

Abstract
Duchenne muscular dystrophy (DMD) is the most common inherited lethal muscle degenerative disease. Currently there is no cure. Highly abbreviated microdystrophin cDNAs were developed recently for adeno-associated virus (AAV)-mediated DMD gene therapy. Among these, a C-terminal-truncated ΔR4-R23/ΔC microgene (ΔR4/ΔC) has been considered as a very promising therapeutic candidate gene. In this study, we packaged a CMV.ΔR4/ΔC cassette in AAV-5 and evaluated the transduction and muscle contractile profiles in the extensor digitorum longus muscles of young (7-week-old) and adult (9-month-old) mdx mice. At ∼3 months post-gene transfer, 50–60% of the total myofibers were transduced in young mdx muscle and the percentage of centrally nucleated myofibers was reduced from ∼70% in untreated mdx muscle to ∼22% in microdystrophin-treated muscle. Importantly, this level of transduction protected mdx muscle from eccentric contraction-induced damage. In contrast, adult mdx muscle was more resistant to AAV-5 transduction, as only ∼30% of the myofibers were transduced at 3 months postinfection. This transduction yielded marginal protection against eccentric contraction-induced injury. The extent of central nucleation was also more difficult to reverse in adult mdx muscle (from ∼83% in untreated to ∼58% in treated). Finally, we determined that the ΔR4/ΔC microdystrophin did not significantly alter the expression pattern of the endogenous full-length dystrophin in normal muscle. Neither did it have any adverse effects on normal muscle morphology or contractility. Taken together, our results suggest that AAV-mediated ΔR4/ΔC microdystrophin expression represents a promising approach to rescue muscular dystrophy in young mdx skeletal muscle.
Keywords: Duchenne muscular dystrophy, mdx, adeno-associated virus, microdystrophin, muscle contraction
Introduction
Duchenne muscular dystrophy (DMD) is the most common inherited lethal muscle wasting disease. This X-linked disorder affects 0.02–0.03% of newborn boys worldwide [1]. Affected boys are usually diagnosed between 3 and 5 years of age [2]. Early symptoms of delayed walking and unsteady gait rapidly progress to general muscle weakness. By age 12, 95% of patients are confined to a wheelchair and most of them develop severe scoliosis [1]. Improved clinical management has significantly extended the life expectancy of DMD patients in recent years [3]. However, the majority of patients still die before age 20 from respiratory and/or cardiac failure [1]. Current treatment options for DMD patients focus primarily on relief of symptoms. At present, there is no cure.
DMD is caused by mutations in the dystrophin gene [4,5]. Since one-third of the cases are derived from new mutations with no prior family history of the disease, genetic counseling cannot eliminate this fatal disease [6]. Replacing and/or repairing the mutated dystrophin gene by gene therapy is perhaps the only way to cure DMD at the molecular level. However, DMD gene therapy is challenged by several technical difficulties, especially the huge size of the dystrophin gene [7]. One approach to overcoming this size obstacle is to develop smaller, but functional, dystrophin isoforms. The therapeutic potential of this approach has been demonstrated in many affected Becker muscular dystrophy (BMD) patients, who carry internally deleted minidystrophin genes [8]. In an extreme case, a patient lacking 43% of dystrophin coding sequences has remained ambulant past age 61 [9].
The dystrophin protein has four distinctive functional domains including the N-terminal, central rod, cysteine-rich (CR), and C-terminal domains. The N-terminal domain and portions of the rod domain interact with cytoskeletal F-actin. The rod domain is composed of 24 spectrin-like repeats and four hinge regions. Together with a WW motif at hinge 4 of the rod domain, the CR domain links dystrophin to the transmembrane dystroglycan complex. The C-terminal domain interacts with several signaling molecules such as syntrophin, dystrobrevin, and nNOS [10,11]. Systemic dissection of each dystrophin domain has revealed the most critical regions that are essential to muscle function [7]. Engineered deletion of less important regions (such as the majority of the rod domain and the C-terminal domain) seems to have minimal effect on overall function. Importantly, transgenic expression of these novel truncated dystrophin isoforms reversed pathological changes in skeletal muscle in mdx mice, a model for DMD [12-18].
Based on these results, a series of highly abbreviated microdystrophin cDNAs was developed recently [17,19-22]. One of the microdystrophin cDNAs, ΔR4-R23, contains only four spectrin-like repeats (the first three and the last one). Transgenic expression of this microgene in mdx mice remarkably ameliorated mdx mouse skeletal muscle pathology. The percentage of central nucleation, a hallmark for muscle degeneration and regeneration, was reduced to less than 1% in both limb muscle and the diaphragm [17]. More importantly, the ΔR4-R23 microdystrophin protected tibialis anterior (TA) muscle from contraction-induced damage [17]. To explore the therapeutic potential of this microgene, we removed the C-terminal domain from the ΔR4-R23 microgene [16] and packaged the C-terminal truncated ΔR4-R23/ΔC micro-gene (ΔR4/ΔC) expression cassette in a type-2 adeno-associated viral vector (AAV-2) [17]. Despite the fact that the ΔR4/ΔC microgene carried only ∼30% of the dystrophin cDNA coding sequence, AAV-2-mediated expression of ΔR4/ΔC microgene reversed several pathological changes in the gastrocnemius muscle of 1-month-old mdx mice. Notably, the treated muscle displayed only 14% centrally nucleated myofibers, while the age-matched control mdx muscle showed 68% central nucleation. Furthermore, the fiber size diversity was significantly reduced in the treated muscle [17]. These encouraging results suggest that the ΔR4/ΔC microgene may serve as an excellent therapeutic candidate gene for DMD gene therapy.
Several important questions remain to be answered regarding the functional competence of the ΔR4/ΔC microgene. In particular, whether the ΔR4/ΔC microdystrophin improves the mdx muscle-specific force and protects mdx muscle from contraction-induced injury. To address these issues, we delivered the ΔR4/ΔC microgene by AAV-5 vector to the extensor digitorum longus (EDL) muscle of young (7-week-old) and adult (9-month-old) mdx mice and performed functional assays on muscle contractility. Consistent with our transgenic study of the C-terminal-inclusive ΔR4-R23 microgene, ΔR4/ΔC microdystrophin did not improve the limb muscle-specific force at most of the tested stimulation frequencies (80, 120, and 150 Hz). However, AAV-5-mediated ΔR4/ΔC microdystrophin expression in slightly more than 50% of myofibers protected mdx muscle from contraction-induced damage in the EDL muscle of young mdx mice. Surprisingly, AAV-5 transduction was limited in the EDL muscle of older mdx mice and only marginal protection was observed in older muscle. Additional morphological studies demonstrated that the ΔR4/ΔC microdystrophin is more effective in reducing central nucleation in young mdx muscle. To extend our observations further, we also delivered the ΔR4/ΔC microgene to the EDL muscle of normal mice. Interestingly, AAV-mediated ΔR4/ΔC expression did not disrupt the endogenous full-length dystrophin gene expression pattern. In addition, forced ΔR4/ΔC expression had no deleterious effects on muscle contraction in the normal C57BL/10 (BL10) EDL muscle.
Results
AAV Infection Alone Does Not Alter EDL Muscle Contraction Profile
To test whether AAV-5-mediated transgene expression in the EDL muscle affected muscle contraction, we infected the left EDL muscle of 6-week-old mice with 1 × 1010 genome particles of AV.RSV.AP and delivered the same volume of Hepes-buffered saline to the right EDL muscle (Fig. 1A, Table 1). Despite the dramatic morphological difference between the BL10 and the mdx EDL muscle, we observed efficient transgene expression in both strains (Fig. 1A). On average, transduction efficiency reached 57% for the BL10 EDL muscle and 54% for the mdx EDL muscle (Table 1). Interestingly, there appeared to be great fiber-to-fiber variations in the level of alkaline phosphatase (AP) expression. Both intensely and lightly stained myofibers were seen in every muscle section (Fig. 1A).
FIG. 1.
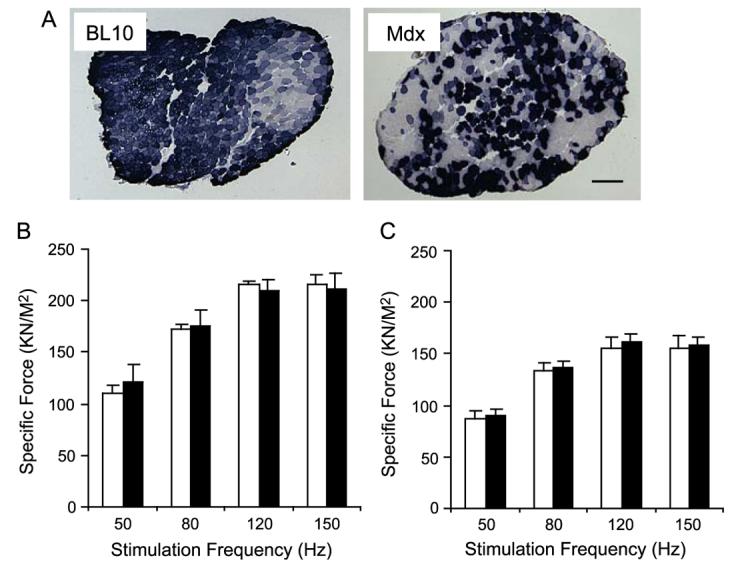
AV.RSV.AP infection did not reduce isometric tetanic force in the EDL muscle. The left EDL muscles of 6-week-old mice were infected with AV.RSV.AP and the contralateral right EDL muscles were injected with equal volumes of saline. Transgene expression and tetanic force were evaluated at 4 weeks postinfection. (A) Efficient AAV transduction was observed in both normal (BL10) and dystrophic (mdx) EDL muscles. Scale bar, 200 μm. (B) Force–frequency relationship in BL10 EDL muscles. N = 4 pairs. (C) Force–frequency relationship in mdx EDL muscles. N = 5 pairs. Open bar, without AAV infection; filled bar, infected with AV.RSV.AP. AAV infection did not alter specific force production in BL10 and mdx EDL muscles.
TABLE 1.
Characteristics of mice and EDL muscles infected by AV.RSV.AP
| EDL characteristics at harvest | |||||||
|---|---|---|---|---|---|---|---|
| Strain | AV.RSV.AP | Na | Age at infection | Age at harvest | Weight (mg) | CSA (mm2)b | Transduction (%) |
| BL10 | No | 4 | N/Ac | 77 days | 8.00 ± 0.29 | 1.51 ± 0.05 | N/Ac |
| BL10 | Yes | 4 | 43 days | 77 days | 8.30 ± 0.16 | 1.56 ± 0.04 | 56.75 ± 5.88d |
| mdx | No | 5 | N/Ac | 74 days | 11.34 ± 0.33 | 2.05 ± 0.06 | N/Ac |
| mdx | Yes | 5 | 42 days | 74 days | 11.36 ± 0.16 | 2.02 ± 0.07 | 53.60 ± 3.88d |
Study was performed in paired legs. The left EDL muscle was infected with AV.RSV.AP, the right EDL muscle of the same mouse was not infected with AAV.
Cross-sectional area (calculated according to fiber length).
Not applicable.
There was no statistical difference in transduction efficiency between BL10 and mdx.
Consistent with previous publications (reviewed in [23,24]), mdx muscle generated much less specific tetanic force than BL10 muscle (Fig. 1). However, irrespective of genetic background, there was no significant difference in specific tetanic force between the left (AAV infected) and the right (saline only) muscles when they were stimulated at 50, 80, 120, and 150 Hz (Fig. 1). In addition, AAV infection did not induce any detectable changes in muscle mass or cross-sectional area (CSA) (Table 1).
To confirm these observations further, we measured the EDL muscle response to eccentric contraction-induced injury. This assay is very sensitive in revealing minor mechanical defects [25]. Eccentric contraction occurs when a contracting muscle is lengthened by force. Such forced lengthening damages the muscle contractile apparatus and reduces subsequent tetanic force development. As shown in Fig. 2A, the EDL muscle was continuously stimulated for 700 ms at 150 Hz. During the last 200 ms stimulation, the EDL muscle was stretched from its optimal length (Lo) to 110% of Lo. A total of 10 repeated stretch cycles were applied to each EDL muscle. Consistent with previous reports [26,27], the mdx EDL muscle was more susceptible to eccentric contraction-induced damage. After the first round of stretching, tetanic force dropped by 10% in mdx EDL muscles (Figs. 2C and 2E), whereas only a minor (0 to 5%) drop was detected in BL10 EDL muscles (Figs. 2B and 2D). By the end of the 10th stretch, tetanic force was reduced to about half of the starting level in mdx muscles (Fig. 2E). Yet, in BL10 muscles, it retained approximately 70% of the starting force (Fig. 2D). Nevertheless, when we compared responses between AAV-infected muscle and uninfected muscle in the same strain, we did not see any significant difference. Taken together, our results suggest that AAV infection alone has minor effect on muscle contraction.
FIG. 2.
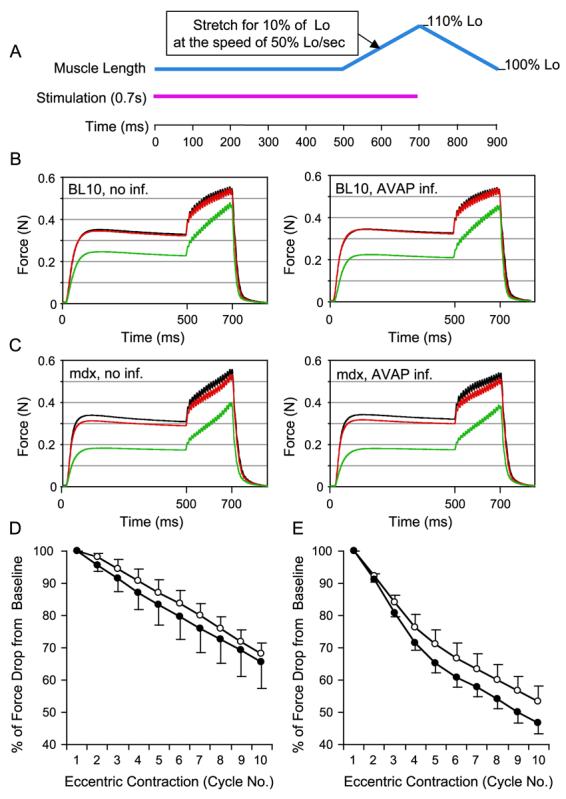
AV.RSV.AP infection did not aggravate eccentric contraction-induced injury in the EDL muscle. (A) Schematic outline of the eccentric contraction protocol used in this study. Muscle was stimulated at 150 Hz for 700 ms (pink line). At the beginning of the stimulation, muscle length (blue line) was adjusted to the optimal length (Lo). At the end of 500 ms stimulation, muscle length was stretched to 110% of Lo at the speed of 50% Lo/s. At the end of stimulation, muscle length was returned to Lo at the same speed. A total of 10 stretch (eccentric contraction) cycles were performed in each muscle. An isometric tetanic force was developed during the first 500 ms stimulation. The change of this tetanic force between each eccentric contraction cycle reflected the degree of muscle injury. (B) Representative force tracing in BL10 EDL muscles. Left, without AAV infection; right, infected with AV.RSV.AP. Black line, force tracing from the first cycle. Red line, force tracing from the second cycle. Green line, force tracing from the 10th cycle. (C) Representative force tracing in mdx EDL muscles. Left, without AAV infection; right, infected with AV.RSV.AP. Black line, force tracing from the first cycle. Red line, force tracing from the second cycle. Green line, force tracing from the 10th cycle. The isometric tetanic force drop was more significant in mdx muscles. However, there was no significant difference between the AV.RSV.AP-infected muscle and the saline-injected control. (D) Relative change in tetanic force during 10 cycles of eccentric contraction in BL10 EDL muscles (N = 4 pairs). The tetanic tension developed during the first cycle was designated as 100%. Open circle, without AAV infection (mean + SEM). Closed circle, infected with AV.RSV.AP (mean − SEM). (E) Relative change of tetanic force during 10 cycles of eccentric contraction in mdx EDL muscles (N = 5 pairs). The isometric tetanic tension developed during the first cycle was designated as 100%. Open circle, without AAV infection (mean + SEM). Closed circle, infected with AV.RSV.AP (mean − SEM). AV.RSV.AP infection resulted in a slightly bigger, but not statistically significant, force deficit in the last few cycles. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
AAV-Mediated Microdystrophin Expression is More Efficient in Correcting Central Nucleation in Young mdx Muscle
mdx muscle is characterized by abundant centrally nucleated, regenerated myofibers. We have previously shown that AAV-mediated ΔR4/ΔC expression reduced central nucleation from 68 to 14% in 1-month-old mdx mice [17]. It remained to be determined whether the ΔR4/ΔC could also halt pathological central nucleation in older mice. To address this question, we delivered AV.CMV.ΔR4/ΔC virus to the left EDL muscles of 7-week-old and 9-month-old mdx mice. In 7-week-old mice, the right EDL muscles were infected with AV.RSV.AP. In 9-month-old mice, the right EDL muscles were not infected. We evaluated transgene expression and central nucleation in the mice infected at 7 weeks of age at age 5 months, while we evaluated those infected at 9 months of age at age 12 months. As shown in Fig. 3, infection at the younger age was more effective in reducing the number of centrally nucleated myofibers. At 5 months of age, the percentage of centrally nucleated myofibers in untreated EDL muscles was 70 ± 2.3%. However, in EDL muscles that were infected with AV.ΔR4/ΔC at 7 weeks of age, the percentage of centrally nucleated myofibers decreased to 21.5 ± 1.3% ( P < 0.05) in microdystrophin-positive myofibers. In older mdx mice, AAV-mediated ΔR4/ΔC expression also resulted in limited, but significant, reduction of central nucleation in transduced myofibers. At 12 months of age, 82.5 ± 2.6% of the untreated mdx EDL muscle myofibers contained centrally located nuclei. This number was reduced to 57.4 ± 1.9% in myofibers that were transduced by AV.ΔR4/ΔC at 9 months of age ( P < 0.05).
FIG. 3.
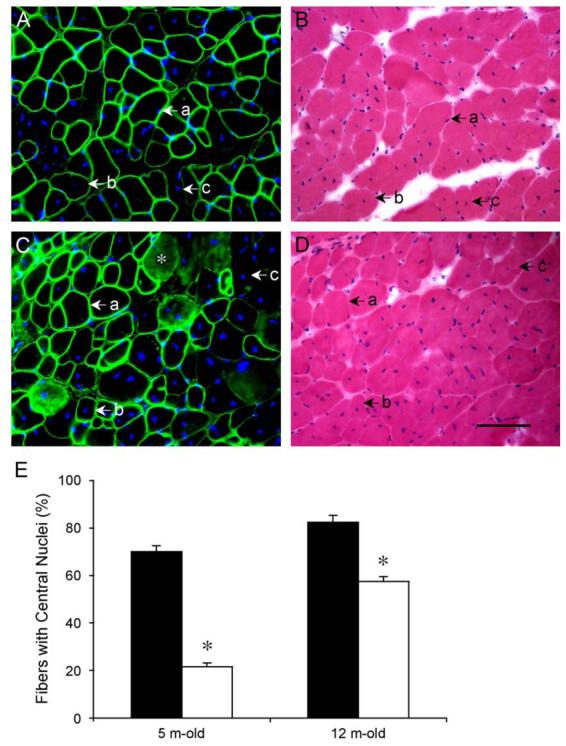
AAV-mediated microdystrophin expression was more efficient in reducing central nucleation in young (7-week-old) mdx EDL muscles. AV.ΔR4/ΔC was delivered to EDL muscles of 7-week-old (young) and 9-month-old (adult) mdx mice. Three months later, the percentage of centrally nucleated myofibers in all transduced (ΔR4/ΔC-positive) myofibers was quantified in each infected EDL muscle. (A and B) Representative immunofluorescence (A, with microdystrophin-specific antibody) and HE (B) staining of an EDL muscle infected at 7 weeks of age. (a) A microdystrophin-positive fiber with a peripherally located nucleus. (b) A microdystrophin-positive fiber with a centrally located nucleus. (c) A microdystrophin-negative fiber with a centrally located nucleus. (C and D) Representative immunofluorescence (C, with microdystrophin-specific antibody) and HE (D) staining of an EDL muscle infected at 9 months of age. (a) A microdystrophin-positive fiber with a peripherally located nucleus. (b) A microdystrophin-positive fiber with a centrally located nucleus. (c) A microdystrophin-negative fiber with a centrally located nucleus. Nonspecific immunoreactivity was observed in cytosol of some myofibers in old mdx muscle (*). In A and C, nuclei were stained with DAPI. Scale bar for D (100 μm) applies to all the photomicrographs. (E) Quantitative evaluation of central nucleation in young (infected at 7 weeks of age and examined at 5 months of age) and older (infected at 9 months of age and examined at 12 months of age) EDL muscles. Filled bar, percentage of fibers with centrally located nuclei in untreated mdx muscles. Open bar, percentage of fibers with centrally located nuclei in microdystrophin-positive myofibers in AV.ΔR4/ΔC-infected mdx muscles. N = 5 for all groups except for 5-month-old AV.ΔR4/ΔC-infected group (N = 9). There was a statistically significant difference between untreated and treated muscles (*P < 0.05). Muscles infected at the younger age were better protected than those infected at the older age.
Microdystrophin Expression in Slightly More Than Half of the EDL Myofibers Results in Better Protection against Eccentric Contraction-Induced Injury in Young mdx Mice
To determine whether the ΔR4/ΔC microdystrophin could improve muscle contractility, we delivered AV.ΔR4/ΔC virus to EDL muscles of 7-week-old mdx mice. On average, 58% of the myofibers were transduced (Fig. 4, Table 2). We first examined the force–frequency relationship between the treated and the untreated EDL muscles. Despite a trend toward an increase in specific force in AV.ΔR4/ΔC virus-infected muscles, we saw statistically significant improvement only at 50 Hz stimulation frequency (Fig. 4B). Compared with muscles that were transduced by a reporter gene AAV vector, AAV-mediated ΔR4/ΔC expression provided better protection against eccentric contraction-induced injury. We saw statistically significant improvements following the second to the eighth eccentric contraction ( P < 0.05) (Fig. 4C).
FIG. 4.
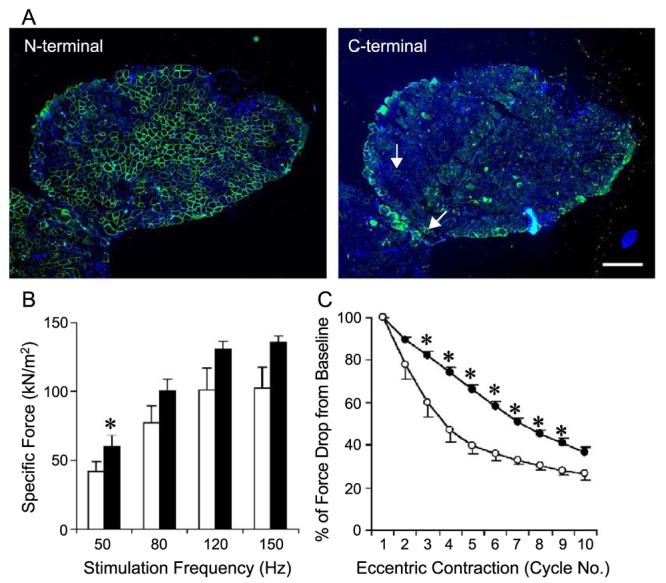
AAV-mediated ΔR4/ΔC microdystrophin expression protected young (7 weeks old at infection) EDL muscles from eccentric contraction-induced injury. The left EDL muscles of 7-week-old mdx mice were infected with AV.ΔR4/ΔC. The contralateral right EDL muscles were infected with AV.RSV.AP. Viral transduction and muscle physiology were examined when mice were 5 months of age. (A) Representative photomicrographs of immunofluorescence staining with the N-terminal-specific (for ΔR4/ΔC microdystrophin, left) and the C-terminal-specific (for endogenous revertant murine dystrophin, right) antibodies. Nuclei were stained with DAPI. Scale bar, 300 μm. On average, approximately 58% of EDL myofibers were transduced by AAV (Table 2). Revertant myofibers were occasionally seen with the antibody against the dystrophin C-terminus (arrow, right). (B) Effect of partial microdystrophin transduction on specific tetanic force in the mdx EDL muscle. Open bar, EDL muscles infected with AV.RSV.AP; filled bar, EDL muscles infected with AV.ΔR4/ΔC. N = 5 pairs. *The difference between AV.ΔR4/ΔC- and AV.RSV.AP-infected muscles was statistically significant ( P < 0.05). (C) Partial microdystrophin expression protected the EDL muscle from the majority of eccentric contraction-induced injuries (from the third to the ninth cycle). Open circle, EDL muscles infected with AV.RSV.AP (mean − SEM); closed circle, EDL muscles infected with AV.ΔR4/ΔC (mean + SEM). N = 5 pairs. *The difference between AV.ΔR4/ΔC- and AV.RSV.AP-infected muscles was statistically significant ( P < 0.05).
TABLE 2.
Characteristics of mdx mice and mdx EDL muscles infected by AV.ΔR4
| EDL characteristics at harvest | ||||||
|---|---|---|---|---|---|---|
| AAV | Na | Age at infection | Age at harvest | Weight (mg) | CSA (mm2)b | Transduction (%) |
| AV.AP | 5 | 47 days (7 wk) | 158 days (5 m) | 14.32 ± 0.82 | 2.81 ± 0.16 | 55.88 ± 2.51d |
| AV.ΔR4 | 5 | 47 days (7 wk) | 158 days (5 m) | 13.95 ± 0.79 | 2.74 ± 0.15 | 57.68 ± 4.29d |
| No infection | 4 | N/Ac | 374 days (12 m) | 14.05 ± 0.72 | 2.86 ± 0.12 | N/Ac |
| AV.ΔR4 | 4 | 276 days (9 m) | 374 days (12 m) | 14.73 ± 0.81 | 3.08 ± 0.15 | 31.77 ± 2.20e |
Study was performed in paired legs. The left EDL muscle was infected with AV.ΔR4, the right EDL muscle of the same mouse was either infected with AV.AP or not infected with AAV.
Cross-sectional area (calculated according to fiber length).
Not applicable.
There was no statistical difference in transduction efficiency between AV.AP and AV.ΔR4.
AV. ΔR4 transduction efficiency in 9-month-old mdx EDL was significantly lower than that in 7-week-old mdx EDL.
AAV-Mediated Microdystrophin Transduction is Less Optimal in the Older EDL Muscle and Results in Limited Protection
Morphometric quantification suggested that central nucleation in the older EDL muscle was more difficult to reverse by ΔR4/ΔC expression (Fig. 3). To evaluate the physiological effect of ΔR4/ΔC on older mdx muscle, we delivered AV.ΔR4/ΔC to the left EDL muscles of 9-month-old mdx mice. We used the contralateral right EDL muscles as sham-infected controls. Unlike 7-week-old EDL muscles, 9-month-old mdx EDL muscles were less efficiently transduced by AAV-5. The average transduction efficiency was 32 ± 2% (Table 2, Fig. 5). In addition, there were also more revertant fibers in the older muscles (Fig. 5). We measured tetanic force generation and response to eccentric contraction-induced injury at 3 months postinjection. We did not see any improvement in the specific force in the treated muscles. The only significant change was the muscle force preservation after the second and the third eccentric contraction ( P < 0.05) (Fig. 5C).
FIG. 5.
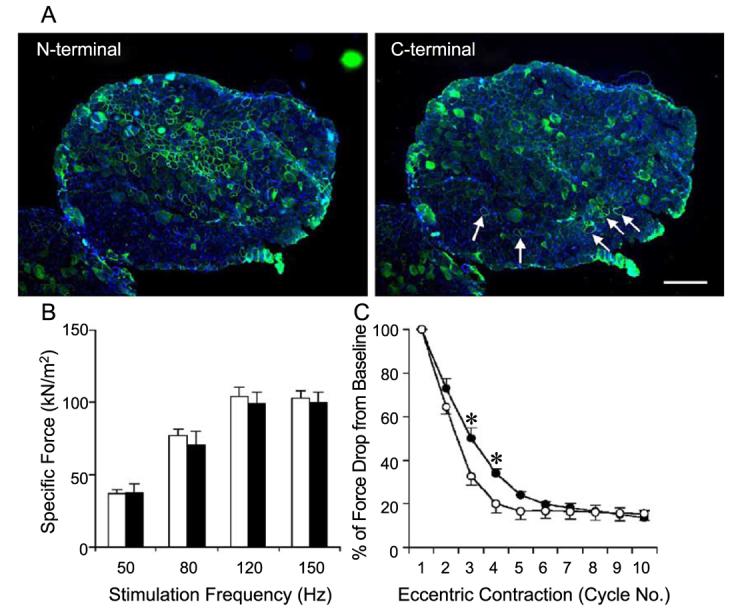
Adult (9-month-old) mdx EDL muscles were poorly transduced by AV.ΔR4/ΔC and minimally protected from eccentric contraction-induced injury. The left EDL muscles of 9-month-old mdx mice were infected with AV.ΔR4/ΔC. The contralateral right EDL muscles served as uninfected controls. Viral transduction and muscle contraction were examined at 3 months postinfection. (A) Representative photomicrographs of immunofluorescence staining with the N-terminal-specific (for ΔR4/ΔC microdystrophin, left) and the C-terminal-specific (for endogenous revertant murine dystrophin, right) antibodies. Scale bar, 300 μm. On average, approximately 32% of the EDL myofibers were transduced by AV.ΔR4/ΔC (Table 2). Revertant myofibers (arrow) were seen more frequently in older mice (right). (B) Limited microdystrophin expression in older mdx EDL muscles did not improve specific tetanic force. Open bar, uninfected EDL muscles; filled bar, AV.ΔR4/ΔC-infected muscles. N = 4 pairs. (C) Limited microdystrophin expression resulted in marginal protection against eccentric contraction-induced injury in the mdx EDL muscle (significant difference was seen only after the second and third stretch cycle). Open circle, uninfected EDL muscles (mean − SEM); closed circle, EDL muscles infected with AV.ΔR4/ΔC (mean + SEM). N = 4 pairs. *The difference between uninfected and AV.ΔR4/ΔC-infected muscles was statistically significant ( P < 0.05).
Ectopically Expressed Microdystrophin Coexists with the Endogenous Full-Length Dystrophin in Normal Muscle and Does Not Alter Muscle Morphology or Contractility
To determine whether AAV-mediated microdystrophin expression could displace the full-length endogenous dystrophin, we delivered AV.ΔR4/ΔC to EDL muscles of 4½-month-old BL10 mice. After an additional 4½ months, the transduction efficiency reached ∼60% (Table 3, Fig. 6). However, there were no significant changes in muscle mass or CSA (Table 3). When we performed immunofluorescence staining with antibodies that were either specific to the mouse endogenous full-length dystrophin or specific to the human microdystrophin, all the myofibers that expressed the microdystrophin also expressed the endogenous full-length dystrophin (Fig. 7A). Forced microdystrophin expression seemed to have no apparent effect on endogenous dystrophin expression pattern. On HE staining, we did not detect any morphological difference between uninfected and infected EDL muscles (Fig. 6B). Finally, we measured specific force and the force drop following eccentric contraction. In these physiological assays, we also did not see any significant difference between the infected and the uninfected muscles (Fig. 7).
TABLE 3.
Characteristics of BL10 mice and BL10 EDL muscles infected by AV.ΔR4
| EDL characteristics at harvest | ||||||
|---|---|---|---|---|---|---|
| AAV | Na | Age at harvest | Age at infection | Weight (mg) | CSA (mm2)b | Transduction (%) |
| No infection | 3 | N/Ac | 272 days (9 m) | 9.29 ± 0.02 | 1.95 ± 0.05 | N/Ac |
| AV.ΔR4 | 3 | 135 days (4.5 m) | 272 days (9 m) | 9.31 ± 0.02 | 1.98 ± 0.03 | 59.67 ± 3.48 |
Study was performed in paired legs. The left EDL muscle was infected with AV.ΔR4, the right EDL muscle of the same mouse was not infected with AAV.
Cross-sectional area (calculated according to fiber length).
Not applicable.
FIG. 6.
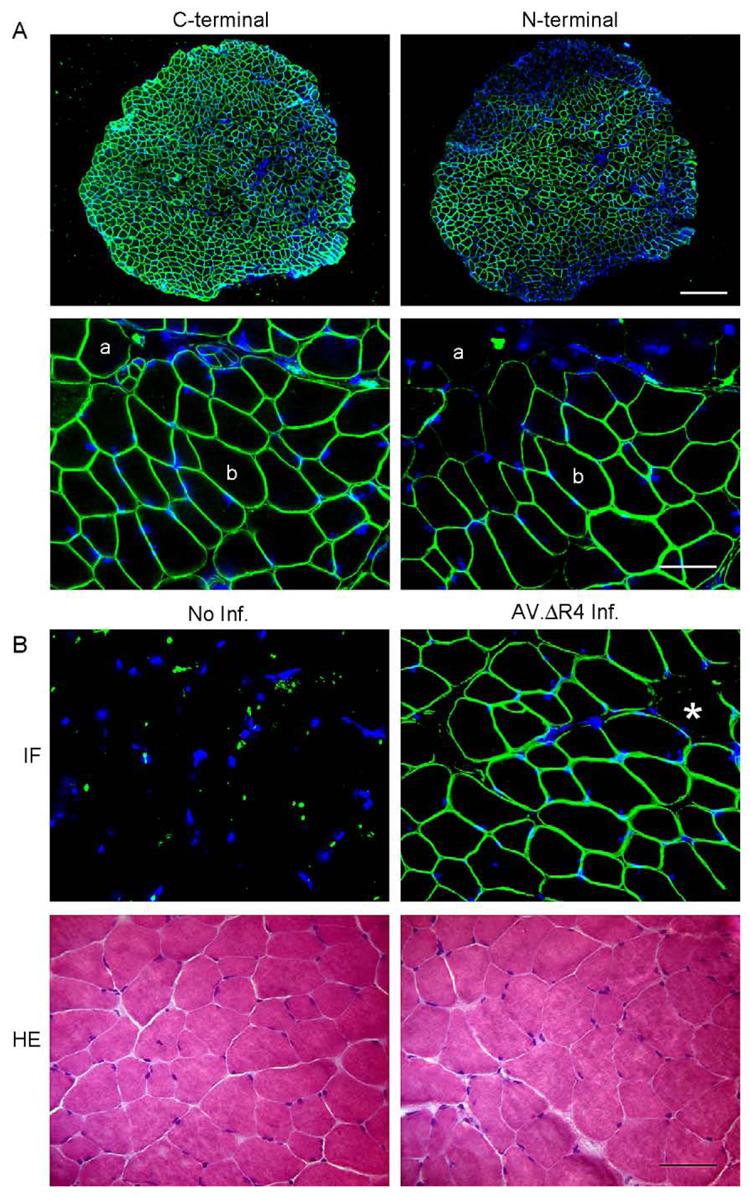
AAV-mediated ΔR4/ΔC microdystrophin was coexpressed with the endogenous full-length dystrophin and did not alter normal EDL muscle morphology. (A) Representative photomicrographs of immunofluorescence staining with the C-terminal-specific (for murine endogenous full-length dystrophin, left) and the N-terminal-specific (for ΔR4/ΔC microdystrophin, right) antibodies. Scale bar for lower power magnification photomicrographs represents 300 μm. Scale bar for higher power magnification photomicrographs represents 50 μm. (a) A myofiber that expressed only the murine full-length dystrophin; (b) a myofiber that expressed both the murine full-length dystrophin and the ΔR4/ΔC microdystrophin. (B) Representative photomicrographs of immunofluorescence (IF, top) and HE (bottom) staining of uninfected (left) and AV.ΔR4/ΔC-infected (right) BL10 EDL muscles. Immunofluorescence staining was performed with anti-dystrophin N-terminal antibody (specific for ΔR4/ΔC microdystrophin, top). *A myofiber that was not transduced by AV.ΔR4/ΔC. Scale bar, 50 μm.
FIG. 7.

AAV-mediated ΔR4/ΔC microdystrophin expression did not compromise contractile properties of the normal EDL muscle. The left EDL muscles of 4.5-month-old mdx mice were infected with AV.ΔR4/ΔC. The contralateral right EDL muscles served as sham-infected controls. Muscle contractile assays were performed when mice were 9 months of age. (A) Force–frequency relationship between sham-infected (open bar) and AV.ΔR4/ΔC-infected (filled bar) EDL muscles. No statistically significant difference was seen between the two groups ( P > 0.05). (B) Relative tetanic force drop during 10 cycles of eccentric contraction. Open circles, sham-infected EDL muscles (mean − SEM); closed circles, EDL muscles infected with AV.ΔR4/ΔC (mean + SEM). N = 3 pairs. There was no statistical difference between AV.ΔR4/ΔC-infected and sham-infected groups ( P > 0.05).
Discussion
The EDL muscle has often been used for in vitro measurement of intact skeletal muscle function. Knowledge of the contractile properties of dystrophin-null muscle is largely derived from experimentation on the mdx EDL muscle [26-31]. In this study, we delivered the ΔR4/ΔC microdystrophin gene to the EDL muscle by AAV-5 viral vector. More than 50% of the myofibers were consistently transduced in the BL10 EDL muscle. In contrast to the uniform structure in BL10 skeletal muscle, mdx skeletal muscle is severely damaged by repeated cycles of degeneration and regeneration, inflammation, and fibrosis. Surprisingly, these morphological alterations did not affect AAV transduction in the EDL muscle of young mdx mice (less than 7 weeks of age). In these mice, we achieved a transduction efficiency similar to that of the BL10 mice. However, the transduction efficiency was reduced in 9-month-old mdx mice. The exact mechanism(s) for this decrease is not clear, but may relate to the increased fibrosis, muscle fiber deformation, and/or fiber type shifting in older mdx muscle [31-37]. Alternatively, the levels of AAV-5 receptor (α-2,3-linked sialic acid) and/or coreceptor (platelet-derived growth factor receptor) in aged muscle may have decreased and therefore reduced transduction. Additional studies will be needed to explore our observation further.
We have recently developed a series of microdystrophin cDNAs for DMD gene therapy. Among these microgenes, the ΔR4/ΔC gene demonstrated superior properties as a potential therapeutic gene. Transgenic overexpression of the C-terminal-inclusive DR4-R23 gene protected TA muscles from contraction-induced injury [17]. AAV-mediated ΔR4/ΔC gene expression also halted progression of dystrophic pathology and maintained sarcolemma integrity [17]. To study further the therapeutic relevance of the ΔR4/ΔC microgene, we delivered AV.ΔR4/ΔC virus to the adult EDL muscle. Our goal was to determine whether AAV-mediated microdystrophin expression was sufficient to restore muscle function and prevent contraction-induced injury.
AAV has been used extensively to deliver reporter and/ or therapeutic genes to mouse skeletal muscle. However, no study has examined the functional consequence of AAV transduction itself in muscle. It is possible that vector administration alone may impair muscle contractility. Alternatively, viral transduction may lead to certain unexpected beneficial effects. For example, adenoviral vectors that carry reporter genes have been shown to alleviate dystrophic pathology by immune-mediated utrophin up-regulation [38]. To exclude the potential compounding influence from AAV infection itself, we first compared specific force and eccentric contraction response profiles between the saline-injected and the AV.RSV.AP-treated EDL muscles. Our results suggest that delivering AAV to the EDL muscle does not affect muscle contractility.
To determine the therapeutic efficacy of AAV-mediated ΔR4/ΔC expression in young (7-week-old) and adult (9-month-old) EDL muscles, we first examined central nucleation in transduced myofibers. Consistent with our previous report [17], administration of AV.ΔR4/ΔC at a younger age was more effective in reducing central nucleation. However, in older mdx myofibers, centrally located nuclei were more resistant to peripheral mobilization following AAV-mediated ΔR4/ΔC expression. Centrally positioned nuclei are indicative of myofiber regeneration. Our results suggest that early intervention may be necessary to stop the pathological degeneration–regeneration process in mdx muscle. Alternatively, young muscle may carry more molecular cues needed for peripheral relocation of nuclei.
The ultimate goal of DMD gene therapy is to enhance force production in dystrophic muscle. In this study, we examined whether AAV-mediated ΔR4/ΔC expression could protect muscle from contraction-induced injury. Clinical studies suggest that a 50% mosaic expression of the full-length dystrophin gene is sufficient to prevent severe skeletal muscle weakness [39,40]. However, mosaic expression of the full-length dystrophin at 30% level in transgenic mdx mice only partially corrected histopathology [13]. Transgenic mosaic expression of a partial C-terminal-deleted dystrophin (Δexon 71–74) in 20% of the mdx diaphragm myofibers resulted in no visible morphology improvement at all. Surprisingly, a slightly higher than 50% expression of the ΔR4/ΔC microgene in 7-week-old mdx skeletal muscle resulted in significant improvement in tetanic force generation following eccentric contraction injury. In our previous studies, transgenic expression of a similar but slightly larger C-terminal-inclusive microdystrophin gene offered similar protection in skeletal muscle [17]. However, in the case of transgenic mice, functional deficiency was prevented, rather than treated, by persistent expression in every myofiber starting at the embryonic stage. Functional improvement from AAV-mediated ΔR4/ΔC expression extends our earlier finding and suggests that the ΔR4/ΔC microgene is not only capable of preventing muscle damage, more importantly, it is also capable of treating the existing pathology. Furthermore, therapeutic effect can be achieved without correcting every single myo-fiber. In support of our observation, Xiao and colleagues have recently demonstrated that a 30–60% expression of a different isoform of microdystrophin in mdx TA muscle can enhance resistance to contraction-induced injury in 2-month-old mice [41]. Taken together, these results have clearly demonstrated the therapeutic potential of the microdystrophin gene in DMD gene therapy.
In addition to young mdx mice, we also explored the therapeutic efficacy of the ΔR4/ΔC gene in 9-month-old mdx mice. In the absence of gene therapy, the older mdx EDL muscle was much more vulnerable to contraction-induced injury (Figs. 4 and 5). Following ΔR4/ΔC expression, we observed only limited protection against contraction-induced damage in older mice. Since transduction efficiency was lower in the older mdx EDL muscle, the marginal effect could be due to inefficient gene transfer. On the other hand, since dystrophic pathology was much more severe in older mdx muscles, these muscles might be less responsive to gene therapy. Dystrophic pathology in older mdx muscle is closer to that in human patients [35,36]. Our results in older muscle highlight the challenge of microdystrophin-mediated therapy. We envision that the C-terminal-truncated microdystrophin may help to slow down the ongoing dystrophic process, but it may not lead to a complete function recovery.
A potential application of AAV-mediated microdystrophin gene therapy is to improve muscle function in BMD patients. In these patients, either the total amount of dystrophin is reduced or a truncated, but partially functional, dystrophin isoform is expressed. Furthermore, gene therapy of DMD may require repeated gene transfer to achieve life-long correction. Under these circumstances, we may likely need to deliver the ΔR4/ΔC microgene to muscles that are already expressing certain levels of dystrophin. It is therefore important to determine whether the ΔR4/ΔC expression interferes with the existing dystrophin. To address this issue, we intentionally delivered AV.ΔR4/ΔC virus to the EDL muscle of normal mice. In these studies, we observed coexpression of both the endogenous full-length dystrophin and the ΔR4/ΔC microdystrophin in the same myofiber. Importantly, such coexpression had no damaging effect on muscle morphology or contractility.
The development of novel AAV microdystrophin vector brings an exciting new approach to DMD gene therapy. In this study, we have systematically evaluated the physiological effect of an AAV microdystrophin vector in the mdx mouse model for DMD. Despite a less than optimal correction in older mdx muscle, the microdystrophin gene protected young mdx muscle from contraction-induced damage. These results support further experimentation with AAV microdystrophin in a large animal model of DMD and, we hope, its eventual application in human patients.
Materials and Methods
Recombinant AAV Production
The cis plasmids, pcisRSVAP and pcisCMVΔR4/ΔC, for rAAV production have been previously described [42]. pcisRSVAP was used to generate AV.RSV.AP, a virus expressing the heat-resistant AP. pcisCMVΔR4/ΔC was used to generate AV.ΔR4/ΔC, a virus carrying the C-terminal-truncated microdystrophin gene [17]. The ΔR4/ΔC microgene contains the N-terminal actin binding domain; the first three and the last spectrin-like repeats; the hinges 1, 2, and 4; and the CR domain [17]. The AAV-5 capsid-pseudotyped viral stocks were prepared according to our published protocols [43]. Briefly, 60% confluent 293 cells were cotransfected with cis plasmid, AAV-2 Rep plasmid, AAV-5 helper plasmid, and adenoviral helper plasmid at a ratio of 1:1:1:3. Crude viral lysate was harvested at 60 h posttransfection and purified through three rounds of CsCl isopycnic ultracentrifugation. The viral titer determination and quality control were carried out as described previously [43].
Recombinant AAV Delivery
All animal experiments were approved by the Animal Care and Use Committee at the University of Missouri and were in accordance with NIH guidelines. The mdx mouse and its parental strain BL10 were originally purchased from The Jackson Laboratory (Bar Harbor, ME, USA). The colonies were subsequently established by in-house breeding at the University of Missouri and mice (including experimental mice and breeding pairs) were housed in a specific-pathogen-free animal facility at 20–23°C with a 12–h light–12-h dark cycle. To minimize interanimal differences, all comparisons were made between the left and the right EDL muscle of the same mouse.
To evaluate gene transfer in the EDL muscle by direct injection, we first anesthetized mice with an intraperitoneal (ip) injection of an anesthetic cocktail (25 mg/ml ketamine, 2.5 mg/ml xylazine, 0.5 mg/ml acepromazine) at 4 μl/g body weight. A 0.5 × 5-mm-long incision was then made along the longitudinal axis in the lateral surface of the distal hind limb. After the EDL tendon was separated from the TA tendon, the TA muscle was gently pulled aside with a Guthrie double hook retractor (Fine Science Tools, Inc., Foster City, CA, USA, Catalog No. 17021-13) and the entire EDL muscle was exposed for injection. Two injections were performed from the proximal and the distal ends, respectively, with a custom-tailored 33-gauge gas-tight Hamilton syringe (Hamilton Co., Reno, NV, USA, Catalog No. 7654-01 for syringe, 7803-05 for needle). In each injection, 5 μl rAAV virus (5 × 109 genome particles in Hepes-buffered saline) was injected directly into the muscle body. Following injection, the wound was closed with a 5-O Sofsilk suture (Auto Suture Co., Norwalk, CT, USA, Catalog No. VS-870) by three or four interrupted stitches.
To prevent gnawing on the suture and improve wound healing, we applied a polyethylene mouse Elizabethan collar (E-collar; Harvard Apparatus, Catalog No. NP 72-0056) around the mouse's neck. This device blocked the head from access to the rest of the body, but still enabled eating, drinking, and comfortable movement. To alleviate pain and discomfort at the operation site, a nonsteroid analgesic drug, banamine (also called Flunixin Meglumine; Schering–Plough Animal Health Corp., Union, NJ, USA, Catalog No. 101-479), was injected subcutaneously at a dose of 3 mg/kg for 3 days after surgery. No adverse reaction to banamine was observed at this dose.
Muscle Contractile Properties
Muscle preparation
Mice were anesthetized ip as noted above. The EDL muscle was carefully dissected and immediately mounted vertically in a jacketed organ bath. The proximal tendon was secured in a stationary clamp at the base of the bath with a 5-O suture (twisted cotton, Sutupak SC-72H; Ethicon). The distal tendon was connected via a 5-O suture to either a 300B or a 305B dual-mode servomotor transducer (Aurora Scientific, Inc., Aurora, ON, Canada) via a custom-made noncompliant fine-wire S hook (Small Parts, Inc., Miami Lakes, FL, USA). The servomotors provided control of force and positioning of the motor arm so that both dynamic and isometric muscle contractions could be elicited. Muscles were incubated with oxygenated Ringer's solution containing (in mM) 137 NaCl, 4.83 KCl, 2 CaCl2, 24 NaHCO3, 1.2 MgSO4, 1.2 NaH2PO4, 10 glucose, pH 7.5. The muscle bath was maintained at 30°C by a Lauda RM6-B circulating water bath (Brinkmann, Westbury, NY, USA). This temperature has been shown to yield the maximal and most stable isometric force in the EDL muscle [44]. Based on our preliminary studies, identical performances for twitch force, isometric force–frequency responses, and eccentric contraction profiles were obtained from either the 300B system or the 305B system when the same strain, age, sex, and body-weight-matched EDL muscles were compared.
Force measurements
Control of and data acquisition from the servomotors were conducted on a Pentium III PC running the Lab-View-based DMC program (Dynamic Muscle Control and Data Acquisition, Version 3.12; Aurora Scientific, Inc.). The interface between the motors and the computer included a multifunctional digital acquisition card (DAQ PCI-6036E; National Instruments, Austin, TX, USA) and an Aurora 604B dual-system analog/digital interface (Aurora Scientific, Inc.). Length and force data were analyzed by the LabView-based DMA program (Dynamic Muscle Data Analysis, Version 3.12; Aurora Scientific, Inc.).
The muscle was initially set at a resting tension of 1 g for 10 min without electrical stimulation. For all experiments, electrical stimuli of 600 mA (20 V) with a 200-μs pulse duration (701A Stimulator; Aurora Scientific, Inc.) were delivered by a pair of platinum electrodes closely flanking either side of the muscle (∼0.5 cm). Lo was determined as the muscle length at which the maximal twitch force (∼50–70 mN) was elicited. In our system, the resting tension at Lo was typically 1 g. After a 3-min rest, the muscle was subjected to a series of three isometric tetanic stimulations at 150 Hz, each for 500 ms with a 1-min rest between each stimulation. These preliminary tetanic contractions stabilized the muscle for subsequent measurements [45]. Resting muscle tension was adjusted to 1 g as required.
After another 5-min rest, the muscle was subjected to a force–frequency measurement protocol. Briefly, the muscle was stimulated for 500 ms at 50, 80, 120, and 150 Hz, respectively, with 1-min rest between each contraction. At the end of the force–frequency protocol, the muscle length at Lo was measured with an electronic digital caliper (±0.01 mm; Control Co., Friendswood, TX, USA). The optimal fiber length (Lf) was determined by multiplying Lo by an Lf/Lo ratio of 0.44 [31,46]. The maximal force response at each stimulation frequency (absolute tetanic force) was normalized to muscle CSA (kN/m2). Muscle CSA was calculated according to the following equation: CSA = (muscle mass, in g)/[(optimal fiber length, in cm) × (muscle density, in g/cm3)]. A muscle density of 1.06 g/cm3 was used [47].
Eccentric contraction protocol
After the determination of the force–frequency relation, the muscle was rested for 10 min and then was subjected to a modified eccentric contraction protocol [26,45]. Briefly, the muscle was stimulated at 150 Hz for 700 ms. After 500 ms stimulation, the muscle was lengthened by 10% Lo at 0.5 Lo/s for 200 ms (Fig. 2A). When stimulation ended, the muscle length was reset to Lo at −0.5 Lo/s (Fig. 2A). This stimulation–stretch cycle was repeated every 2 min for a total of 10 cycles. The maximal isometric tetanic force developed during the first 500 ms of stimulation of the first cycle was designated 100%. This force was identical to the tetanic force observed at 150 Hz during the force–frequency determination. The percentage of tetanic force loss at each cycle was determined according to the formula force drop % = ( F1 − Fn)/F1, where F1 was the tetanic force obtained during the first cycle and Fn represented the tetanic force obtained during the nth cycle.
Morphology Studies
Alkaline phosphatase expression evaluation
A previously described histochemical staining protocol was used to detect AAV-mediated heat-resistant AP expression in the EDL muscle [42]. Briefly, endogenous heat-labile AP was first inactivated by incubating at 65°C for 30 min. Staining was carried out at 37°C for 10 min in a solution containing 75 mg/ml nitroblue tetrazolium chloride, 50 mg/ml 5-bromo-4-chloro-3-indolyl phosphate p-toluidine salt, 0.24 mg/ml levamisole, 100 mM Tris (pH 9.5), 50 mM MgCl2, and 100 mM NaCl.
Indirect immunofluorescence staining for dystrophin
The ΔR4/ΔC microgene was derived from the human dystrophin gene, and its expression was evaluated with the human dystrophin N-terminal-specific monoclonal antibody Dys-3 (1:10 dilution; Novocastra, Newcastle, UK) according to our previously published protocol [42]. The revertant dystrophin-positive myofibers in mdx mice and the full-length dystrophin expression in BL10 mice were detected with a monoclonal anti-dystrophin C-terminal antibody, Dys-2 (1:30 dilution; Novocastra, Newcastle, UK) as previously described [48]. To visualize nuclei and reduce photobleaching, slides were mounted using the SlowFade Light Antifade Kit with DAPI (Molecular Probes, Eugene, OR, USA, Catalog No. S-24636). Photomicrographs were taken with a Qimage Retiga 1300 camera using a Nikon E800 fluorescence microscope.
Central nucleation quantification
The percentage of centrally nucleated myofibers in the untreated mdx mouse EDL muscle was determined by manually counting the total number of myofibers and the total number of myofibers carrying centrally located nuclei in an 8-μm HE-stained section with an electronic colony counter. Equal or more than four representative muscle sections were quantified for each muscle sample. The percentage of central nucleation was calculated with the formula % central nucleation = (total number of myofibers carrying centrally located nuclei)/(total number of myofibers). The percentage of centrally nucleated myofibers in AV.ΔR4/ΔC-infected mdx EDL muscles was determined only in the transduced myofibers (Dys-3-positive myofibers). In this case, % central nucleation = (total number of ΔR4/ΔC-positive myofibers that carry centrally located nuclei)/(total number of ΔR4/ΔC-positive myofibers).
Statistical Analysis
Data are expressed as means ± SEM (or as indicated). A one-way ANOVA was used to determine whether there was statistical difference among groups. In the case of significant statistical difference, a post hoc Tukey analysis was used to determine further differences between means. In studies that involved only a comparison between the left and the right EDL muscle, a paired t test was used to determine statistical significance. Significance level was set at 0.05.
Acknowledgments
We thank Dr. Wei Ding, Mr. Zheng Sun, Ms. Tara Greer, Dr. Gordon Lynch, and Dr. Paul Gregorevic for their help in developing the in vitro muscle contraction assay. We thank Dr. Scott Korte for his help in developing the EDL muscle gene delivery technique. This work was supported by grants from the National Institutes of Health (AR-49419, D.D.), the Muscular Dystrophy Association (D.D. and J.C.), the University of Missouri Research Board (D.D.), and the ChildrenTs Miracle Network (D.D.).
References
- 1.Emery AEH, Muntoni F. Duchenne Muscular Dystrophy. Oxford Univ. Press; Oxford/New York: 2003. [Google Scholar]
- 2.Bushby KM, Hill A, Steele JG. Failure of early diagnosis in symptomatic Duchenne muscular dystrophy. Lancet. 1999;353:557–558. doi: 10.1016/s0140-6736(98)05279-9. [DOI] [PubMed] [Google Scholar]
- 3.Eagle M, et al. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002;12:926–929. doi: 10.1016/s0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- 4.Hoffman EP, Brown RH, Jr., Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 5.Koenig M, et al. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50:509–517. doi: 10.1016/0092-8674(87)90504-6. [DOI] [PubMed] [Google Scholar]
- 6.Chamberlain JS, Gibbs RA, Ranier JE, Nguyen PN, Caskey CT. Deletion screening of the Duchenne muscular dystrophy locus via multiplex DNA amplification. Nucleic Acids Res. 1988;16:11141–11156. doi: 10.1093/nar/16.23.11141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain JS. Gene therapy of muscular dystrophy. Hum. Mol. Genet. 2002;11:2355–2362. doi: 10.1093/hmg/11.20.2355. [DOI] [PubMed] [Google Scholar]
- 8.Love DR, et al. Characterization of deletions in the dystrophin gene giving mild phenotypes. Am. J. Med. Genet. 1990;37:136–142. doi: 10.1002/ajmg.1320370132. [DOI] [PubMed] [Google Scholar]
- 9.England SB, et al. Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature. 1990;343:180–182. doi: 10.1038/343180a0. [DOI] [PubMed] [Google Scholar]
- 10.Bredt DS. Knocking signalling out of the dystrophin complex. Nat. Cell Biol. 1999;1:E89–91. doi: 10.1038/12085. [DOI] [PubMed] [Google Scholar]
- 11.Rando TA. The dystrophin–glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- 12.Rafael JA, et al. Prevention of dystrophic pathology in mdx mice by a truncated dystrophin isoform. Hum. Mol. Genet. 1994;3:1725–1733. doi: 10.1093/hmg/3.10.1725. [DOI] [PubMed] [Google Scholar]
- 13.Phelps SF, et al. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum. Mol. Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- 14.Rafael JA, et al. Forced expression of dystrophin deletion constructs reveals structure–function correlations. J. Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corrado K, et al. Transgenic mdx mice expressing dystrophin with a deletion in the actin-binding domain display a “mild Becker” phenotype. J. Cell Biol. 1996;134:873–884. doi: 10.1083/jcb.134.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford GE, et al. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J. Cell Biol. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harper SQ, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 18.Warner LE, et al. Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin–glycoprotein complex and partially prevents dystrophy. Hum. Mol. Genet. 2002;11:1095–1105. doi: 10.1093/hmg/11.9.1095. [DOI] [PubMed] [Google Scholar]
- 19.Wang B, Li J, Xiao X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA. 2000;97:13714–13719. doi: 10.1073/pnas.240335297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Scott J, et al. Viral vectors for gene transfer of micro-, mini-, or full-length dystrophin. Neuromuscul. Disord. 2002;12(Suppl):S23. doi: 10.1016/s0960-8966(02)00078-0. [DOI] [PubMed] [Google Scholar]
- 21.Fabb SA, Wells DJ, Serpente P, Dickson G. Adeno-associated virus vector gene transfer and sarcolemmal expression of a 144 kDa micro-dystrophin effectively restores the dystrophin-associated protein complex and inhibits myofibre degeneration in nude/mdx mice. Hum. Mol. Genet. 2002;11:733–741. doi: 10.1093/hmg/11.7.733. [DOI] [PubMed] [Google Scholar]
- 22.Sakamoto M, et al. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem. Biophys. Res. Commun. 2002;293:1265–1272. doi: 10.1016/S0006-291X(02)00362-5. [DOI] [PubMed] [Google Scholar]
- 23.Gillis JM, Deconinck N. The physiological evaluation of gene therapies of dystrophin-deficient muscles. Adv. Exp. Med. Biol. 1998;453:411–416. doi: 10.1007/978-1-4684-6039-1_45. [DOI] [PubMed] [Google Scholar]
- 24.Watchko JF, O'Day TL, Hoffman EP. Functional characteristics of dystrophic skeletal muscle: insights from animal models. J. Appl. Physiol. 2002;93:407–417. doi: 10.1152/japplphysiol.01242.2001. [DOI] [PubMed] [Google Scholar]
- 25.Warren GL, Lowe DA, Armstrong RB. Measurement tools used in the study of eccentric contraction-induced injury. Sports Med. 1999;27:43–59. doi: 10.2165/00007256-199927010-00004. [DOI] [PubMed] [Google Scholar]
- 26.Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moens P, Baatsen PH, Marechal G. Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil. 1993;14:446–451. doi: 10.1007/BF00121296. [DOI] [PubMed] [Google Scholar]
- 28.Williams DA, Head SI, Lynch GS, Stephenson DG. Contractile properties of skinned muscle fibres from young and adult normal and dystrophic (mdx) mice. J. Physiol. 1993;460:51–67. doi: 10.1113/jphysiol.1993.sp019458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pastoret C, Sebille A. Time course study of the isometric contractile properties of mdx mouse striated muscles. J. Muscle Res. Cell Motil. 1993;14:423–431. doi: 10.1007/BF00121294. [DOI] [PubMed] [Google Scholar]
- 30.Lynch GS, Hinkle RT, Faulkner JA. Power output of fast and slow skeletal muscles of mdx (dystrophic) and control mice after clenbuterol treatment. Exp. Physiol. 2000;85:295–299. [PubMed] [Google Scholar]
- 31.Lynch GS, Hinkle RT, Chamberlain JS, Brooks SV, Faulkner JA. Force and power output of fast and slow skeletal muscles from mdx mice 6–28 months old. J. Physiol. 2001;535:591–600. doi: 10.1111/j.1469-7793.2001.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Webster C, Silberstein L, Hays AP, Blau HM. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell. 1988;52:503–513. doi: 10.1016/0092-8674(88)90463-1. [DOI] [PubMed] [Google Scholar]
- 33.Head SI, Williams DA, Stephenson DG. Abnormalities in structure and function of limb skeletal muscle fibres of dystrophic mdx mice. Proc. R. Soc. London B Biol. Sci. 1992;248:163–169. doi: 10.1098/rspb.1992.0058. [DOI] [PubMed] [Google Scholar]
- 34.Petrof BJ, et al. Adaptations in myosin heavy chain expression and contractile function in dystrophic mouse diaphragm. Am. J. Physiol. 1993;265:C834–841. doi: 10.1152/ajpcell.1993.265.3.C834. [DOI] [PubMed] [Google Scholar]
- 35.Lefaucheur JP, Pastoret C, Sebille A. Phenotype of dystrophinopathy in old mdx mice. Anat. Rec. 1995;242:70–76. doi: 10.1002/ar.1092420109. [DOI] [PubMed] [Google Scholar]
- 36.Pastoret C, Sebille A. mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci. 1995;129:97–105. doi: 10.1016/0022-510x(94)00276-t. [DOI] [PubMed] [Google Scholar]
- 37.Dellorusso C, Crawford RW, Chamberlain JS, Brooks SV. Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J. Muscle Res. Cell Motil. 2001;22:467–475. doi: 10.1023/a:1014587918367. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto K, et al. Immune response to adenovirus-delivered antigens upregulates utrophin and results in mitigation of muscle pathology in mdx mice. Hum. Gene Ther. 2000;11:669–680. doi: 10.1089/10430340050015572. [DOI] [PubMed] [Google Scholar]
- 39.Hoffman EP, et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne's or Becker's muscular dystrophy. N. Engl. J. Med. 1988;318:1363–1368. doi: 10.1056/NEJM198805263182104. [DOI] [PubMed] [Google Scholar]
- 40.Arahata K, et al. Mosaic expression of dystrophin in symptomatic carriers of Duchenne's muscular dystrophy. N. Engl. J. Med. 1989;320:138–142. doi: 10.1056/NEJM198901193200302. [DOI] [PubMed] [Google Scholar]
- 41.Watchko J, et al. Adeno-associated virus vector-mediated minidystrophin gene therapy improves dystrophic muscle contractile function in mdx mice. Hum. Gene Ther. 2002;13:1451–1460. doi: 10.1089/10430340260185085. [DOI] [PubMed] [Google Scholar]
- 42.Yue Y, et al. Microdystrophin gene therapy of cardiomyopathy restores dystrophin–glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duan D, Yan Z, Yue Y, Ding W, Engelhardt JF. Enhancement of muscle gene delivery with pseudotyped AAV-5 correlates with myoblast differentiation. J. Virol. 2001;75:7662–7671. doi: 10.1128/JVI.75.16.7662-7671.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Segal SS, Faulkner JA. Temperature-dependent physiological stability of rat skeletal muscle in vitro. Am. J. Physiol. 1985;248:C265–270. doi: 10.1152/ajpcell.1985.248.3.C265. [DOI] [PubMed] [Google Scholar]
- 45.Grange RW, Gainer TG, Marschner KM, Talmadge RJ, Stull JT. Fast-twitch skeletal muscles of dystrophic mouse pups are resistant to injury from acute mechanical stress. Am. J. Physiol. Cell Physiol. 2002;283:C1090–1101. doi: 10.1152/ajpcell.00450.2001. [DOI] [PubMed] [Google Scholar]
- 46.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J. Physiol. 1988;404:71–82. doi: 10.1113/jphysiol.1988.sp017279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mendez J, Keys A. Density and composition of mammalian muscle. Metabolism. 1960;9:184–188. [Google Scholar]
- 48.Yue Y, Skimming JW, Liu M, Strawn T, Duan D. Full-length dystrophin expression in half of the heart cells ameliorates beta-isoproterenol-induced cardiomyopathy in mdx mice. Hum. Mol. Genet. 2004;13:1669–1675. doi: 10.1093/hmg/ddh174. [DOI] [PMC free article] [PubMed] [Google Scholar]