAAC) and utrophin knockout (DL240, 5′-TGCCAAGTTCTAATTCCATCAGAAGCTG), respectively. A 640-bp band is diagnostic for wild-type utrophin. The utrophin knockout band is at 450 bp.
Only male mice were used in the gene transfer study. To determine the gender in newborn mice, we performed a four-primer PCR. One primer set was used to amplify an X-chromosome-specific gene, Smcx (forward primer DL501, 5′-GTGAAGGAGGAATTAGGTGG; reverse primer DL502, 5′-GATGTGGTAATTGTCATCAC) [45]. A 1.1-kb band is diagnostic for the presence of the X chromosome. Another primer set was used to amplify a Y-chromosome-specific gene, Sry (forward primer DL420, 5′- TCTTAAACTCTGAAGAAGAGAC; reverse primer DL421, 5′- GTCTTGCCTGTATGTGATGG) [46]. A 404-bp band is diagnostic for the presence of the Y chromosome. Male mice showed both 1.1-kb and 404-bp bands.
Recombinant AAV Stock and gene delivery
The ΔR4/ΔC microgene was a gift from Dr. Jeffrey S. Chamberlain at the University of Washington (Seattle, WA, USA) [5]. Recombinant AAV-6 packaging plasmids (pMTrep2 and pCMVcap6) were gifts from Dr. A. Dusty Miller at the Fred Hutchinson Cancer Research Center (Seattle, WA, USA) [47]. The cis plasmid for AAV packaging, pcisCMV.ΔR4/ΔC, has been described before [6]. Recombinant AAV-6 stock was generated by quadruple plasmid transfection with the cis plasmid, pMTrep2, pCMVcap6, and pHelper (Stratagene, La Jolla, CA, USA) at a ratio of 1:1:3:3 in 293 cells [19]. Crude viral lysate was purified by three rounds of CsCl isopycnic ultracentrifugation as we described before [8].
To deliver AAV to neonatal mouse muscle, 3-day-old pups were anesthetized by hypothermic shock as we described before [6]. Since the EDL muscle cannot be identified in newborn mice, 30 μl of AV.ΔR4/ΔC (6 × 1010 vg viral particles) was injected into the anterior muscle compartment of the left hind limb with a custom-designed 33-gauge gas-tight Hamilton syringe. The contralateral right limb muscle was injected with an equal volume of saline.
Morphological studies
The ΔR4/ΔC microgene is derived from the human dystrophin gene. AAV-mediated ΔR4/ΔC microdystrophin expression was revealed with an anti-human dystrophin N-terminus monoclonal antibody (Dys-3, 1:10 dilution; from Novocastra, Newcastle, UK) [6]. The same immunostaining protocol was used to detect utrophin (1:20; Vector Laboratories, Burlingame, CA, USA) and other components in the DGC including β-dystroglycan (1:50; Novocastra), β-sarcoglycan (1:50; Novocastra), dystrobrevin (1:200; BD Biosciences, San Diego, CA, USA), and syntrophin (1:200; Abcam, Cambridge, MA, USA). A polyclonal anti-nNOS C-terminus antibody (1:2000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) was used to evaluate nNOS expression. Briefly, 8-μm muscle cryosections were blocked in 5% goat serum KPBS at room temperature for 30 min. After being washed in 0.2% gelatin KPBS, muscle sections were incubated with the primary antibody for 12 h at 4°C. After a wash in 0.2% gelatin KPBS, nNOS staining was revealed with an Alexa 594-conjugated goat anti-rabbit secondary antibody. Neuromuscular junctions were highlighted with Alexa 594-conjugated α-bungarotoxin (1:500; Molecular Probes, Eugene, OR, USA).
General muscle histology was revealed by hematoxylin and eosin (HE) staining. Transduction efficiency was determined by dividing the total number of Dys-3-positive cells (immunostaining) with the total number of myofibers (HE staining of adjacent section). At least two montage composites of the entire cross sections in the midbelly were quantified for each muscle sample. The percentage of centrally nucleated myofibers was determined as described before [8]. Masson trichrome staining was used to examine fibrosis. Briefly, 8-μm muscle cryosections were sequentially stained in Weigerts iron hematoxylin (10 min), 1% Ponceau–acetic acid (5 min), and 1% aniline blue (5 s). Fibrous tissue stained blue, nuclei stained dark brown, and muscle stained dark red. Nonspecific esterase (α-naphthyl butyrate esterase) staining was used to identify macrophage infiltration. Briefly, 8-μm muscle cryosections were stained for 1 h at room temperature in a staining solution containing 200 mM phosphate buffer, pH 7.4, 0.023% α-naphthyl acetate, and 0.145% azotized pararosanilin. Macrophages stained dark brown in the interstitial region.
Muscle force measurements
Muscle contractile properties were examined in the EDL muscle in 2-month-old mice. The EDL muscle was prepared for contractile measurements as we described previously [8]. Briefly, the EDL muscle was carefully dissected out and vertically mounted in a 30°C jacket organ bath in oxygenated Ringer's solution (95% O2, 5% CO2). Muscle tendons were attached to a 300B dual-mode servomotor transducer (Aurora Scientific, Inc., Aurora, ON, Canada). Optimal muscle length (Lo) was determined as the muscle length at which the maximal twitch force was elicited by supramaximal stimulation. After all the myofibers were activated with three 500-ms tetanic stimulations at 150 Hz, the absolute twitch force was determined. Tetanic muscle force was then measured at 50, 80, 120, and 150 Hz as we described before [8]. The specific force (kN/m2) was calculated after normalizing the absolute twitch or tetanic force with muscle cross-sectional area (CSA). Muscle CSA was calculated according to the equation CSA = (muscle mass)/[(0.44 × optimal muscle length) × (1.06 g/cm3)], in which 0.44 represents the ratio of fiber length to optimal muscle length (Lf/Lo) for the EDL muscle and 1.06 g/cm3 is the muscle density. In the eccentric contraction assay, the EDL muscle was stimulated at 150 Hz for 700 ms. During the last 200 ms, the muscle was lengthened by 10% Lo at 0.5 Lo/s for 200 ms. A total of 10 cycles of eccentric contraction were applied to the EDL muscle. The maximal isometric tetanic force developed during the first 500 ms of stimulation of the first cycle was designated as the baseline tension (100%). The percentage of tetanic force loss at each cycle was determined by comparing with the baseline tension. Data acquisition and analysis were conducted with the DMC/DMA software (version 3.12; Aurora Scientific).
Statistical analysis
Data are presented as means ± standard error of the mean. Statistical analysis was performed with the SPSS software. Statistical significance was determined using one-way ANOVA followed by Bonferroni post hoc analysis among different groups. A paired t test was also performed to determine statistical significance between the left EDL (AV.ΔR4/ΔC infected) and the right EDL (uninfected) in the u-dko mice. The difference was considered significant when P < 0.05.
Acknowledgments
We thank Dr. Mark Grady for sharing the utrophin/dystrophin double-knockout mouse model. We thank Dr. Jeffrey Chamberlain for providing the ΔR4/ΔC microdystrophin gene and sharing unpublished data. We thank Dr. A. Dusty Miller for the AAV-6 packaging plasmids. This work was supported by grants from the National Institutes of Health (AR-49419, D.D.) and the Muscular Dystrophy Association (D.D.).
Appendix A. Supplementary Data
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.ymthe.2006. 01.007.
References
- 1.Emery AEH, Muntoni F. Duchenne Muscular Dystrophy. Oxford Univ. Press; Oxford; New York: 2003. [Google Scholar]
- 2.England SB, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343:180–182. doi: 10.1038/343180a0. [DOI] [PubMed] [Google Scholar]
- 3.Beggs AH, et al. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am J Hum Genet. 1991;49:54–67. [PMC free article] [PubMed] [Google Scholar]
- 4.Chamberlain JS. Gene therapy of muscular dystrophy. Hum Mol Genet. 2002;11:2355–2362. doi: 10.1093/hmg/11.20.2355. [DOI] [PubMed] [Google Scholar]
- 5.Harper SQ, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 6.Yue Y, et al. Microdystrophin gene therapy of cardiomyopathy restores dystrophin–glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gregorevic P, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu M, et al. Adeno-associated virus-mediated micro-dystrophin expression protects young mdx muscle from contraction-induced injury. Mol Ther. 2005;11:245–256. doi: 10.1016/j.ymthe.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abmayr S, Gregorevic P, Allen JM, Chamberlain JS. Phenotypic improvement of dystrophic muscles by rAAV/microdystrophin vectors is augmented by Igf1 codelivery. Mol Ther. 2005;12:441–450. doi: 10.1016/j.ymthe.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 10.Sicinski P, et al. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 11.Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82:291–329. doi: 10.1152/physrev.00028.2001. [DOI] [PubMed] [Google Scholar]
- 12.Deconinck AE, et al. Utrophin–dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 13.Grady RM, et al. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 14.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc Natl Acad Sci USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crawford GE, et al. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J Cell Biol. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harper SQ, Crawford RW, DelloRusso C, Chamberlain JS. Spectrin-like repeats from dystrophin and alpha-actinin-2 are not functionally interchangeable. Hum Mol Genet. 2002;11:1807–1815. doi: 10.1093/hmg/11.16.1807. [DOI] [PubMed] [Google Scholar]
- 17.Brenman JE, Chao DS, Xia H, Aldape K, Bredt DS. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell. 1995;82:743–752. doi: 10.1016/0092-8674(95)90471-9. [DOI] [PubMed] [Google Scholar]
- 18.Chang WJ, et al. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc Natl Acad Sci USA. 1996;93:9142–9147. doi: 10.1073/pnas.93.17.9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai Y, et al. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol. 2005;23:1435–1439. doi: 10.1038/nbt1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan D, Yue Y, Engelhardt JF. Adeno-associated virus. In: Albelda SM, editor. Lung Biology in Health and Disease, Gene Therapy in Lung Disease. Dekker; New York: 2002. pp. 51–92. [Google Scholar]
- 21.Lefaucheur JP, Pastoret C, Sebille A. Phenotype of dystrophinopathy in old mdx mice. Anat Rec. 1995;242:70–76. doi: 10.1002/ar.1092420109. [DOI] [PubMed] [Google Scholar]
- 22.Grange RW, et al. Functional and molecular adaptations in skeletal muscle of myoglobin-mutant mice. Am J Physiol Cell Physiol. 2001;281:C1487–C1494. doi: 10.1152/ajpcell.2001.281.5.C1487. [DOI] [PubMed] [Google Scholar]
- 23.Blankinship MJ, et al. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther. 2004;10:671–678. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh A, Yue Y, Duan D. Viral serotype and the transgene sequence influence overlapping adeno-associated viral (AAV) vector-mediated gene transfer in skeletal muscle. J Gene Med. doi: 10.1002/ jgm.835. in press. electronic publication ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sealock R, et al. Localization of dystrophin relative to acetylcholine receptor domains in electric tissue and adult and cultured skeletal muscle. J Cell Biol. 1991;113:1133–1144. doi: 10.1083/jcb.113.5.1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peters MF, Adams ME, Froehner SC. Differential association of syntrophin pairs with the dystrophin complex. J Cell Biol. 1997;138:81–93. doi: 10.1083/jcb.138.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peters MF, et al. Differential membrane localization and intermolecular associations of alpha-dystrobrevin isoforms in skeletal muscle. J Cell Biol. 1998;142:1269–1278. doi: 10.1083/jcb.142.5.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn AH, Kunkel LM. Syntrophin binds to an alternatively spliced exon of dystrophin. J Cell Biol. 1995;128:363–371. doi: 10.1083/jcb.128.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newey SE, Benson MA, Ponting CP, Davies KE, Blake DJ. Alternative splicing of dystrobrevin regulates the stoichiometry of syntrophin binding to the dystrophin protein complex. Curr Biol. 2000;10:1295–1298. doi: 10.1016/s0960-9822(00)00760-0. [DOI] [PubMed] [Google Scholar]
- 30.Sadoulet-Puccio HM, Rajala M, Kunkel LM. Dystrobrevin and dystrophin: an interaction through coiled-coil motifs. Proc Natl Acad Sci USA. 1997;94:12413–12418. doi: 10.1073/pnas.94.23.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–1286. doi: 10.1016/0092-8674(93)90615-w. [DOI] [PubMed] [Google Scholar]
- 32.Chao DS, Silvagno F, Bredt DS. Muscular dystrophy in mdx mice despite lack of neuronal nitric oxide synthase. J Neurochem. 1998;71:784–789. doi: 10.1046/j.1471-4159.1998.71020784.x. [DOI] [PubMed] [Google Scholar]
- 33.Crosbie RH, et al. mdx muscle pathology is independent of nNOS perturbation. Hum Mol Genet. 1998;7:823–829. doi: 10.1093/hmg/7.5.823. [DOI] [PubMed] [Google Scholar]
- 34.Thomas GD, et al. Impaired metabolic modulation of alpha-adrenergic vasoconstriction in dystrophin-deficient skeletal muscle. Proc Natl Acad Sci USA. 1998;95:15090–15095. doi: 10.1073/pnas.95.25.15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sander M, et al. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci USA. 2000;97:13818–13823. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas GD, Shaul PW, Yuhanna IS, Froehner SC, Adams ME. Vasomodulation by skeletal muscle-derived nitric oxide requires alpha-syntrophin-mediated sarcolemmal localization of neuronal nitric oxide synthase. Circ Res. 2003;92:554–560. doi: 10.1161/01.RES.0000061570.83105.52. [DOI] [PubMed] [Google Scholar]
- 37.Wehling M, Spencer MJ, Tidball JG. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J Cell Biol. 2001;155:123–132. doi: 10.1083/jcb.200105110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum Mol Genet. 2005;14:1921–1933. doi: 10.1093/hmg/ddi197. [DOI] [PubMed] [Google Scholar]
- 39.Grady RM, et al. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol. 1999;1:215–220. doi: 10.1038/12034. [DOI] [PubMed] [Google Scholar]
- 40.Adams ME, et al. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol. 2000;150:1385–1398. doi: 10.1083/jcb.150.6.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kramarcy NR, Sealock R. Syntrophin isoforms at the neuromuscular junction: developmental time course and differential localization. Mol Cell Neurosci. 2000;15:262–274. doi: 10.1006/mcne.1999.0823. [DOI] [PubMed] [Google Scholar]
- 42.Duan D, Yue Y, Engelhardt JF. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol Ther. 2001;4:383–391. doi: 10.1006/mthe.2001.0456. [DOI] [PubMed] [Google Scholar]
- 43.Grady RM, Merlie JP, Sanes JR. Subtle neuromuscular defects in utrophin-deficient mice. J Cell Biol. 1997;136:871–882. doi: 10.1083/jcb.136.4.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grange RW, Gainer TG, Marschner KM, Talmadge RJ, Stull JT. Fast-twitch skeletal muscles of dystrophic mouse pups are resistant to injury from acute mechanical stress. Am J Physiol Cell Physiol. 2002;283:C1090–C1101. doi: 10.1152/ajpcell.00450.2001. [DOI] [PubMed] [Google Scholar]
- 45.Xu J, Burgoyne PS, Arnold AP. Sex differences in sex chromosome gene expression in mouse brain. Hum Mol Genet. 2002;11:1409–1419. doi: 10.1093/hmg/11.12.1409. [DOI] [PubMed] [Google Scholar]
- 46.Kunieda T, et al. Sexing of mouse preimplantation embryos by detection of Y chromosome-specific sequences using polymerase chain reaction. Biol Reprod. 1992;46:692–697. doi: 10.1095/biolreprod46.4.692. [DOI] [PubMed] [Google Scholar]
- 47.Halbert CL, Allen JM, Miller AD. Adeno–associated virus type 6 (aav6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of aav2 vectors. J Virol. 2001;75:6615–6624. doi: 10.1128/JVI.75.14.6615-6624.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]