

Abstract
Limited packaging capacity has hampered adeno-associated virus (AAV)-mediated gene therapy for many common genetic diseases such as cystic fibrosis (CF) and Duchenne muscular dystrophy (DMD). Trans-splicing AAV (tsAAV) vectors double AAV packaging capacity but their transduction efficiency has been too low to be useful. We have recently overcome this hurdle by rational vector design. We have shown that a pair of optimized mini-dystrophin tsAAV vectors can reach the same transduction efficiency as that of a single AAV vector after local injection in dystrophic muscle. However, global gene transfer is required to treat diseases like DMD. To test whether systemic delivery can be achieved with tsAAV vectors, we generated a set of optimized alkaline phosphatase (AP) tsAAV vectors. We delivered AAV serotype 9 pseudotyped AP tsAAV intravenously to newborn mice. Six weeks later, we observed high-level transduction in all body skeletal muscle and the heart, the tissues that are affected in DMD. We also detected efficient transduction in the lung, the primary organ affected in CF. Our results provide the first evidence of whole-body transduction with tsAAV vectors and further raise the hope of tsAAV gene therapy for DMD and CF.
INTRODUCTION
Many genetic diseases affect multiple tissues and/or organs. The challenge in gene therapy for these diseases lies in delivering the therapeutic gene to all the affected tissues/organs. Systemic gene transfer has been a daunting task for many years until recent success with adeno-associated virus (AAV).1-11 AAV is a single-stranded DNA virus and recombinant AAV vector has become a popular gene delivery vehicle in the last two decades.12 Earlier interest in serotype-2 AAV (AAV-2) has now expanded to a large number of newly isolated serotypes.13 The divergence of the capsid sequence and/or structure in the new serotypes has resulted in striking differences in their transduction profile. Although initial comparisons of different AAV serotypes were made in single tissues following local delivery, it was soon realized that many new serotypes could reach remote tissues after intra-vascular injection. Body-wide transduction has been reported for AAV-6, -8, and -9.1-3,6,9,10 Several groups have capitalized on this property and have achieved whole-body amelioration in the mouse models of several inherited diseases, including Duchenne muscular dystrophy (DMD), congenital muscular dystrophy, limb-girdle muscular dystrophy, Pompe disease, and type II glycogen storage disease.1,4,5,7,8,10,11
AAV has many features that render it a useful gene transfer vehicle. However, AAV is a small DNA virus with an extremely limited packaging capacity. The wild-type AAV genome is ∼4.8 kb. In recombinant AAV, transduction competent viruses are produced when the vector genome is 5 kb or less. The small packaging capacity poses a challenge for AAV-mediated gene therapy in many inherited diseases such as cystic fibrosis (CF), DMD, and hemophilia A. In these diseases, the therapeutic expression cassette exceeds the viral packaging limit. Trans-splicing AAV (tsAAV) vectors are a newly developed technology to double AAV packaging capacity. In this approach, the therapeutic gene is split into two parts and each part is carried by an AAV virion. After coinfection, the full-length gene is reconstituted through head-to-tail vector genome recombination. Therapeutic protein is expressed after the viral junction is removed from the mature mRNA by the cellular splicing machinery (reviewed in ref. 14). We have recently demonstrated efficient transduction by tsAAV vectors in a mouse model of DMD by local injection.15 More than 90% of myofibers in a single muscle were transduced by tsAAV. This efficiency is quite comparable to that of a single intact AAV vector.15-17
Despite the success in local gene transfer, it is not clear whether one can achieve efficient whole-body transduction with tsAAV vectors. A cure for many genetic diseases requires global transduction of all the affected tissues and/or organs. In the case of DMD, morbidity and mortality result from severe pathology not only in skeletal muscle, but also in the heart. Achieving systemic gene transfer will likely bring tsAAV closer to clinical application. In this study, we explored the potential of systemic gene transfer with AAV serotype 9 (AAV-9) trans-splicing vectors. Using a pair of optimized alkaline phosphatase (AP) gene tsAAV vectors, we detected a therapeutic level of expression in all skeletal muscle and the heart. We also observed high-level transduction in the lung. This is the first demonstration of successful whole-body transduction with tsAAV. Our results have set the foundation for applying this promising vector technology to human gene therapy.
RESULTS
Developing optimized reporter gene tsAAV vectors
tsAAV vectors have been used to express LacZ, erythropoietin, factor VIII, and mini-dystrophin.15,18-22 Besides our recently reported mini-dystrophin tsAAV, none of the published tsAAV vectors achieved the same transduction efficiency as that of a single AAV. To evaluate systemic transduction by tsAAV vectors, we first constructed a pair of rationally designed reporter gene tsAAVs that express heat-resistant human placental AP (AV.AP.Donor and AV.AP. Acceptor). We split the AP gene at the junction of exons 8 and 9.23,15 The splicing signals are obtained from a synthetic intron as we have described.23,24 As a control, we also included a single intact AAV vector (AV.AP) in our study.16,25 Gene expression in both tsAAV and single AAV is controlled by the the RSV promoter and the SV40 pA signal.
We first examined the transduction efficiency of AP tsAAV vectors in cell culture and by local muscle injection in the tibialis anterior muscle of C57BL10 (BL10) mice. Efficient AP expression was observed only in AV.AP.Donor and AV.AP.Acceptor coinfected cells and muscle. There was no leaky expression from singly infected vector (AV.AP.Donor alone or AV.AP.Acceptor alone) (data not shown). Furthermore, we observed comparable in vivo transduction efficiency (by counting the percentage of positive myofibers) between tsAAV and single AAV (data not shown). To our knowledge, this is the first set of reporter gene tsAAV vectors that has reached the transduction efficiency of a single intact AAV vector.
Systemic delivery of AP tsAAV resulted in body-wide transduction of striated muscle but not smooth muscle
As our goal is to achieve whole-body transduction, we delivered AP tsAAV vectors through the facial vein in newborn mice. Control groups included mock-infection (negative control) and single intact AV.AP infection (positive control). The AV.AP group represented the maximal achievable level of expression. Transgene expression was examined 6 weeks later.
In single intact vector-infected mice, we obtained widespread high-level expression in all striated muscles including all skeletal muscle and the heart (Figures 1, 2, and 4a). When AP trans-splicing vector-infected mice were evaluated, we also observed broad AP expression in all skeletal muscles throughout the entire body (Figures 1, 2, and 4a). Although the tsAAV group showed reduced staining intensity (Figure 4a), the total numbers of transduced skeletal myofibers were comparable between the tsAAV and single AAV groups. Overall, we achieved ≥90% transduction in both groups (Figure 4a). In a tissue lysate assay, the total AP activity was not significantly lower in tsAAV-infected skeletal muscle, but there was a trend towards reduced activity (Figure 4a). High-level AP expression was also observed in the hearts of tsAAV-infected mice (Figures 2 and 4a). Morphometric quantification revealed that more than 50% of cardiomyocytes were transduced. The AP activity in the tsAAV infected heart reached ∼60% of that in the single AAV infected heart.
Figure 1. Wide-spread transduction of skeletal muscle following intravenous delivery of trans-splicing AAV vectors in newborn mice.
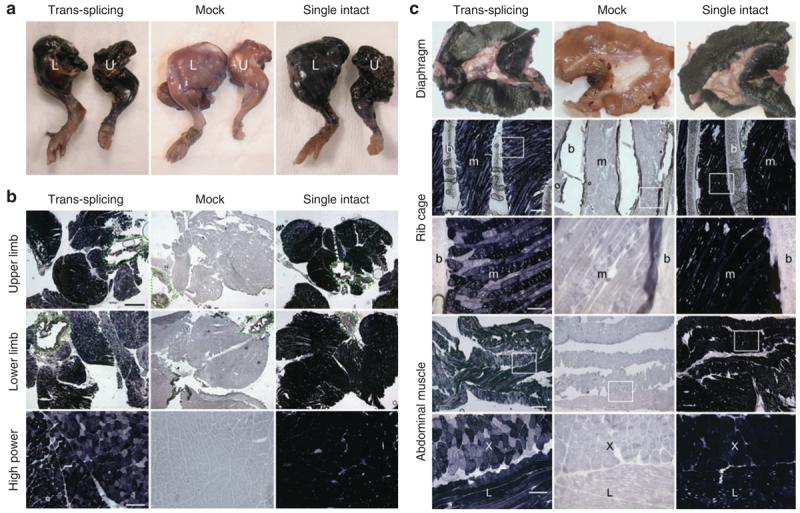
Representative histochemical staining photomicrographs from each group (N = 3–5 mice/group) are shown. (a) Full-view of the whole mount limb muscle (L, lower limb; U, upper limb). (b) Cross-sections of the entire limb (top two rows; bar, 1 mm) or high magnification view of representative areas of muscles in the lower limb (bottom row; bar, 200 μm), respectively. Dotted lines in low magnification photomicrographs mark the location of bone. (c) AP expression in respiratory muscles including the diaphragm (top row, whole mount images), inter-costal muscles (middle two rows, cross sections), and abdominal muscles (bottom two rows, cross-sections), respectively. Photomicrographs in high magnification (bar, 100 μm) are the closer view of the boxed areas in the respective low magnification (bar, 400 μm) photomicrographs. b, bone (rib); m, muscle; X, regions where myofibers are in cross-sectional orientation; L, regions where myofibers are in longitudinal orientation.
Figure 2. Trans-splicing AAV vectors efficiently transduce cardiac muscle after neonatal systemic delivery.
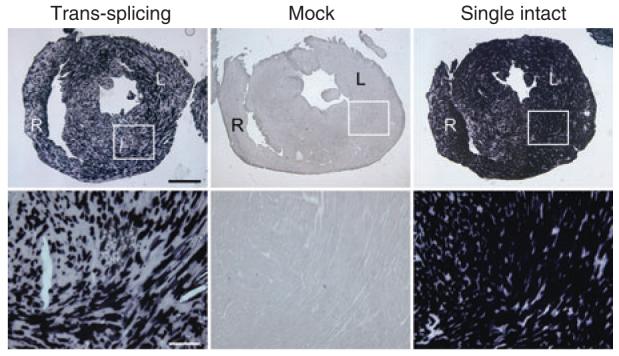
Photomicrographs are representative images of tsAAV, mock and single AAV-infected hearts, respectively. Top panels, full view images of the entire heart sections. R, right ventricular wall; L, left ventricular wall. Scale bar, 1 mm (low magnification). Bottom panels, closer view of the boxed areas in the top panels. Bar, 200 μm.
Figure 4. Quantitative comparison of the transduction efficiency of the trans-splicing vectors and that of the single intact vector.
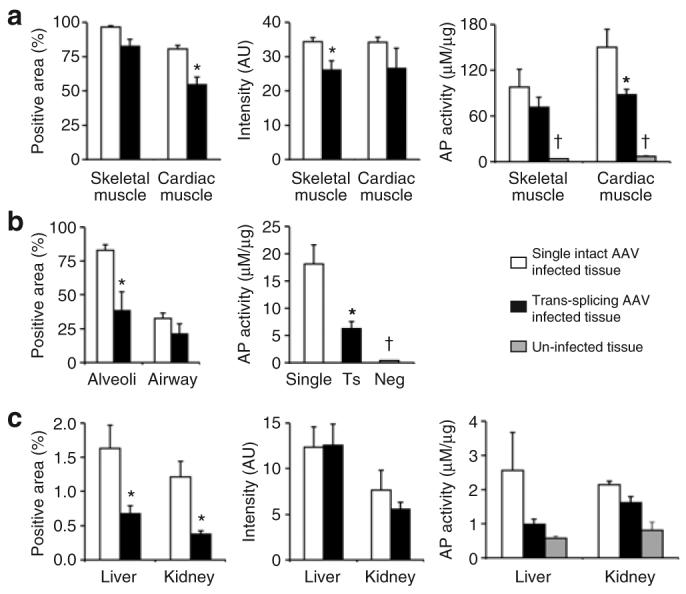
(a) AP-positive area, relative intensity and total tissue lysate activity in skeletal muscle and the heart. (b) Percentage of AP-positive cells in alveoli and conducting airway, respectively (quantified by manual counting). Also shown is the total tissue lysate AP activity. Single, AV.AP-infected lung; Ts, tsAAV infected lung; Neg, mock-treated lung. (c) AP-positive area, relative intensity and total tissue lysate activity in the liver and the kidney. AU, artificial unit. Asterisk, statistically different between the trans-splicing vector group and the single intact vector group. Cross, the AP activity in uninfected tissues is significantly lower than that in AAV-infected tissues.
Despite robust transduction in skeletal and cardiac muscle, we observed limited AP expression in smooth muscle (Supplementary Figure S1). Because surrounding cells (such as skeletal muscle myofibers, cardiomyocytes, and endothelial cells) were efficiently transduced, it is very likely that the poor transduction in smooth muscle is due to intrinsic biological properties of these cells rather than vector spreading and/or distribution.
Systemic delivery of AP tsAAV resulted in therapeutic level transduction in the lung but not in the liver and the kidney
We next evaluated transduction efficiency in several internal organs including the lung, liver and kidney (Figures 3b and 3c and 4c). In the lung, both alveolar and bronchiolar epithelial cells were transduced by tsAAV (Figures 3a and 4b). However, the overall AP activity in the tsAAV-infected lung was lower than that of the AV.AP-infected lung (Figure 4b). Despite high-level transduction in the lung, we detected only limited transduction in the liver and the kidney with the single intact AAV (Figures 3b, 3c, and 4c). Interestingly, when we delivered the same batch of the AV.AP vector to adult mice via tail vein injection, we observed high-level transduction in the adult liver and kidney (Supplementary Figure S2). Consistent with the poor transduction by AV.AP, tsAAV did not yield strong transduction either in the liver or the kidney after systemic injection in neonates (Figures 3b, 3c, and 4c).
Figure 3. Systemic delivery of a single intact AAV or a pair of trans-splicing AAV vectors results in efficient transduction in the lung but only minimal transduction in the liver and the kidney.
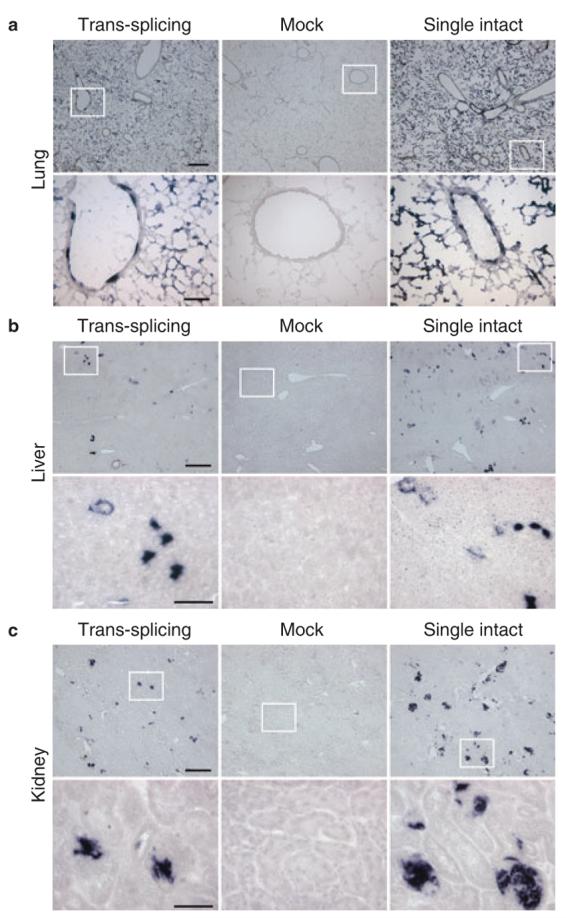
Photomicro-graphs are representative sections in (a) the lung, (b) the liver, and (c) the kidney. Higher power magnification of the boxed areas in the top row is shown in the corresponding panels in the bottom row. Scale bar in a, 400 μm (top panel, low magnification) and 100 μm (bottom panel, high magnification), respectively; bar in b, 200 μm (top panel, low magnification) and 50 μm (bottom panel, high magnification), respectively; bar in c, 200 μm (top panel, low magnification), and 50 μm (bottom panel, high magnification), respectively.
DISCUSSION
The development of the tsAAV vectors has revolutionized our perception of the small packaging capacity of the AAV vector (reviewed in ref. 14). However, the therapeutic potential of tsAAVs will be significantly limited if we cannot achieve bodywide transduction. In this proof-of-principle study, we examined whether whole-body transduction is feasible with AAV-9-pseudotyped tsAAV.
We first examined striated muscle. AAV-9 was recently reported to mediate strong expression in both skeletal and cardiac muscle.9,10 Consistent with these reports, we also obtained efficient transduction with AV.AP. Encouragingly, tsAAV resulted in an impressive transduction of skeletal muscle. We observed that tsAAV transduction approached that of a single intact AAV (Figure 4a). In the heart, the transduction efficiency of tsAAV did not reach that of the single intact vector. Nevertheless, it reached 50% of the threshold needed for treating cardiomyopathy in diseases such as DMD.26 We previously packaged the same expression cassette of AV.AP in AAV-2 and AAV-5 and achieved little (for AAV-2) or very limited expression in selected regions of the myocardium (for AAV-5) following direct intra-cavity injection in newborn mice.27 AAV-9 apparently was more efficient than AAV-2 and AAV-5 (Figure 2). The cardiac tropism of AAV-9 may have contributed to the high-level of tsAAV transduction in the heart.
AAV-9 has been shown to be superior for lung gene transfer following direct intra-tracheal injection.28,29 It also resulted in moderate transduction of the lung following systemic delivery in mice.9,10 Consistent with these reports, we also detected numerous AP-positive cells in the lungs from AV.AP-infected mice. Strikingly, tsAAV also resulted in broad transduction of both alveolar cells and conducting airway epithelial cells (Figures 3a and 4b). Although the single vector was significantly more efficient than tsAAV vectors in alveolar cells, both groups showed similar efficiency in the conducting airway. It is possible that the difference may have resulted from different transduction properties in these two cell types. Importantly, levels of gene transfer in both alveoli and airways were higher than 10%, a level thought to be sufficient for CF gene therapy.30
In adult mice, AAV-9 has been shown to transduce the liver following portal vein or tail vein injection.9,28 We have obtained similar results in adult mice (Supplementary Figure S2). However, in neonatal mice, both the liver and the kidney were poorly transduced (Figures 3b and c, 4c). This observation is consistent with a recent study using AAV-8.3 AAV-8 is considered a superior serotype for liver gene transfer.31 Systemic delivery of AAV-8 in adult mice resulted in strong expression in the liver.6 However, systemic delivery of AAV-8 in newborn mice failed to maintain persistent transduction.3 After 2 weeks, liver transduction drops to the undetectable levels.3 It is currently thought that the loss of expression after neonatal infection is mainly due to the loss of vector genome during postnatal development in these organs.3
In summary, we have demonstrated for the first time that tsAAV can be used for whole-body gene transfer. Importantly, the level of transduction in several tissues is quite suitable for therapeutic applications. CF and DMD are two lethal systemic diseases affecting mainly the lung and muscle, respectively. Traditionally, the large size of the disease genes has precluded the possibility of delivering a full-length cDNA expression cassette with a single intact AAV vector. The development of the trans-splicing vectors has raised the hope of solving the size issue. The demonstration of efficient gene expression in all body muscle as well as in the lung has made it possible to capitalize on the large packaging capacity of tsAAV to treat these diseases. One remaining issue is expression in tissues that are not affected by the disease. This represents an important safety concern for any systemic gene delivery approach. It is possible that the expanded packaging capacity in tsAAV may allow inclusion of large tissue-specific regulatory elements (such as a tissue-specific promoter) to minimize the risk associated with systemic delivery.
MATERIALS AND METHODS
AAV production
The cis plasmid for the single AP intact vector (pcisAV.AP) has been reported before.16,25 The cis plasmids for AP tsAAV were generated by a multistep cloning procedure. We first inserted a synthetic intron from pCI (Promega, Madison, WI) at the junction of exons 8 and 9 in pcisAV.AP. Next, we created a unique KpnI site at the center of the synthetic intron in this intermediate construct by inactivating a second KpnI site at the backbone. There are two SalI sites in the original pcisAV.AP plasmid. One is located right before the RSV promoter and another is located right after the polyA. We then generated two additional intermediate constructs by selectively inactivating one of the SalI sites. Finally, pcisAV.AP. Donor was generated by KpnI/SalI double digestion from one of the last step intermediate to remove the second half of the intron, the 3′ end of the AP gene and polyA. pcisAV.AP.Acceptor was generated by SalI/KpnI digestion from the other last step intermediate to remove the RSV promoter, the 5′ portion of the AP gene and the first half of the synthetic intron.
AAV-9 vectors were produced by triple plasmid transfection using a pRep2/Cap9 helper plasmid generously provided by Drs. Gao and Wilson.28,32 Recombinant viral stocks were purified through two rounds of isopycnic CsCl2 ultracentrifugation as described before.15 Viral titration and quality control were performed according to our previous publications.16,23
In vivo gene delivery
All animal experiments were approved by the Animal Care and Use Committee at the University of Missouri and were in accordance with NIH guidelines. In this study, we used newborn C57Bl/10 (BL10) mice. Adult breeders were purchased from The Jackson Laboratory (Bar Harbor, Maine). Breeding was carried out in a specific-pathogen-free facility. We first optimized the injection condition with tattoo dye (time, route, and volume). Best results were achieved at 36 to 48 h after mice were born by facial vein injection. The maximal deliverable volume was 70 μl. For AV.AP infection, 3.5 × 1011 vg AAV-9 particles were delivered in 70 μl 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer with a custom-designed 33-gauge gas-tight Hamilton syringe (Hamilton, Reno, NV). For tsAAV infection, 3.5 × 1011 vg particles of each vector (AV.AP.Donor and AV.AP.Acceptor) were mixed thoroughly in 70 μl 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer and then delivered using similar method. In mock-treated animals, we injected 70 μl 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid buffer. After injection, mice were returned to the cage for recovery. More than 95% neonatal mice survived the procedure.
Gene expression evaluation
At 6 weeks postinfection, mice were killed and various tissues/organs were examined for AP expression using our published histochemical staining protocol25 and an AP enzyme activity assay (StemTAG AP Activity Assay kit; Cell biolabs http://www.cellbiolabs.com; Cat. No. CBA-301). To compare transduction efficiency among experimental groups, we used two different quantification methods. We first performed morphometric analyses on digitized tissue sectional images. In skeletal and cardiac muscle, the liver and the kidney, we used NIH Image J software (version 1.36b) to quantify the percentage of AP-positive area (AP-positive area/total tissue area) and relative AP staining intensity (the mean intensity of AP-positive area in AAV-infected tissue normalized by the average mean intensity in mock-infected tissue) according to previous publications.33-36 In morphometric study, three to seven representative cross-section images were quantified for each tissue sample. Images from three to eight independent samples were examined for each tissue. In lung gene therapy studies, transduction efficiency is usually quantified by counting the number of positive cells in conducting airway and alveoli, respectively.25,29,37 We have used similar manual quantification method for the lung. Besides morphometric quantification, we also measured the total AP activity in tissue lysate using the StemTAG AP kit. The assay was performed essentially as described in the manufacturer's manual except that we first incubated tissue lysate at 65°C for 1 h to inactivate endogenous heat-labile AP. In lysate assay, the AP activity is defined as μM of para-nitrophenol being generated per microgram of total protein in the tissue lysate (one molecule of para-nitrophenol is generated when one molecule of phosphate is released from the substrate).
Statistical analysis
Data are presented as mean±s.e.m. Statistical analysis was performed with the SPSS software (SPSS, Chicago, IL). In two-group comparison (such as these in morphometric quantification studies), we used two tailed independent student t-test. In three-group comparison (such as tissue lysate AP activity assay), we used one-way analysis of variance followed by Tukey post hoc analysis. Difference was considered significant when P<0.05.
Supplementary Material
Supplementary Figure 1. Systemic delivery of AAV-9 in neonatal mice does not lead to efficient smooth muscle transduction. 3.5 × 10e11 vg particles of AV.AP (pseudopackaged in AAV-9 capsid) were delivered to newborn mice via facial vein. AP expression in smooth muscle was evaluated at six weeks post-infection. A, AP expression in the esophagus is robust in the skeletal muscle layer but poor in the smooth muscle layer. Serial sections of the middle one third of the esophagus was examined by AP staining (counter-stained with eosin) and immunostaining with antibodies specific for smooth muscle (monoclonal anti-α smooth muscle actin; catalog # A 2547; Sigma, St. Louis, MO) and skeletal muscle (polyclonal anti-skeletal muscle myosin; catalog # M 7523; Sigma, St. Louis, MO), respectively. Top panel, full view photomicrographs of the entire esophagus cross-section; Middle and bottom panels, high power photomicrographs of the boxed areas (box 1 and box 2, respectively) in the top panel. Arrowheads mark smooth muscle in the middle and bottom panels; Asterisks mark skeletal muscle in the middle and bottom panels. Scale bar in the top panel, 400 μm; scale bar in the middle and bottom panel, 100 μm. B, AP expression in the aorta. Some expression was seen in endothelial cells (arrows) but smooth muscle was scarcely transduced (arrowheads). Scale bar in the top panel, 200 μm; scale bar in the bottom panel, 50 μm. C, AP expression in small vessels in the heart (top panel) and skeletal muscle (bottom panel). Expression was detected in skeletal muscle myofibers, cardiomycytes and endothelial cells but not in smooth muscle. Serial tissue sections were stained by AP (counter-stained with eosin) and smooth muscle α-actin antibody, respectively. Scale bar, 100 μm.
Supplementary Figure 2. AAV-9 efficiently transduces the liver and the kidney in adult mice but not in neonatal mice. To compare the relative transduction efficiency, the same batch of AAV-9 virus (AV.AP) was delivered to neonatal mice (via facial vein; 3.5 × 10e11 vg particles/mouse = 2.33 ×10e11 vg particles/gram body weight) and 6-week-old adult mice (via tail vein; 1 ×10e12 vg particles/mouse = 5 × 10e10 vg particles/gram body weight), respectively. Transgene expression was examined at six weeks later by histochemical staining of the tissue sections. The liver and the kidney in adult mice were transduced much more efficiently than the corresponding organs in neonatal mice, respectively. Scale bar, 200 μm.
ACKNOWLEDGMENTS
We thank Drs. Guangping Gao and Jim Wilson for providing the AAV-9 packaging plasmid pRep2/Cap9. We also thank Dr. Gang Yao for the help with image quantification and Mr. Hyun-Hee Lee for sharing facial vein injection technique. This work is supported by grants from the National Institutes of Health (AR-49419, DD), the Muscular Dystrophy Association (DD), and the Cystic Fibrosis Foundation (DD).
REFERENCES
- 1.Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG, et al. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat Med. 2004;10:828–834. doi: 10.1038/nm1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blankinship MJ, Gregorevic P, Allen JM, Harper SQ, Harper H, Halbert CL, et al. Efficient transduction of skeletal muscle using vectors based on adeno-associated virus serotype 6. Mol Ther. 2004;10:671–678. doi: 10.1016/j.ymthe.2004.07.016. [DOI] [PubMed] [Google Scholar]
- 3.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 4.Zhu T, Zhou L, Mori S, Wang Z, McTiernan CF, Qiao C, et al. Sustained whole-body functional rescue in congestive heart failure and muscular dystrophy hamsters by systemic gene transfer. Circulation. 2005;112:2650–2659. doi: 10.1161/CIRCULATIONAHA.105.565598. [DOI] [PubMed] [Google Scholar]
- 5.Qiao C, Li J, Zhu T, Draviam R, Watkins S, Ye X, et al. Amelioration of laminin-alpha2-deficient congenital muscular dystrophy by somatic gene transfer of miniagrin. Proc Natl Acad Sci USA. 2005;102:11999–12004. doi: 10.1073/pnas.0502137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakai H, Fuess S, Storm TA, Muramatsu S, Nara Y, Kay MA. Unrestricted hepatocyte transduction with adeno-associated virus serotype 8 vectors in mice. J Virol. 2005;79:214–224. doi: 10.1128/JVI.79.1.214-224.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mah C, Cresawn KO, Fraites TJ, Jr, Pacak CA, Lewis MA, Zolotukhin I, et al. Sustained correction of glycogen storage disease type II using adeno-associated virus serotype 1 vectors. Gene Ther. 2005;12:1405–1409. doi: 10.1038/sj.gt.3302550. [DOI] [PubMed] [Google Scholar]
- 8.Denti MA, Rosa A, D'Antona G, Sthandier O, De Angelis FG, Nicoletti C, et al. Body-wide gene therapy of Duchenne muscular dystrophy in the mdx mouse model. Proc Natl Acad Sci USA. 2006;103:3758–3763. doi: 10.1073/pnas.0508917103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inagaki K, Fuess S, Storm TA, Gibson GA, Mctiernan CF, Kay MA, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006;14:45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DF, et al. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006;99:e3–e9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- 11.Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L, et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med. 2006;12:787–789. doi: 10.1038/nm1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter BJ. Adeno-associated virus and the development of adeno-associated virus vectors: a historical perspective. Mol Ther. 2004;10:981–989. doi: 10.1016/j.ymthe.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 13.Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther. 2005;5:285–297. doi: 10.2174/1566523054065057. [DOI] [PubMed] [Google Scholar]
- 14.Duan D, Yan Z, Engelhardt JF. Expanding the capacity of AAV vectors. In: Bloom ME, et al., editors. Parvoviruses. Hodder Arnold; Distributed in the USA by Oxford University Press; London, New York: 2006. pp. 525–532. [Google Scholar]
- 15.Lai Y, Yue Y, Liu M, Ghosh A, Engelhardt JF, Chamberlain JS, et al. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol. 2005;23:1435–1439. doi: 10.1038/nbt1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M, Yue Y, Harper SQ, Grange RW, Chamberlain JS, Duan D. Adeno-associated virus-mediated micro-dystrophin expression Protects young Mdx muscle from contraction-induced injury. Mol Ther. 2005;11:245–256. doi: 10.1016/j.ymthe.2004.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yue Y, Liu M, Duan D. C-terminal truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double knock-out mice. Mol Ther. 2006;14:79–87. doi: 10.1016/j.ymthe.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun L, Li J, Xiao X. Overcoming adeno-associated virus vector size limitation through viral DNA heterodimerization. Nat Med. 2000;6:599–602. doi: 10.1038/75087. [DOI] [PubMed] [Google Scholar]
- 19.Yan Z, Zhang Y, Duan D, Engelhardt JF. From the cover: trans-splicing vectors expand the utility of adeno-associated virus for gene therapy. Proc Natl Acad Sci USA. 2000;97:6716–6721. doi: 10.1073/pnas.97.12.6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duan D, Yue Y, Engelhardt JF. Expanding AAV packaging capacity with trans-splicing or overlapping vectors: a quantitative comparison. Mol Ther. 2001;4:383–391. doi: 10.1006/mthe.2001.0456. [DOI] [PubMed] [Google Scholar]
- 21.Chao H, Sun L, Bruce A, Xiao X, Walsh CE. Expression of human factor VIII by splicing between dimerized AAV vectors. Mol Ther. 2002;5:716–722. doi: 10.1006/mthe.2002.0607. [DOI] [PubMed] [Google Scholar]
- 22.Reich SJ, Auricchio A, Hildinger M, Glover E, Maguire AM, Wilson JM, et al. Efficient trans-splicing in the retina expands the utility of adeno-associated virus as a vector for gene therapy. Hum Gene Ther. 2003;14:37–44. doi: 10.1089/10430340360464697. [DOI] [PubMed] [Google Scholar]
- 23.Xu Z, Yue Y, Lai Y, Ye C, Qiu J, Pintel DJ, et al. Trans-splicing adeno-associated viral vector-mediated gene therapy is limited by the accumulation of spliced mRNA but not by dual vector coinfection efficiency. Hum Gene Ther. 2004;15:896–905. doi: 10.1089/hum.2004.15.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lai Y, Yue Y, Liu M, Duan D. Synthetic intron improves transduction efficiency of the trans-splicing adeno-associated viral vectors. Hum Gene Ther. 2006;17:1036–1042. doi: 10.1089/hum.2006.17.ft-246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan D, Yue Y, Yan Z, Yang J, Engelhardt JF. Endosomal processing limits gene transfer to polarized airway epithelia by adeno-associated virus. J Clin Invest. 2000;105:1573–1587. doi: 10.1172/JCI8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yue Y, Skimming JW, Liu M, Strawn T, Duan D. Full-length dystrophin expression in half of the heart cells ameliorates beta-isoproterenol-induced cardiomyopathy in mdx mice. Hum Mol Genet. 2004;13:1669–1675. doi: 10.1093/hmg/ddh174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin-glycoprotein complex and improves sarcolemma integrity in the Mdx mouse heart. Circulation. 2003;108:1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao GP, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–6388. doi: 10.1128/JVI.78.12.6381-6388.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Limberis MP, Wilson JM. Adeno-associated virus serotype 9 vectors transduce murine alveolar and nasal epithelia and can be readministered. Proc Natl Acad Sci USA. 2006;103:12993–12998. doi: 10.1073/pnas.0601433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson LG, Olsen JC, Sarkadi B, Moore KL, Swanstrom R, Boucher RC. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 31.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci USA. 2002;99:11854–11859. doi: 10.1073/pnas.182412299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao G, Alvira MR, Somanathan S, Lu Y, Vandenberghe LH, Rux JJ, et al. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc Natl Acad Sci USA. 2003;100:6081–6086. doi: 10.1073/pnas.0937739100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan Z, Zak R, Zhang Y, Ding W, Godwin S, Munson K, et al. Distinct classes of proteasome-modulating agents cooperatively augment recombinant adeno-associated virus type 2 and type 5-mediated transduction from the apical surfaces of human airway epithelia. J Virol. 2004;78:2863–2874. doi: 10.1128/JVI.78.6.2863-2874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grider MH, Mamounas LA, Le W, Shine HD. In situ expression of brain-derived neurotrophic factor or neurotrophin-3 promotes sprouting of cortical serotonergic axons following a neurotoxic lesion. J Neurosci Res. 2005;82:404–412. doi: 10.1002/jnr.20635. [DOI] [PubMed] [Google Scholar]
- 35.Ficklin MB, Zhao S, Feng G. Ubiquilin-1 regulates nicotine-induced up-regulation of neuronal nicotinic acetylcholine receptors. J Biol Chem. 2005;280:34088–34095. doi: 10.1074/jbc.M506781200. [DOI] [PubMed] [Google Scholar]
- 36.Chan P, Yuen T, Ruf F, Gonzalez-Maeso J, Sealfon SC. Method for multiplex cellular detection of mRNAs using quantum dot fluorescent in situ hybridization. Nucleic Acids Res. 2005;33:e161. doi: 10.1093/nar/gni162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Halbert CL, Allen JM, Miller AD. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat Biotechnol. 2002;20:697–701. doi: 10.1038/nbt0702-697. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Systemic delivery of AAV-9 in neonatal mice does not lead to efficient smooth muscle transduction. 3.5 × 10e11 vg particles of AV.AP (pseudopackaged in AAV-9 capsid) were delivered to newborn mice via facial vein. AP expression in smooth muscle was evaluated at six weeks post-infection. A, AP expression in the esophagus is robust in the skeletal muscle layer but poor in the smooth muscle layer. Serial sections of the middle one third of the esophagus was examined by AP staining (counter-stained with eosin) and immunostaining with antibodies specific for smooth muscle (monoclonal anti-α smooth muscle actin; catalog # A 2547; Sigma, St. Louis, MO) and skeletal muscle (polyclonal anti-skeletal muscle myosin; catalog # M 7523; Sigma, St. Louis, MO), respectively. Top panel, full view photomicrographs of the entire esophagus cross-section; Middle and bottom panels, high power photomicrographs of the boxed areas (box 1 and box 2, respectively) in the top panel. Arrowheads mark smooth muscle in the middle and bottom panels; Asterisks mark skeletal muscle in the middle and bottom panels. Scale bar in the top panel, 400 μm; scale bar in the middle and bottom panel, 100 μm. B, AP expression in the aorta. Some expression was seen in endothelial cells (arrows) but smooth muscle was scarcely transduced (arrowheads). Scale bar in the top panel, 200 μm; scale bar in the bottom panel, 50 μm. C, AP expression in small vessels in the heart (top panel) and skeletal muscle (bottom panel). Expression was detected in skeletal muscle myofibers, cardiomycytes and endothelial cells but not in smooth muscle. Serial tissue sections were stained by AP (counter-stained with eosin) and smooth muscle α-actin antibody, respectively. Scale bar, 100 μm.
Supplementary Figure 2. AAV-9 efficiently transduces the liver and the kidney in adult mice but not in neonatal mice. To compare the relative transduction efficiency, the same batch of AAV-9 virus (AV.AP) was delivered to neonatal mice (via facial vein; 3.5 × 10e11 vg particles/mouse = 2.33 ×10e11 vg particles/gram body weight) and 6-week-old adult mice (via tail vein; 1 ×10e12 vg particles/mouse = 5 × 10e10 vg particles/gram body weight), respectively. Transgene expression was examined at six weeks later by histochemical staining of the tissue sections. The liver and the kidney in adult mice were transduced much more efficiently than the corresponding organs in neonatal mice, respectively. Scale bar, 200 μm.