

Abstract
The last decade has evidenced unprecedented progress in gene therapy of Duchenne and Becker muscular dystrophy (DMD and BMD) skeletal muscle disease. Cardiomyopathy is a leading cause of morbidity and mortality in both patients and carriers of DMD, BMD and X-linked dilated cardiomyopathy. However, there is little advance in heart gene therapy. The gene, the vector, vector delivery, the target tissue and animal models are five fundamental components in developing an effective gene therapy. Intensive effort has been made in optimizing gene transfer vectors and methods. Systemic and/or local delivery of recombinant adeno-associated viral vector have resulted in widespread transduction in the rodent heart. The current challenge is to define other parameters that are essential for a successful gene therapy such as the best candidate gene(s), the optimal expression level and the target tissue. This review focuses on these long-ignored aspects and points out future research directions. In particular, we need to address whether all or only some of the recently developed mini- and microgenes are protective in the heart, whether partial correction can lead to whole heart function improvement, whether over-expression is hazardous and whether correcting skeletal muscle disease can slow down or stop the progression of cardiomyopathy. Discussion is also made on whether the current mouse models can meet these research needs.
INTRODUCTION
Dystrophin-deficient cardiomyopathy refers to the cardiac manifestation of three closely related diseases including Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD) and X-linked dilated cardiomyopathy (XLDC). The common genetic defect in these diseases is dystrophin gene mutation. The 427 kD muscle dystrophin protein has four functional domains including the N-terminal, central rod, cysteine-rich (CR) and C-terminal domains (Fig. 1). Together with dystroglycan and sarcoglycan, dystrophin and its partners form the dystrophin-associated glycoprotein complex (DGC) (1). The DGC discharges mechanical stress to the extra-cellular matrix and therefore stabilizes the sarcolemma during muscle contraction [reviewed in (2)]. The DGC also acts as a critical hub in several signal transduction pathways [reviewed in (3)].
Figure 1.
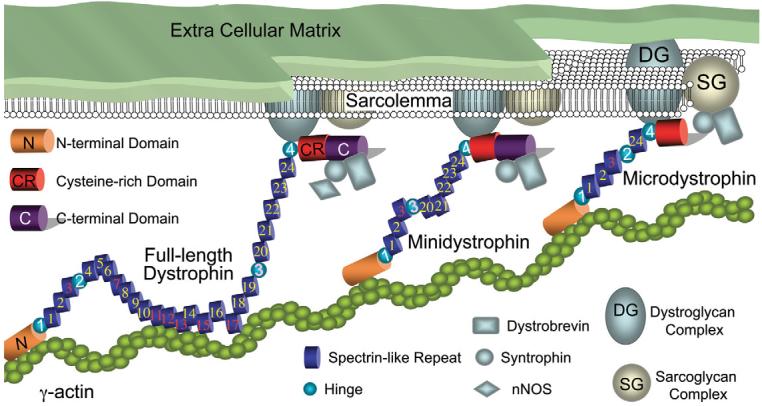
Schematic outline of full-length dystrophin, minidystrophin and microdystrophin and their interaction with other cellular proteins. Spectrin-like repeats are numbered from 1 to 24 (positively charged repeats are in red color, other repeats are in yellow color). Proline-rich hinges are numbered from 1 to 4. Hinge 3 is in pink color to indicate that it can be cleaved by viral protease. Hinge 2 to repeat 19 are deleted in minidystrophin. Repeat 4–23 and the C-terminal domain are deleted in microdystrophin. Not drawn to scale.
DMD results from a complete loss of dystrophin. BMD is due to reduced protein expression and/or expression of a truncated but partially functional protein (4,5). XLDC is caused by selective loss of dystrophin in the heart (6,7). One-third of DMD patients show signs of cardiac dysfunction by mid-teenage and virtually all DMD patients develop cardiac damage by the end of their life. DMD patients (10–40%) eventually die from heart failure (8,9). It is estimated that cardiomyopathy can shorten the life expectancy of DMD patients by at least 2 years (10). Cardiac involvement is more prevalent in BMD patients. By age 40, more than 90% of BMD patients display some signs of heart disease (11-14). The high incidence in BMD is considered a natural consequence of a longer lifespan in these patients. It may have allowed more time for clinical signs to develop (12,13). In addition to patients, female carriers are also at high risk. Nearly half of them have electrocardiography (ECG) changes (15) and 7–18% suffer from heart function defect (11,14,16). Besides heart transplantation (17-20), symptom-relieving medicines (such as angiotensin-converting enzyme-inhibitors and β-blockers) are the only available treatment now (9). Currently, there is no cure.
HEART AND SKELETAL MUSCLE HAVE DIFFERENT NEEDS FOR DYSTROPHIN
To develop an effective gene therapy for dystrophin-null cardiomyopathy, one should always bear in mind that the heart is different from skeletal muscle. First, the anatomic structure of the contractile unit is different between cardiac and skeletal muscle. In contrast to the multinucleated syncytial myotube in skeletal muscle, cardiomyocytes are mainly single nucleated and are separated from each other by intercalated discs. Second, there is subtle difference in force development. In the absence of dystrophin, cardiomyocytes develop a higher than normal stretch-tension (21), but skeletal muscle develops an equal or lower than normal stretch-tension (22,23). Third, dystrophin shows distinctive expression patterns in two tissues. In skeletal muscle, dystrophin is concentrated at the costamere (24-27). Costameres are vinculin-rich subsarcolemmal rib-like structures. They anchor the Z-line to the sarcolemma and thus provide a mechanical linkage between contracting myofibrils and the extra-cellular matrix [reviewed in (27)]. Costameres are found in both skeletal and cardiac muscle (28,29). Interestingly, in the rodent heart, dystrophin is uniformly distributed along the sarcolemma and there is no preference for the costamere (30). [Although in the human heart dystrophin partially co-localizes with the costamere (31).] Another striking finding is the presence of dystrophin in the T-tubule membrane in the heart, but not in skeletal muscle (30-33). T-tubules are involved in excitation–contraction coupling but not in force transmission. The association of dystrophin in cardiac T-tubules suggests that dystrophin may play additional roles in the heart. Taken together, these subtle, but fundamental differences in dystrophin distribution emphasize the necessity of developing heart-specific gene therapy.
CHALLENGES IN HEART GENE THERAPY
The current success in skeletal muscle gene therapy is built on accumulative efforts from years' of transgenic studies (34-44). These experiments demonstrate that a level of 20% normal dystrophin is sufficient to maintain muscle function (36,37) and a 50-fold over-expression is not toxic (45). Unfortunately, similar studies have not been performed in the heart. A top priority in cardiac gene therapy is to address these fundamental questions.
Which gene(s) to use?
Theoretically, one would consider the endogenous full-length dystrophin cDNA as the ideal therapeutic gene. However, there are two potential problems. First, the full-length dystrophin cDNA is beyond the packaging capacity of most available viral vectors. Second, the wild-type dystrophin gene may not be the best gene in preventing/treating cardiomyopathy. It has been shown that the hinge 3 region is a cleavage site for coxsackievirus protease (46,47). Although an epidemic linkage remains to be established between coxsackievirus infection and heart failure in DMD/BMD/XLDC patients, Xiong et al. (48) have clearly shown that dystrophin deficiency is a predisposing factor to viral myocarditis and heart failure in mice. There is also anecdotal case report describing heart failure in BMD patient after a flu-like illness (49). It is possible that a modified gene may be therapeutically advantageous to the wild-type dystrophin gene. In support of this notion, a recent study suggests that mutations in the hinge 3 region may protect DMD/BMD patients from dilated cardiomyopathy (50).
A 6 kb minigene and a 3.8 kb microgene are the two most promising candidate genes for skeletal muscle gene therapy. The minigene is identical to the full-length dystrophin cDNA except for a perfectly phased deletion in the rod domain (from hinge 2 to spectrin-like repeat 19). As this gene provides full protection in skeletal muscle, it may also yield a good protection in the heart. The only limitations are: (i) it requires two AAV virions for delivery (41,51); (ii) it carries the hinge 3 region and may be susceptible to coxsackieviral protease cut. The microgene does not carry the C-terminal domain and it also has a much larger deletion in the rod domain (from repeat 4 to 23). The microgene can be packaged in a single AAV virion but only provides basic protection in skeletal muscle (41,52,53). In a preliminary study, we found that AAV-mediated microgene expression can restore the DGC in the heart and protect the heart from stress-induced sarcolemmal injury (54) (Fig. 2). We also found a non-discriminative expression of microdystrophin along the entire sarcolemma in cardiomyocytes but a punctate expression in skeletal muscle myofibers (Fig. 2). This distribution profile is similar to the full-length protein (27,30,31). Despite the encouraging results, the inherent limitation in the microgene itself may prevent it from offering a full protection. In particular, comparing with other dystrophin isoforms, the microgene carries the least amount of genetic information. Furthermore, the consequence of the C-terminal domain deletion remains to be explored in the heart. Taking together, comprehensive studies are needed to determine whether the abbreviated mini- and microgenes can reduce heart pathology and maintain heart function.
Figure 2.
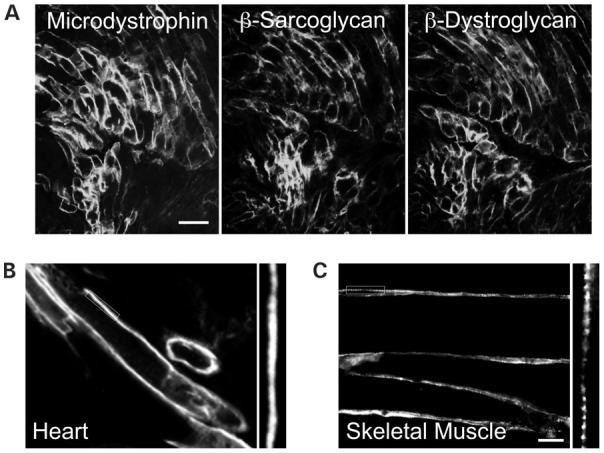
Microdystrophin restores the DGC and displays a no-interrupted expression pattern along the sarcolemma in the mdx heart. Microdystrophin was delivered to the neonatal mdx heart by AAV vector. Transgene expression was examined when mice were 10-month-old. (A) Serial sections showing co-localization of microdystrophin with two other major components of the DGC, β-sarcoglycan and β-dystroglycan. Scale bar: 50 μm. Adapted from Yue et al. (2003) Circulation, 108, 1626. (B) Continuous labeling of microdystrophin along the sarcolemma in the heart. (C) To study microdystrophin expression in skeletal muscle, an AAV virus carrying the microgene was injected into the limb muscle in 2-month-old mdx mice. Immunofluorescence staining was performed 3 months later. Microdystrophin displays a punctate staining pattern in skeletal muscle. Scale bar: 20 μm (this bar also applies to B). In B and C, high magnification photomicrographs of the boxed areas are highlighted in the sides, respectively.
How much dystrophin is enough?
Despite rapid progress in gene delivery technique, it is unrealistic to expect a 100% transduction of every single cell in the heart. New methods have been developed to deliver transgene to more than 50% heart cells in several rodent models with AAV vectors (55-58). The question is whether this is sufficient to protect the heart. To address this issue, we have produced genetically defined heterozygous female mice by crossing mdx and BL10 (59). Because of random X-chromosome inactivation, only half cardiomyocytes express dystrophin in heterozygous mice. In the hearts of heterozygous mice, some areas have more dystrophin positive cells, whereas other areas only have a few positive cells (Fig. 3). Heart function was examined in 3--month-old mice after β-isoproterenol challenge. Despite a partial dystrophin expression, stress-induced cardiomyopathy was significantly attenuated (Fig. 3). The most impressive correction was seen in the hemodynamic assay. In all parameters we examined, we observed a 100% protection (59). Although this is an encouraging finding, the lack of clinical heart disease in young mdx mice has limited the significance of the study. It remains to be determined whether dystrophin expression in half cardiomyocytes can protect the heart in symptomatic animals such as aged mdx mice.
Figure 3.
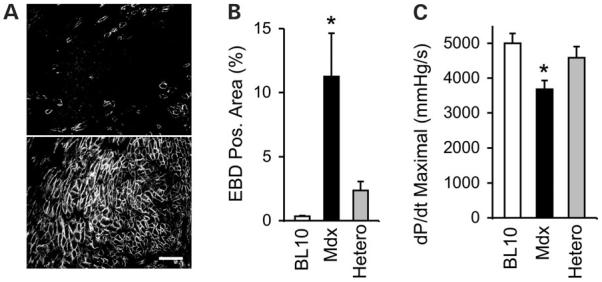
Mosaic dystrophin expression in heterozygous mdx mice protects the heart from stress-induced cardiomyopathy. (A) Dystrophin expression in the heart of heterozygous female mice. Top and bottom panels depict regions of low and high dystrophin expression, respectively. Scale bar: 100 μm. (B) Quantification of sarcolemma damage (EBD positive area) in the hearts of different mouse strains following β-isoproterenol challenge. (C) Correction of hemodynamic defect in heterozygous mice. Bar graph shows the dP/dt maximal. Asterisk denotes results in mdx were significantly different from these in BL10 or heterozygous mice. EBD, Evans blue dye; Hetero, heterozygous mice. Adapted from Yue et al. (2004) Hum. Mol. Genet., 13, 1669.
A more relevant issue to clinical gene therapy is to determine the therapeutic threshold for the mini- and microgenes. These abbreviated synthetic genes may be less competent than the full-length gene. Furthermore, achieving ≥50% transduction efficiency in the human hearts is clearly a daunting task. Cardiac transgenic mice carrying mosaic expression of the mini- and microgenes and/or viral delivery will be needed to address this important question.
Will too much dystrophin be toxic?
An important finding in skeletal muscle transgenic study is that dystrophin over-expression is not toxic (45). However, in the reported study, dystrophin was only moderately over-expressed in the heart (45). Although the heart seems to be able to accommodate a limited range of transgene expression, numerous reports have revealed apparent cardiotoxicity when a transgene is over-expressed. This is true not only for transgenes that have biological activity in the heart (60-63), it is also true for commonly used reporter proteins such as GFP (64) and proteins that are considered harmless such as the Gal4 yeast transcription factor (65).
Cardiac toxicity of dystrophin over-expression has not been studied. It is very likely that there exists an optimal range. The heart is known to display different levels of tolerance to different proteins. A six-fold higher expression of adenosine receptor causes dilated cardiomyopathy (63). However, a 16-fold over-expression of myosin light chain 1 is not toxic. Toxicity is observed only when the myosin light chain 1 level is 27-fold higher than normal (62). The toxic threshold is not known for dystrophin. Considering the high likelihood of cardiotoxicity from over-expression, one should be cautious in moving experimental heart gene therapy to patients before the tolerable range is determined and before the methods are developed to control gene expression. In the meantime, an urgent issue is to find out how much dystrophin is too much.
Where should the therapeutic gene(s) be delivered?
In general, the goal of gene therapy is to deliver the therapeutic gene to the place where it is missing. A logical assumption for heart gene therapy will be to restore dystrophin expression in cardiac muscle. However, some studies suggest that there are also skeletal muscle and vascular smooth muscle components in dystrophic heart diseases. The question is whether we need to restore dystrophin expression in all three muscles, including cardiac, skeletal and smooth muscle, in order to cure dystrophin-deficient heart diseases.
The pathogenesis of dystrophic heart disease is not completely understood. Several studies suggest that vasospasm from the disruption of the sarcoglycan complex is responsible for cardiomyopathy in certain types of limb girdle muscular dystrophy (such as in β- or δ-sarcoglycan-deficient mice) (66-68). However, recent studies by Wheeler et al. (69,70) suggest that vasospasm is not due to sarcoglycan deficiency. Rather it is caused by cytokines released from the injured cardiomyocytes and/or inflammatory cells in γ- or δ-sarcoglycan-deficient mice. Although it is tempting to hypothesize that vascular dysfunction may have contributed to the heart pathology in dystrophin-deficient disease, this hypothesis is not supported by experimental data. First, the sarcoglycan complex is not disturbed in mdx vascular smooth muscle (68). Second, a vasodilating drug (verapamil) that has effectively reduced cardiomyopathy in δ-sarcoglycan null mice does not protect the mdx mouse heart (68). Importantly, verapamil may trigger death in DMD patients (71). The most compelling evidence against the smooth muscle theory comes from a recent gene rescue experiment (70). In this study, wheeler et al. showed that rescuing the missing DGC component in vascular smooth muscle cannot stop heart disease in sarcoglycan-deficient mice. But restoring expression in cardiomyocytes can ameliorate heart disease (70). Taken together, rescuing smooth muscle dystrophin expression may not be necessary nor will it be protective in dystrophin-deficient heart disease.
Controversy exists as to whether skeletal muscle disease contributes to cardiomyopathy. Dystrophin-null mdx mouse is the most commonly used model for DMD. Adult mdx mice display mild skeletal muscle weakness and subtle heart pathology (72,73). However, when myoD (a skeletal muscle-specific transcription factor) is inactivated, mdx mice develop serious skeletal muscle disease. Interestingly, these dystrophin/myoD double-null (m-dko) mice also develop severe cardiomyopathy (74). As myoD is expressed only in skeletal muscle (not in the heart), it has been speculated that a secondary response to the severe skeletal muscle disease, rather than the loss of dystrophin itself, may be crucial for severe cardiomyopathy in these mice. This finding is reminiscent of an earlier patient study that showed a correlation between the severity of skeletal muscle disease and the development of heart disease (75). If, indeed, dystrophic cardiomyopathy is secondary to severe skeletal muscle damage, the therapy should then focus on treating skeletal muscle pathology.
This hypothesis is challenged by several studies. First, it has been demonstrated that the loss of dystrophin in the heart alone is sufficient to cause severe heart disease in XLDC patients. As dystrophin expression is not disturbed in skeletal muscle in these patients, the lack of cardiac dystrophin is apparently the primary cause of the heart pathology (7,76-78). Second, a study in γ-sarcoglycan-deficient mice suggests that transgenic rescue of skeletal muscle pathology has no beneficial effect on the heart (79). Heart disease is ameliorated only when the γ-sarcoglycan gene is expressed in cardiomyocytes (70).
In summary, the primary target tissue in gene therapy should be cardiac muscle. However, ameliorating skeletal muscle disease may further improve heart function.
MOUSE MODELS TO DEVELOP HEART GENE THERAPY
A number of mouse models have been developed to mimic human DMD/BMD. Among these, dystrophin-null mdx mice, m-dko and dystrophin/utrophin double knockout (u-dko) mice and m-dko mice are particularly attractive for heart gene therapy studies.
Mdx mice
Naturally occurring mdx mice result from a nonsense mutation in exon 23 (80,81). Skeletal muscle pathology in mdx mice has been extensively characterized. The heart in young mdx mice (<3-month-old) display infrequent and mild changes such as small degenerative foci and limited inflammation (72,73,82-85). By 6–8 months of age, the mdx heart starts to show moderate myocardial necrosis and fibrosis (68,72). Prominent heart pathology does not appear until mice are about 12-month-old (86) and it gets worse as mice age (86-88).
Several recent studies have begun to evaluate physiological changes in the mdx heart. Despite a lack of significant histology alteration in the young mdx heart, direct ex vivo measurement of myocardium contraction has revealed a substantial force reduction in 8–14-week-old mdx mice (89,90). However, this force decline is not sufficient to alter hemodynamic function unless mice are stressed (21,59,83,87,91,92). In contrast to young mdx mice, the features of dilated cardiomyopathy can be more easily detected in old mice. In one study, investigators reported echo-cardiography change in 10-month-old mdx mice (87). ECG study in old mdx mice also revealed similar findings as have been reported in DMD/BMD patients such as deep Q wave, increased R/S ratio, frequent premature ventricular contraction and reduced heart rate variability (86).
Despite a relatively mild cardiac phenotype at young age, mdx mice do develop heart disease when they get old. As mdx mice share exactly the same genetic defect as human patients and are readily available, they represent a valuable model for heart gene therapy studies.
U-dko mice
Utrophin is an ubiquitously expressed autosomal paralogue of dystrophin and it is up-regulated in mdx mice (45,92-96).
To test whether utrophin up-regulation compensates for the loss of dystrophin, u-dko mice were generated (73,97). In sharp contrast to the mild phenotype in mdx mice, u-dko mice are weaker and smaller. They show progressive muscle wasting, growth retardation, weight loss, scoliosis and contractures (Fig. 4). They die prematurely between the age of 8 and 10 weeks. As the catastrophic clinical progression in u-dko mice closely reproduces the severe phenotype of DMD patients, these mice are considered as excellent models for DMD gene therapy studies (53).
Figure 4.

Utrophin/dystrophin (u-dko) and myoD/dystrophin (m-dko) double knock-out mice recapitulate the clinical phenotype in DMD patients. (A) U-dko mice are significantly smaller than age and sex-matched mdx mice (left panel). U-dko mice also display abnormal hind limb contracture (arrow, right panel). (B) Inflammation in the heart of a 2-month-old Grady strain u-dko mouse. HE, hematoxylin–eosin stain; NSE, non-specific esterase stain for macrophage. Arrows, macrophage. Scale bar: 50 μm. (C) Scoliosis (arrow) is a common feature in 5-month-old m-dko mice, but it is absent in age- and sex-matched mdx mice. (D) Representative heart pathology in m-dko mice. EBD uptake in 4-month-old m-dko heart; scale bar: 1 mm. MT and AR, Masson trichrome (for fibrosis, arrow) and Alizarin red (for calcification, arrow) stain, respectively; scale bar: 100 μm.
Two strains of u-dko mice were developed independently in two laboratories (73,97). Although the majority of mice in the strain reported by Grady et al. (73) show severe cardiomyopathy between 8 and 10 weeks of age (Fig. 4), the strain reported by Deconinck et al. (97) seems to display a relatively milder cardiac pathology. The heart weight/body weight ratio in 8–10-week-old Deconinck u-dko strain is not different from that of wild-type littermates (90). In a semi-quantitative assay, the histopathology of 10-week-old Deconinck strain u-dko heart also scores similarly to the mdx heart (85). The exact mechanism for the different heart phenotype in these two strains is not clear. It may relate to the difference in the genetic background on which these mice are generated. Alternatively, it may reflect the difference in gene knockout strategy. All utrophin isoforms are inactivated in the Grady strain, whereas only the largest utrophin is inactivated in the Deconinck strain.
M-dko mice
The mild phenotype in mdx mice has been partially accredited to extremely efficient muscle regeneration as demonstrated by muscle hypertrophy and the prevalence of centrally located nuclei (82,98-100). The activated satellite cells are responsible for skeletal muscle regeneration. The absolute number of satellite cells is not altered in mdx mice, but their regenerative capacity is enhanced (101,102).
MyoD is a basic helix-loop-helix myogenic transcription factor that is specifically involved in satellite cell activation (103). To test whether an increased muscle regenerative capacity has contributed to the relatively healthy appearance of mdx mice, m-dko mice were generated by crossing myoD knockout mice and mdx mice (101). As expected, m-dko mice develop severe dystrophic signs as seen in patients, such as substantially reduced muscle mass, pronounced scoliosis, abnormal waddling gait and premature death at ∼ 12 months of age (74) (Fig. 4). Surprisingly, m-dko mice also develop severe dilated cardiomyopathy (74) (Fig. 4). As myoD is only required in skeletal muscle development (104), it is thought that cardiomyopathy in m-dko mice is secondary to severe skeletal muscle disease (74). M-dko mice provide a unique variant to mdx and u-dko mice. Genetically, m-dko heart is identical to human patients. Phenotypically, m-dko heart displays the exactly same progressive dilated cardiomyopathy as seen in patients.
In summary, mdx mice (especially old mice), u-dko and m-dko mice constitute a great resource to study dystrophin-null cardiomyopathy and to develop gene therapy. Because of the difference in gene mutation and the degree of the pathology, a comprehensive study including all three models will likely overcome the limitations of the single model and yield useful information to guide human gene therapy.
SUMMARY AND FUTURE DIRECTION
The enthusiasm for gene therapy of dystrophin-deficient cardiomyopathy has reached a record high level. Tremendous progress has been made in developing heart gene delivery vehicles, in optimizing heart gene transfer methods and in generating novel synthetic mini- and microgenes. Now the challenges are to understand the basic parameters. In particular, we need to know whether we should correct every single cardiomyocyte in order to achieve organ level improvement. We need to know whether treating the heart is sufficient to alleviate the cardiac phenotype. Most importantly, we need to know which candidate dystrophin gene(s) will work in the heart and how much protein will be required. It is now the golden time to systemically address these important questions in experimental animal models. Knowledge in these areas will pave the way to eventually curing dystrophin-null cardiomyopathy in human patients.
ACKNOWLEDGEMENTS
This work is supported by grants from the National Institutes of Health (AR-49419, DD) and the Muscular Dystrophy Association (DD).
Footnotes
Conflict of Interest statement. None declared.
REFERENCES
- 1.Campbell KP, Kahl SD. Association of dystrophin and an integral membrane glycoprotein. Nature. 1989;338:259–262. doi: 10.1038/338259a0. [DOI] [PubMed] [Google Scholar]
- 2.Petrof BJ. The molecular basis of activity-induced muscle injury in Duchenne muscular dystrophy. Mol. Cell Biochem. 1998;179:111–123. doi: 10.1023/a:1006812004945. [DOI] [PubMed] [Google Scholar]
- 3.Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24:1575–1594. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- 4.Beggs AH, Hoffman EP, Snyder JR, Arahata K, Specht L, Shapiro F, Angelini C, Sugita H, Kunkel LM. Exploring the molecular basis for variability among patients with Becker muscular dystrophy: dystrophin gene and protein studies. Am. J. Hum. Genet. 1991;49:54–67. [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffman EP. Genotype/phenotype correlations in Duchenne/ Becker dystrophy. Mol. Cell Biol. Hum. Dis. Ser. 1993;3:12–36. doi: 10.1007/978-94-011-1528-5_2. [DOI] [PubMed] [Google Scholar]
- 6.Towbin JA, Bowles KR, Bowles NE. Etiologies of cardiomyopathy and heart failure. Nat. Med. 1999;5:266–267. doi: 10.1038/6474. [DOI] [PubMed] [Google Scholar]
- 7.Cohen N, Muntoni F. Multiple pathogenetic mechanisms in X-linked dilated cardiomyopathy. Heart. 2004;90:835–841. doi: 10.1136/hrt.2003.023390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox GF, Kunkel LM. Dystrophies and heart disease. Curr. Opin. Cardiol. 1997;12:329–343. [PubMed] [Google Scholar]
- 9.Baxter P. Treatment of the heart in Duchenne muscular dystrophy. Dev. Med. Child Neurol. 2006;48:163. doi: 10.1017/S0012162206000351. [DOI] [PubMed] [Google Scholar]
- 10.Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002;12:926–929. doi: 10.1016/s0960-8966(02)00140-2. [DOI] [PubMed] [Google Scholar]
- 11.Politano L, Nigro V, Nigro G, Petretta VR, Passamano L, Papparella S, Di Somma S, Comi LI. Development of cardiomyopathy in female carriers of Duchenne and Becker muscular dystrophies. JAMA. 1996;275:1335–1338. [PubMed] [Google Scholar]
- 12.Melacini P, Fanin M, Danieli GA, Villanova C, Martinello F, Miorin M, Freda MP, Miorelli M, Mostacciuolo ML, Fasoli G, et al. Myocardial involvement is very frequent among patients affected with subclinical Becker's muscular dystrophy. Circulation. 1996;94:3168–3175. doi: 10.1161/01.cir.94.12.3168. [DOI] [PubMed] [Google Scholar]
- 13.Saito M, Kawai H, Akaike M, Adachi K, Nishida Y, Saito S. Cardiac dysfunction with Becker muscular dystrophy. Am. Heart J. 1996;132:642–647. doi: 10.1016/s0002-8703(96)90250-1. [DOI] [PubMed] [Google Scholar]
- 14.Grain L, Cortina-Borja M, Forfar C, Hilton-Jones D, Hopkin J, Burch M. Cardiac abnormalities and skeletal muscle weakness in carriers of Duchenne and Becker muscular dystrophies and controls. Neuromuscul. Disord. 2001;11:186–191. doi: 10.1016/s0960-8966(00)00185-1. [DOI] [PubMed] [Google Scholar]
- 15.Hoogerwaard EM, van der Wouw PA, Wilde AA, Bakker E, Ippel PF, Oosterwijk JC, Majoor-Krakauer DF, van Essen AJ, Leschot NJ, de Visser M. Cardiac involvement in carriers of Duchenne and Becker muscular dystrophy. Neuromuscul. Disord. 1999;9:347–351. doi: 10.1016/s0960-8966(99)00018-8. [DOI] [PubMed] [Google Scholar]
- 16.Nolan MA, Jones OD, Pedersen RL, Johnston HM. Cardiac assessment in childhood carriers of Duchenne and Becker muscular dystrophies. Neuromuscul. Disord. 2003;13:129–132. doi: 10.1016/s0960-8966(02)00197-9. [DOI] [PubMed] [Google Scholar]
- 17.Quinlivan RM, Dubowitz V. Cardiac transplantation in Becker muscular dystrophy. Neuromuscul. Disord. 1992;2:165–167. doi: 10.1016/0960-8966(92)90002-n. [DOI] [PubMed] [Google Scholar]
- 18.Melacini P, Fanin M, Angelini A, Pegoraro E, Livi U, Danieli GA, Hoffman EP, Thiene G, Dalla Volta S, Angelini C. Cardiac transplantation in a Duchenne muscular dystrophy carrier. Neuromuscul. Disord. 1998;8:585–590. doi: 10.1016/s0960-8966(98)00071-6. [DOI] [PubMed] [Google Scholar]
- 19.Ruiz-Cano MJ, Delgado JF, Jimenez C, Jimenez S, Cea-Calvo L, Sanchez V, Escribano P, Gomez MA, Gil-Fraguas L, Saenz de la Calzada C. Successful heart transplantation in patients with inherited myopathies associated with end-stage cardiomyopathy. Transplant Proc. 2003;35:1513–1515. doi: 10.1016/s0041-1345(03)00515-3. [DOI] [PubMed] [Google Scholar]
- 20.Patane F, Zingarelli E, Attisani M, Sansone F. Successful heart transplantation in Becker's muscular dystrophy. Eur. J. Cardiothorac. Surg. 2006;29:250. doi: 10.1016/j.ejcts.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 21.Yasuda S, Townsend D, Michele DE, Favre EG, Day SM, Metzger JM. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature. 2005;436:1025–1029. doi: 10.1038/nature03844. [DOI] [PubMed] [Google Scholar]
- 22.Pasternak C, Wong S, Elson EL. Mechanical function of dystrophin in muscle cells. J. Cell Biol. 1995;128:355–361. doi: 10.1083/jcb.128.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grange RW, Gainer TG, Marschner KM, Talmadge RJ, Stull JT. Fast-twitch skeletal muscles of dystrophic mouse pups are resistant to injury from acute mechanical stress. Am. J. Physiol. Cell Physiol. 2002;283:C1090–C1101. doi: 10.1152/ajpcell.00450.2001. [DOI] [PubMed] [Google Scholar]
- 24.Porter GA, Dmytrenko GM, Winkelmann JC, Bloch RJ. Dystrophin colocalizes with beta-spectrin in distinct subsarcolemmal domains in mammalian skeletal muscle. J. Cell Biol. 1992;117:997–1005. doi: 10.1083/jcb.117.5.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Straub V, Bittner RE, Leger JJ, Voit T. Direct visualization of the dystrophin network on skeletal muscle fiber membrane. J. Cell Biol. 1992;119:1183–1191. doi: 10.1083/jcb.119.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rybakova IN, Patel JR, Ervasti JM. The dystrophin complex forms a mechanically strong link between the sarcolemma and costameric actin. J. Cell Biol. 2000;150:1209–1214. doi: 10.1083/jcb.150.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ervasti JM. Costameres: the Achilles' heel of Herculean muscle. J. Biol. Chem. 2003;278:13591–13594. doi: 10.1074/jbc.R200021200. [DOI] [PubMed] [Google Scholar]
- 28.Pardo JV, Siliciano JD, Craig SW. Vinculin is a component of an extensive network of myofibril-sarcolemma attachment regions in cardiac muscle fibers. J. Cell Biol. 1983;97:1081–1088. doi: 10.1083/jcb.97.4.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Danowski BA, Imanaka-Yoshida K, Sanger JM, Sanger JW. Costameres are sites of force transmission to the substratum in adult rat cardiomyocytes. J. Cell Biol. 1992;118:1411–1420. doi: 10.1083/jcb.118.6.1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stevenson S, Rothery S, Cullen MJ, Severs NJ. Dystrophin is not a specific component of the cardiac costamere. Circ. Res. 1997;80:269–280. doi: 10.1161/01.res.80.2.269. [DOI] [PubMed] [Google Scholar]
- 31.Kaprielian RR, Stevenson S, Rothery SM, Cullen MJ, Severs NJ. Distinct patterns of dystrophin organization in myocyte sarcolemma and transverse tubules of normal and diseased human myocardium. Circulation. 2000;101:2586–2594. doi: 10.1161/01.cir.101.22.2586. [DOI] [PubMed] [Google Scholar]
- 32.Klietsch R, Ervasti JM, Arnold W, Campbell KP, Jorgensen AO. Dystrophin-glycoprotein complex and laminin colocalize to the sarcolemma and transverse tubules of cardiac muscle. Circ. Res. 1993;72:349–360. doi: 10.1161/01.res.72.2.349. [DOI] [PubMed] [Google Scholar]
- 33.Frank JS, Mottino G, Chen F, Peri V, Holland P, Tuana BS. Subcellular distribution of dystrophin in isolated adult and neonatal cardiac myocytes. Am. J. Physiol. 1994;267:C1707–C1716. doi: 10.1152/ajpcell.1994.267.6.C1707. [DOI] [PubMed] [Google Scholar]
- 34.Cox GA, Sunada Y, Campbell KP, Chamberlain JS. Dp71 can restore the dystrophin-associated glycoprotein complex in muscle but fails to prevent dystrophy. Nat. Genet. 1994;8:333–339. doi: 10.1038/ng1294-333. [DOI] [PubMed] [Google Scholar]
- 35.Rafael JA, Sunada Y, Cole NM, Campbell KP, Faulkner JA, Chamberlain JS. Prevention of dystrophic pathology in mdx mice by a truncated dystrophin isoform. Hum. Mol. Genet. 1994;3:1725–1733. doi: 10.1093/hmg/3.10.1725. [DOI] [PubMed] [Google Scholar]
- 36.Phelps SF, Hauser MA, Cole NM, Rafael JA, Hinkle RT, Faulkner JA, Chamberlain JS. Expression of full-length and truncated dystrophin mini-genes in transgenic mdx mice. Hum. Mol. Genet. 1995;4:1251–1258. doi: 10.1093/hmg/4.8.1251. [DOI] [PubMed] [Google Scholar]
- 37.Wells DJ, Wells KE, Asante EA, Turner G, Sunada Y, Campbell KP, Walsh FS, Dickson G. Expression of human full-length and minidystrophin in transgenic mdx mice: implications for gene therapy of Duchenne muscular dystrophy. Hum. Mol. Genet. 1995;4:1245–1250. doi: 10.1093/hmg/4.8.1245. [DOI] [PubMed] [Google Scholar]
- 38.Rafael JA, Cox GA, Corrado K, Jung D, Campbell KP, Chamberlain JS. Forced expression of dystrophin deletion constructs reveals structure–function correlations. J. Cell Biol. 1996;134:93–102. doi: 10.1083/jcb.134.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corrado K, Rafael JA, Mills PL, Cole NM, Faulkner JA, Wang K, Chamberlain JS. Transgenic mdx mice expressing dystrophin with a deletion in the actin- binding domain display a ‘mild Becker’ phenotype. J. Cell Biol. 1996;134:873–884. doi: 10.1083/jcb.134.4.873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crawford GE, Faulkner JA, Crosbie RH, Campbell KP, Froehner SC, Chamberlain JS. Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain. J. Cell Biol. 2000;150:1399–1410. doi: 10.1083/jcb.150.6.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harper SQ, Hauser MA, DelloRusso C, Duan D, Crawford RW, Phelps SF, Harper HA, Robinson AS, Engelhardt JF, Brooks SV, et al. Modular flexibility of dystrophin: implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002;8:253–261. doi: 10.1038/nm0302-253. [DOI] [PubMed] [Google Scholar]
- 42.Harper SQ, Crawford RW, DelloRusso C, Chamberlain JS. Spectrin-like repeats from dystrophin and alpha-actinin-2 are not functionally interchangeable. Hum. Mol. Genet. 2002;11:1807–1815. doi: 10.1093/hmg/11.16.1807. [DOI] [PubMed] [Google Scholar]
- 43.Warner LE, DelloRusso C, Crawford RW, Rybakova IN, Patel JR, Ervasti JM, Chamberlain JS. Expression of Dp260 in muscle tethers the actin cytoskeleton to the dystrophin-glycoprotein complex and partially prevents dystrophy. Hum. Mol. Genet. 2002;11:1095–1105. doi: 10.1093/hmg/11.9.1095. [DOI] [PubMed] [Google Scholar]
- 44.Judge LM, Haraguchiln M, Chamberlain JS. Dissecting the signaling and mechanical functions of the dystrophin-glycoprotein complex. J. Cell Sci. 2006;119:1537–1546. doi: 10.1242/jcs.02857. [DOI] [PubMed] [Google Scholar]
- 45.Cox GA, Cole NM, Matsumura K, Phelps SF, Hauschka SD, Campbell KP, Faulkner JA, Chamberlain JS. Overexpression of dystrophin in transgenic mdx mice eliminates dystrophic symptoms without toxicity [see comments] Nature. 1993;364:725–729. doi: 10.1038/364725a0. [DOI] [PubMed] [Google Scholar]
- 46.Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat. Med. 1999;5:320–326. doi: 10.1038/6543. [DOI] [PubMed] [Google Scholar]
- 47.Badorff C, Berkely N, Mehrotra S, Talhouk JW, Rhoads RE, Knowlton KU. Enteroviral protease 2A directly cleaves dystrophin and is inhibited by a dystrophin-based substrate analogue. J. Biol. Chem. 2000;275:11191–11197. doi: 10.1074/jbc.275.15.11191. [DOI] [PubMed] [Google Scholar]
- 48.Xiong D, Lee GH, Badorff C, Dorner A, Lee S, Wolf P, Knowlton KU. Dystrophin deficiency markedly increases enterovirus-induced cardiomyopathy: a genetic predisposition to viral heart disease. Nat. Med. 2002;8:872–877. doi: 10.1038/nm737. [DOI] [PubMed] [Google Scholar]
- 49.Varghese A, Pennell DJ. Late gadolinium enhanced cardiovascular magnetic resonance in Becker muscular dystrophy. Heart. 2004;90:e59. doi: 10.1136/hrt.2004.041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jefferies JL, Eidem BW, Belmont JW, Craigen WJ, Ware SM, Fernbach SD, Neish SR, Smith EO, Towbin JA. Genetic predictors and remodeling of dilated cardiomyopathy in muscular dystrophy. Circulation. 2005;112:2799–2804. doi: 10.1161/CIRCULATIONAHA.104.528281. [DOI] [PubMed] [Google Scholar]
- 51.Lai Y, Yue Y, Liu M, Ghosh A, Engelhardt JF, Chamberlain JS, Duan D. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat. Biotechnol. 2005;23:1435–1439. doi: 10.1038/nbt1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu M, Yue Y, Harper SQ, Grange RW, Chamberlain JS, Duan D. Adeno-associated virus-mediated micro-dystrophin expression protects young mdx muscle from contraction-induced injury. Mol. Ther. 2005;11:245–256. doi: 10.1016/j.ymthe.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yue Y, Liu M, Duan D. C-terminal truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double knock-out mice. Mol. Ther. 2006;14:79–87. doi: 10.1016/j.ymthe.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, Russell DW, Chamberlain JS. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, Xiao X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 57.Zhu T, Zhou L, Mori S, Wang Z, McTiernan CF, Qiao C, Chen C, Wang DW, Li J, Xiao X. Sustained whole-body functional rescue in congestive heart failure and muscular dystrophy hamsters by systemic gene transfer. Circulation. 2005;112:2650–2659. doi: 10.1161/CIRCULATIONAHA.105.565598. [DOI] [PubMed] [Google Scholar]
- 58.Mah C, Cresawn KO, Fraites TJ, Jr., Pacak CA, Lewis MA, Zolotukhin I, Byrne BJ. Sustained correction of glycogen storage disease type II using adeno-associated virus serotype 1 vectors. Gene Ther. 2005;12:1405–1409. doi: 10.1038/sj.gt.3302550. [DOI] [PubMed] [Google Scholar]
- 59.Yue Y, Skimming JW, Liu M, Strawn T, Duan D. Full-length dystrophin expression in half of the heart cells ameliorates beta-isoproterenol-induced cardiomyopathy in mdx mice. Hum. Mol. Genet. 2004;13:1669–1675. doi: 10.1093/hmg/ddh174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Milano CA, Dolber PC, Rockman HA, Bond RA, Venable ME, Allen LF, Lefkowitz RJ. Myocardial expression of a constitutively active alpha 1B-adrenergic receptor in transgenic mice induces cardiac hypertrophy. Proc. Natl Acad. Sci. USA. 1994;91:10109–10113. doi: 10.1073/pnas.91.21.10109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Colbert MC, Hall DG, Kimball TR, Witt SA, Lorenz JN, Kirby ML, Hewett TE, Klevitsky R, Robbins J. Cardiac compartment-specific overexpression of a modified retinoic acid receptor produces dilated cardiomyopathy and congestive heart failure in transgenic mice. J. Clin. Invest. 1997;100:1958–1968. doi: 10.1172/JCI119727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.James J, Osinska H, Hewett TE, Kimball T, Klevitsky R, Witt S, Hall DG, Gulick J, Robbins J. Transgenic over-expression of a motor protein at high levels results in severe cardiac pathology. Transgenic Res. 1999;8:9–22. doi: 10.1023/a:1008894507995. [DOI] [PubMed] [Google Scholar]
- 63.Black RG, Jr., Guo Y, Ge ZD, Murphree SS, Prabhu SD, Jones WK, Bolli R, Auchampach JA. Gene dosage-dependent effects of cardiac-specific overexpression of the A3 adenosine receptor. Circ. Res. 2002;91:165–172. doi: 10.1161/01.res.0000028007.91385.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huang WY, Aramburu J, Douglas PS, Izumo S. Transgenic expression of green fluorescence protein can cause dilated cardiomyopathy. Nat. Med. 2000;6:482–483. doi: 10.1038/74914. [DOI] [PubMed] [Google Scholar]
- 65.Habets PE, Clout DE, Lekanne Deprez RH, van Roon MA, Moorman AF, Christoffels VM. Cardiac expression of Gal4 causes cardiomyopathy in a dose-dependent manner. J. Muscle Res. Cell Motil. 2003;24:205–209. doi: 10.1023/a:1026055612227. [DOI] [PubMed] [Google Scholar]
- 66.Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, et al. Disruption of the sarcoglycan–sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98:465–474. doi: 10.1016/s0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- 67.Durbeej M, Cohn RD, Hrstka RF, Moore SA, Allamand V, Davidson BL, Williamson RA, Campbell KP. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol. Cell. 2000;5:141–151. doi: 10.1016/s1097-2765(00)80410-4. [DOI] [PubMed] [Google Scholar]
- 68.Cohn RD, Durbeej M, Moore SA, Coral-Vazquez R, Prouty S, Campbell KP. Prevention of cardiomyopathy in mouse models lacking the smooth muscle sarcoglycan-sarcospan complex. J. Clin. Invest. 2001;107:R1–R7. doi: 10.1172/JCI11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wheeler MT, Korcarz CE, Collins KA, Lapidos KA, Hack AA, Lyons MR, Zarnegar S, Earley JU, Lang RM, McNally EM. Secondary coronary artery vasospasm promotes cardiomyopathy progression. Am. J. Pathol. 2004;164:1063–1071. doi: 10.1016/S0002-9440(10)63193-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wheeler MT, Allikian MJ, Heydemann A, Hadhazy M, Zarnegar S, McNally EM. Smooth muscle cell-extrinsic vascular spasm arises from cardiomyocyte degeneration in sarcoglycan-deficient cardiomyopathy. J. Clin. Invest. 2004;113:668–675. doi: 10.1172/JCI20410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zalman F, Perloff JK, Durant NN, Campion DS. Acute respiratory failure following intravenous verapamil in Duchenne's muscular dystrophy. Am. Heart J. 1983;105:510–511. doi: 10.1016/0002-8703(83)90371-x. [DOI] [PubMed] [Google Scholar]
- 72.Bridges LR. The association of cardiac muscle necrosis and inflammation with the degenerative and persistent myopathy of MDX mice. J. Neurol. Sci. 1986;72:147–157. doi: 10.1016/0022-510x(86)90003-1. [DOI] [PubMed] [Google Scholar]
- 73.Grady RM, Teng H, Nichol MC, Cunningham JC, Wilkinson RS, Sanes JR. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell. 1997;90:729–738. doi: 10.1016/s0092-8674(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 74.Megeney LA, Kablar B, Perry RL, Ying C, May L, Rudnicki MA. Severe cardiomyopathy in mice lacking dystrophin and MyoD. Proc. Natl Acad. Sci. USA. 1999;96:220–225. doi: 10.1073/pnas.96.1.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hunsaker RH, Fulkerson PK, Barry FJ, Lewis RP, Leier CV, Unverferth DV. Cardiac function in Duchenne's muscular dystrophy. Results of 10-year follow-up study and non-invasive tests. Am. J. Med. 1982;73:235–238. doi: 10.1016/0002-9343(82)90184-x. [DOI] [PubMed] [Google Scholar]
- 76.Towbin JA, Hejtmancik JF, Brink P, Gelb B, Zhu XM, Chamberlain JS, McCabe ER, Swift M. X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation. 1993;87:1854–1865. doi: 10.1161/01.cir.87.6.1854. [DOI] [PubMed] [Google Scholar]
- 77.Muntoni F, Wilson L, Marrosu G, Marrosu MG, Cianchetti C, Mestroni L, Ganau A, Dubowitz V, Sewry C. A mutation in the dystrophin gene selectively affecting dystrophin expression in the heart. J. Clin. Invest. 1995;96:693–699. doi: 10.1172/JCI118112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Milasin J, Muntoni F, Severini GM, Bartoloni L, Vatta M, Krajinovic M, Mateddu A, Angelini C, Camerini F, Falaschi A, et al. A point mutation in the 5′ splice site of the dystrophin gene first intron responsible for X-linked dilated cardiomyopathy. Hum. Mol. Genet. 1996;5:73–79. doi: 10.1093/hmg/5.1.73. [DOI] [PubMed] [Google Scholar]
- 79.Zhu X, Wheeler MT, Hadhazy M, Lam MY, McNally EM. Cardiomyopathy is independent of skeletal muscle disease in muscular dystrophy. FASEB J. 2002;16:1096–1098. doi: 10.1096/fj.01-0954fje. [DOI] [PubMed] [Google Scholar]
- 80.Bulfield G, Siller WG, Wight PA, Moore KJ. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl Acad. Sci. USA. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 82.Coulton GR, Morgan JE, Partridge TA, Sloper JC. The mdx mouse skeletal muscle myopathy: I. A histological, morphometric and biochemical investigation. Neuropathol. Appl. Neurobiol. 1988;14:53–70. doi: 10.1111/j.1365-2990.1988.tb00866.x. [DOI] [PubMed] [Google Scholar]
- 83.Kamogawa Y, Biro S, Maeda M, Setoguchi M, Hirakawa T, Yoshida H, Tei C. Dystrophin-deficient myocardium is vulnerable to pressure overload in vivo. Cardiovasc Res. 2001;50:509–515. doi: 10.1016/s0008-6363(01)00205-x. [DOI] [PubMed] [Google Scholar]
- 84.Nakamura A, Yoshida K, Takeda S, Dohi N, Ikeda S. Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett. 2002;520:18–24. doi: 10.1016/s0014-5793(02)02739-4. [DOI] [PubMed] [Google Scholar]
- 85.Hainsey TA, Senapati S, Kuhn DE, Rafael JA. Cardiomyopathic features associated with muscular dystrophy are independent of dystrophin absence in cardiovasculature. Neuromuscul. Disord. 2003;13:294–302. doi: 10.1016/s0960-8966(02)00286-9. [DOI] [PubMed] [Google Scholar]
- 86.Wehling-Henricks M, Jordan MC, Roos KP, Deng B, Tidball JG. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet. 2005;14:1921–1933. doi: 10.1093/hmg/ddi197. [DOI] [PubMed] [Google Scholar]
- 87.Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul. Disord. 2004;14:491–496. doi: 10.1016/j.nmd.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 88.Lefaucheur JP, Pastoret C, Sebille A. Phenotype of dystrophinopathy in old mdx mice. Anat. Rec. 1995;242:70–76. doi: 10.1002/ar.1092420109. [DOI] [PubMed] [Google Scholar]
- 89.Sapp JL, Bobet J, Howlett SE. Contractile properties of myocardium are altered in dystrophin- deficient mdx mice. J. Neurol. Sci. 1996;142:17–24. doi: 10.1016/0022-510x(96)00167-0. [DOI] [PubMed] [Google Scholar]
- 90.Janssen PM, Hiranandani N, Mays TA, Rafael-Fortney JA. Utrophin deficiency worsens cardiac contractile dysfunction present in dystrophin-deficient mdx mice. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H2373–H2378. doi: 10.1152/ajpheart.00448.2005. [DOI] [PubMed] [Google Scholar]
- 91.Danialou G, Comtois AS, Dudley R, Karpati G, Vincent G, Des Rosiers C, Petrof BJ. Dystrophin-deficient cardiomyocytes are abnormally vulnerable to mechanical stress-induced contractile failure and injury. FASEB J. 2001;15:1655–1657. doi: 10.1096/fj.01-0030fje. [DOI] [PubMed] [Google Scholar]
- 92.Wilding JR, Schneider JE, Sang AE, Davies KE, Neubauer S, Clarke K. Dystrophin- and MLP-deficient mouse hearts: marked differences in morphology and function, but similar accumulation of cytoskeletal proteins. FASEB J. 2005;19:79–81. doi: 10.1096/fj.04-1731fje. [DOI] [PubMed] [Google Scholar]
- 93.Khurana TS, Watkins SC, Chafey P, Chelly J, Tome FM, Fardeau M, Kaplan JC, Kunkel LM. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991;1:185–194. doi: 10.1016/0960-8966(91)90023-l. [DOI] [PubMed] [Google Scholar]
- 94.Matsumura K, Ervasti JM, Ohlendieck K, Kahl SD, Campbell KP. Association of dystrophin-related protein with dystrophin-associated proteins in mdx mouse muscle. Nature. 1992;360:588–591. doi: 10.1038/360588a0. [DOI] [PubMed] [Google Scholar]
- 95.Love DR, Morris GE, Ellis JM, Fairbrother U, Marsden RF, Bloomfield JF, Edwards YH, Slater CP, Parry DJ, Davies KE. Tissue distribution of the dystrophin-related gene product and expression in the mdx and dy mouse. Proc. Natl Acad. Sci. USA. 1991;88:3243–3247. doi: 10.1073/pnas.88.8.3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pons F, Robert A, Fabbrizio E, Hugon G, Califano JC, Fehrentz JA, Martinez J, Mornet D. Utrophin localization in normal and dystrophin-deficient heart. Circulation. 1994;90:369–374. doi: 10.1161/01.cir.90.1.369. [DOI] [PubMed] [Google Scholar]
- 97.Deconinck AE, Rafael JA, Skinner JA, Brown SC, Potter AC, Metzinger L, Watt DJ, Dickson JG, Tinsley JM, Davies KE. Utrophin–dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. doi: 10.1016/s0092-8674(00)80532-2. [DOI] [PubMed] [Google Scholar]
- 98.Anderson JE, Ovalle WK, Bressler BH. Electron microscopic and autoradiographic characterization of hindlimb muscle regeneration in the mdx mouse. Anat. Rec. 1987;219:243–257. doi: 10.1002/ar.1092190305. [DOI] [PubMed] [Google Scholar]
- 99.Carnwath JW, Shotton DM. Muscular dystrophy in the mdx mouse: histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci. 1987;80:39–54. doi: 10.1016/0022-510x(87)90219-x. [DOI] [PubMed] [Google Scholar]
- 100.Geissinger HD, Rao PV, McDonald-Taylor CK. ‘mdx’ mouse myopathy: histopathological, morphometric and histochemical observations on young mice. J. Comp. Pathol. 1990;102:249–263. doi: 10.1016/s0021-9975(08)80015-1. [DOI] [PubMed] [Google Scholar]
- 101.Megeney LA, Kablar B, Garrett K, Anderson JE, Rudnicki MA. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996;10:1173–1183. doi: 10.1101/gad.10.10.1173. [DOI] [PubMed] [Google Scholar]
- 102.Reimann J, Irintchev A, Wernig A. Regenerative capacity and the number of satellite cells in soleus muscles of normal and mdx mice. Neuromuscul. Disord. 2000;10:276–282. doi: 10.1016/s0960-8966(99)00118-2. [DOI] [PubMed] [Google Scholar]
- 103.Rudnicki MA, Braun T, Hinuma S, Jaenisch R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- 104.Morkin E. Regulation of myosin heavy chain genes in the heart. Circulation. 1993;87:1451–1460. doi: 10.1161/01.cir.87.5.1451. [DOI] [PubMed] [Google Scholar]