

Abstract
Relatively little is known about the seminal genetic events that trigger the development of low-grade gliomas in children. Genetically engineered mouse models of the neurofibromatosis-1–inherited tumor predisposition syndrome have identified key intracellular growth control pathways, defined the contribution of the tumor microenvironment to glioma growth, and helped researchers understand the genetic basis for glioma susceptibility. In addition, genetically engineered mouse low-grade glioma models have recently been used in preclinical therapeutic studies to evaluate the efficacy of particular biologically based therapies and to define outcome measures.
Keywords: Neurofibromin, astrocytoma, tumor microenvironment, preclinical therapeutics, brain tumor, genetically engineered mice
Introduction
Low-grade gliomas represent the most common brain tumor in the pediatric population, accounting for 30% of all central nervous system primary tumors in individuals younger than 20 years of age.1 Both grade I (pilocytic astrocytomas) and grade II (diffuse fibrillary astrocytomas) tumors are included within the World Health Organization group of low-grade glial neoplasms.2 Pilocytic astrocytomas are the more common histologic subtype (15%-20% of all primary central nervous system neoplasms) and are generally circumscribed, slow-growing tumors composed of neoplastic glial fibrillary acidic protein (GFAP)–immunoreactive cells. These tumors may arise anywhere within the neuroaxis but are most commonly seen in the optic pathway, cerebellum, and brainstem. Microscopically, these tumors are characterized by a biphasic cellular appearance, in which areas composed of compacted bipolar cells with Rosenthal fibers alternate with areas composed of loose textured multipolar cells with microcysts and eosinophilic granular bodies. Consistent with their slow growth rates, pilocytic astrocytomas have rare mitotic activity; however, despite their benign nature, significant microvascular proliferation may be seen.
In contrast to pilocytic astrocytomas, World Health Organization grade II astrocytomas are usually diffusely infiltrative neoplasms. They may develop in any region of the central nervous system but are most commonly located in the cerebral lobes and brainstem. Microscopically, diffuse fibrillary astrocytomas are composed of well-differentiated neoplastic glial fibrillary acidic protein–immunoreactive cells, with moderately increased cellularity and occasional nuclear atypia. Necrosis and microvascular proliferation are not usually found in these tumors. It is worth noting that grade II astrocytomas in adults typically progress to higher grade malignancies over time, whereas in children, grade II astrocytomas exhibit a more generally benign clinical course. In this regard, the 10-year overall survival for children and young adults with pilocytic astrocytoma or fibrillary astrocytoma is greater than 90% and 80%, respectively.1
Treatment for pediatric low-grade glioma has more recently focused on the use of chemotherapy, owing to the neurocognitive and endocrine sequelae associated with radiation therapy.3,4 However, most of the agents currently in clinical use are antineoplastic drugs that have been used for the treatment of adult brain tumors and do not specifically target the unique biochemical or cellular abnormalities found in pediatric low-grade glioma. Unfortunately, efforts to develop such drugs are limited by the relative lack of information regarding the key genetic changes important for pediatric low-grade glioma formation and growth.
One approach to identifying the seminal molecular changes that drive low-grade glioma development and continued growth involves the study of inherited cancer syndromes in which affected individuals are prone to low-grade glioma formation. The most common of these pediatric syndromes is neurofibromatosis type 1, also known as von Recklinghausen's disease5: 15% to 20% of children with neurofibromatosis type 1 develop low-grade gliomas affecting the optic nerves, optic chiasm, and hypothalamus (optic pathway gliomas).6,7 Most of these gliomas are classified as World Health Organization grade I tumors with intense glial fibrillary acidic protein immunostaining and low proliferative indices.8,9 Interestingly, in the context of neurofibromatosis type 1, optic pathway gliomas typically develop in young children (mean age = 4 years), exhibit indolent growth patterns, and have even been reported to regress spontaneously.10
Examination of pilocytic astrocytomas from children with neurofibromatosis type 1 has confirmed biallelic NF1 gene inactivation and loss of NF1 protein (neurofibromin) expression.8 In contrast, sporadic pilocytic astrocytomas do not harbor inactivating NF1 mutations and likely result from different genetic changes.11,12 Despite the fact that only 15% of all pilocytic astrocytomas result from loss of NF1 function, it is highly likely that pilocytic astrocytoma formation in the general population reflects deregulation of growth control pathways similar to those modulated by NF1. Support for this notion derives from studies demonstrating that neurofibromin is a negative regulator of the RAS proto-oncogene in vitro and in vivo, such that NF1 inactivation leads to increased activation of RAS and RAS downstream effectors.13-15 Similar to neurofibromatosis type 1–associated pilocytic astrocytomas, sporadic pilocytic astrocytomas also exhibit increased activation of RAS downstream effectors,16 suggesting that molecular changes that mimic NF1 loss may contribute to gliomagenesis in sporadic pilocytic astrocytomas. One of these changes is mutational activation of RAS, resulting in a RAS molecule that is constitutively activated, similar to the effects of neurofibromin loss. Recently, there have been two reports of KRAS oncogenic mutations in sporadic pilocytic astrocytoma.16,17
In many respects, neurofibromatosis type 1 represents a tractable model system to study the molecular and cellular pathogenesis of pediatric low-grade glioma. Importantly, neurofibromatosis type 1 is an excellent genetic model with which to study low-grade glioma formation and growth, since the initiating molecular event (loss of NF1 function) is well established. Using neurofibromatosis type 1 as a portal into sporadic gliomagenesis, we will discuss how cell-based and genetically engineered mouse studies have defined some of the key intracellular signaling pathways responsible for low-grade glioma growth, elucidated the role of the tumor microenvironment in gliomagenesis, and uncovered the contribution of “modifier genes” to glioma formation (Figure 1). Lastly, we will discuss how genetically engineered Nf1 mouse optic glioma models can be used for both therapeutic discovery and candidate drug evaluation prior to human clinical trials.
Figure 1.
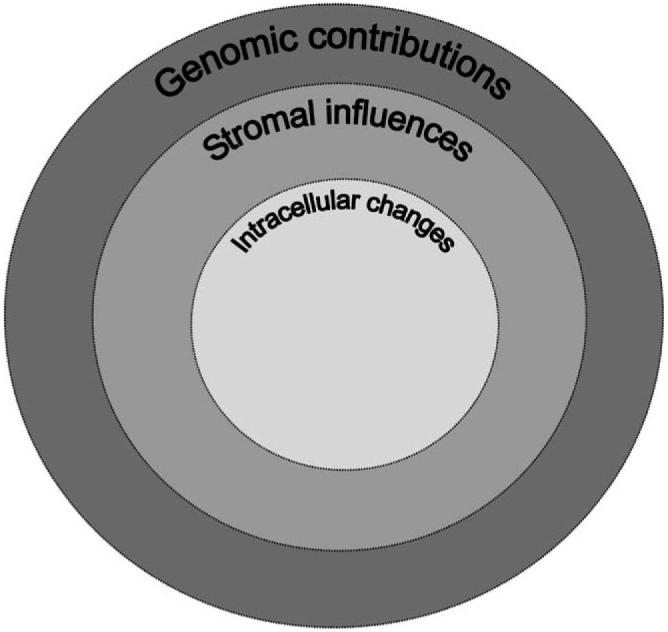
Low-grade glioma formation represents the composite effects of molecular changes that reflect deregulated intracellular growth control pathways (intracellular changes), important cellular and biochemical signals that emanate from the tumor microenvironment (stromal influences), and modifier genes in the genome (genomic contributions).
Intracellular Signaling Pathways
With the identification of the NF1 gene in 1990,18,19 it became possible to define the mechanism underlying NF1 tumor suppressor function. Analysis of the predicted NF1 protein sequence revealed that neurofibromin contains a small domain remarkably similar in structure to the functional domain of a family of proteins that negatively regulate RAS proteins.20 These negative RAS regulators, termed guanosine triphosphatase (GTPase)–activating proteins, inactivate RAS by accelerating the conversion of active, GTP-bound RAS to inactive, GDP-bound RAS.21 Active RAS in many cell types drives cell proliferation by initiating a cascade of protein phosphorylation events that culminate in increased cell proliferation and/or decreased cell death. Moreover, activating RAS mutations are oncogenic and lead to tumor formation in mice. In this regard, loss of neurofibromin expression in neurofibromatosis type 1–associated tumors leads to increased RAS activity, which likely initiates the process of tumor formation.
The observation that RAS hyperactivation results from NF1 inactivation in tumors prompted investigators to inhibit RAS activity in cells and tumors lacking neurofibromin expression. These in vitro experiments clearly demonstrated that RAS inhibition, either pharmacologically or genetically, reversed the growth advantage conferred by NF1 loss. Based on these exciting preliminary preclinical data, a series of human clinical studies using RAS inhibitors to treat neurofibromatosis type 1–associated tumors was initiated. RAS activation requires a posttranslational lipid modification that facilitates the insertion of RAS into the plasma membrane and allows RAS to efficiently activate its downstream effects and promote cell growth.22 This lipid modification (farnesylation) is inhibited by farnesyltransferase inhibitors that had been developed to treat other cancers.23 Unfortunately, treatment of neurofibromatosis type 1 patients harboring peripheral nerve tumors (plexiform neurofibromas) with farnesyltransferase inhibitors has not resulted in reproducible tumor shrinkage to date.24
These seemingly disappointing clinical trial results may have been foreshadowed by elegant genetically engineered mouse studies by Mahgoub and colleagues.25 Using their Nf1 leukemia mouse model, they found that RAS inhibition by farnesyltransferase inhibitors attenuated the growth of Nf1-deficient mouse leukemic cells in vitro, but had no effect on leukemia development in vivo. One possible explanation for these results is preferential regulation of specific RAS isoforms by neurofibromin. Using Nf1-deficient mouse astrocytes, we first demonstrated that neurofibromin loss results only in K-RAS activation, despite the fact that all three RAS isoforms (H-RAS, K-RAS, and N-RAS) are expressed in astrocytes.26 Moreover, optic glioma formation in Nf1+/− mice is induced by K-RAS (but not H-RAS) activation in astroglial cells in vivo. Recent studies have now shown that neurofibromin selectively regulates specific RAS isoforms in other cell types,27,28 suggesting that future therapeutic approaches will need to target the RAS isoform that is specifically hyperactivated in any given cell as a result of neurofibromin loss.
To identify other signaling intermediates activated by neurofibromin loss in primary mouse astrocytes, we used a proteomics-based approach. We found that a large number of proteins involved in ribosomal biogenesis and protein translation control were increased in Nf1−/− astrocytes relative to wild-type controls.29 Consistent with this observation, protein translation was increased fivefold to eightfold in Nf1-deficient astrocytes. One of the major signaling pathways responsible for regulating protein translation is the mammalian target of rapamycin (mTOR) pathway (Figure 2). mTOR is a large serine/threonine protein kinase molecule that integrates a diverse number of extracellular cues (eg, hypoxia, amino acid availability).30,31
Figure 2.
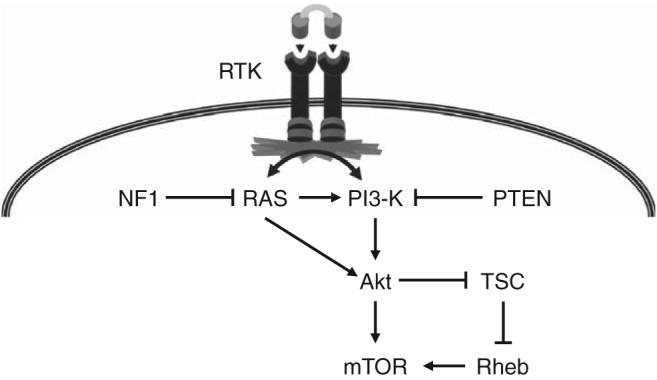
Activation of the mammalian target of rapamycin (mTOR) pathway can result from numerous glioma-associated genetic changes. Inactivation of the NF1 gene results in increased RAS activity, which in turn leads to activation of phosphoinositol-3-kinase and Akt. Increased Akt activity leads to increased mTOR activation, either by direct activation of mTOR or via phosphorylation and inactivation of the TSC signaling complex. Loss of TSC gene function results in increased RAS homolog enriched in brain activity, which in turn results in increased mTOR activity. In addition, the PTEN tumor suppressor gene is frequently mutationally inactivated in gliomas, leading to increased phosphoinositol-3-kinase activity and mTOR activation. Similarly, mutational activation or constitutive signaling through receptor tyrosine kinases, such as EGFR, results in RAS and phosphoinositol-3-kinase hyperactivation and downstream increased mTOR pathway activation.
Recent studies have shown that one way mTOR regulates ribosomal biogenesis and protein translation is by modulating the synthesis of a nucleolar shuttling protein called nucleophosmin.32,33 In part, nucleophosmin functions to chaperone newly synthesized ribosomal subunits from the nucleolus to the cytoplasm, where protein translation occurs.34 In this fashion, mTOR regulation of nucleophosmin controls the rate of protein synthesis at the level of the ribosome. We have shown that neurofibromin regulates nucleophosmin levels in astrocytes in a mTOR-dependent, rapamycin-inhibitable fashion in vitro and in vivo.33 Moreover, inhibition of nucleophosmin nuclear shuttling reverses the abnormal cellular phenotypes (proliferation, motility, and actin cytoskeleton organization) found in Nf1-deficient astrocytes. Studies are ongoing to determine precisely how nucleophosmin regulates protein translation at the ribosome and to identify specific translationally regulated transcripts that underlie the various Nf1-deficient cellular phenotypes.
Similar to deregulated RAS activity, mTOR activation is a common feature of sporadic low-grade glioma. In this regard, several key glioma-associated genetic changes result in increased mTOR activity (Figure 2). The inherited tumor predisposition syndrome, tuberous sclerosis complex, results from inactivating mutations in the tuberous sclerosis complex genes, TSC1 and TSC2.35,36 The gene products of TSC1 (hamartin) and TSC2 (tuberin) form a single signaling complex that functions to negatively regulate the small RAS-like protein, Ras homolog enriched in brain (Rheb), which in turn binds to and activates mTOR.37-39 Loss of TSC function results in increased Rheb activity and high levels of mTOR pathway activation. Moreover, inhibition of mTOR function using rapamycin results in decreased growth of human tuberous sclerosis complex–associated brain tumors.40
Mutational inactivation of the PTEN gene is a common genetic signature of high-grade glioma.41 PTEN is a negative regulator of the phosphoinositol-3-kinase protein,42 such that PTEN loss in human and mouse tumors leads to high levels of Akt activity. Akt can either directly or indirectly activate mTOR by phosphorylating tuberin to result in loss of TSC complex function, high levels of Rheb activity, and mTOR activation.43-45 Lastly, mutational activation of EGFR is also observed in many high-grade gliomas.46 Increased EGFR signaling leads to increased RAS and phosphoinositol-3-kinase activity, which culminates in increased mTOR activation. Taken together, while NF1 loss only accounts for a small fraction of all low-grade glioma-associated genetic changes, the identification of mTOR as a target for neurofibromin growth regulation has expanded our understanding of the key growth control pathways operative in glioma, and suggests that additional low-grade glioma genetic changes will be identified by virtue of their ability to result in increased mTOR pathway activation.
Stromal Influences
As does the developing brain, neoplastic glial cells respond to both positive and negative signals that emanate from cells in the local tumor microenvironment. In addition to non-neoplastic glial cells and neurons, at least two other important cell types (endothelial cells and microglia) that can influence tumorigenesis and growth derive from the normal brain (Figure 3). Endothelial cells lining the tumor vasculature not only produce a vast number of growth-promoting molecules but may also create a specialized cellular niche for progenitor cells important for tumor viability.47 In addition, immune system macrophage-like cells (microglia) are also found in human brain tumors.48,49 These cells likewise elaborate a large number of growth factors and cytokines that could potentially dictate when and where tumors form.50,51 Collectively, these findings support the notion that low-grade gliomas represent complex cellular microcosms composed of neoplastic glial cells nested within an environment rich in growth factors that recapitulate some of the cues that regulate normal brain development during embryogenesis.
Figure 3.
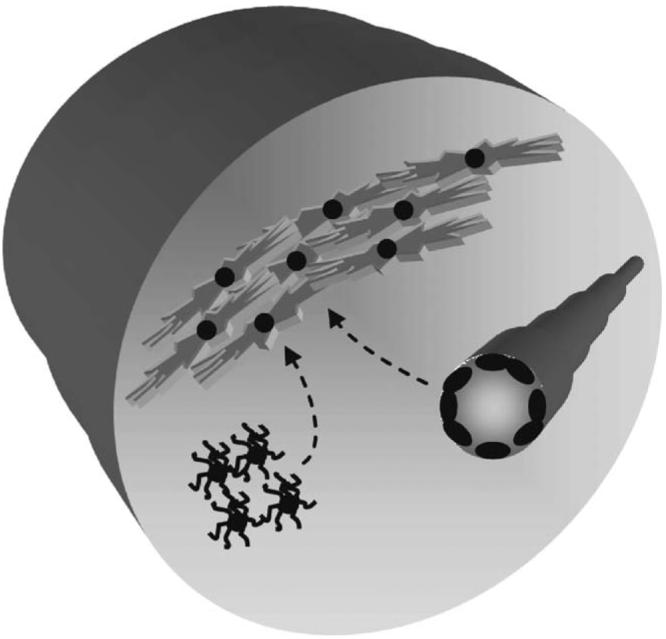
The low-grade glioma microcosm is composed of numerous distinct cell types that each may contribute uniquely to tumorigenesis and continued glioma growth. For example, microglia and endothelial cells provide important stroma-derived signals that promote glioma growth.
Insights into the role of the tumor microenvironment have derived from experiments in which mice were engineered to lack Nf1 expression in astrocytes or Schwann cells. Because optic gliomas and neurofibromas in humans are associated with NF1 loss in glial and Schwann cells, respectively, mice lacking neurofibromin in these cell types might be predicted to develop gliomas and neurofibromas. While these mice had increased numbers of glial and Schwann cells, they did not develop gliomas or neurofibromas.52,53 Because individuals with neurofibromatosis type 1 start life with one mutated (nonfunctional) NF1 gene in all cells of their body and lose the one remaining functional allele only in select glial or Schwann cells to result in glioma or neurofibroma formation, Nf1+/− mice (harboring one mutated and one wild-type Nf1 gene) that lack neurofibromin expression in glial or Schwann cells were developed. Nf1+/− mice with Schwann cell neurofibromin loss developed neurofibromas,52 while Nf1+/− mice with neurofibromin loss in glial cells formed optic gliomas.54 These results strongly suggested that Nf1+/− cells in the tumor microenvironment are crucial for tumor formation in both the central and peripheral nervous system.
Based on the observation that microglia are found in human low-grade gliomas as well as in a genetically engineered Nf1 mouse optic glioma model,55 studies were designed to address the possibility that Nf1+/− microglia have unique properties relevant to promoting the growth of Nf1−/− astrocytes.56 First, Nf1+/− microglia were shown to proliferate faster than wild-type microglia in vitro. Second, inactivation of microglia in Nf1 genetically engineered mice resulted in reduced optic glioma proliferation in vivo. Third, Nf1+/− microglia elaborate paracrine factors that increase the proliferation of Nf1−/− but not wild-type, astrocytes in vitro. One of these factors was hyaluronidase, an enzyme that degrades hyaluronan, a component of the extracellular matrix, and influences both cell proliferation and motility.57 Lastly, hyaluronidase promotes Nf1-deficient astrocyte proliferation by signaling through the neurofibromin-regulated mitogen-activated protein kinase (MAPK) pathway. Taken together, these data support a clear role for microglia in neurofibromatosis type 1–associated glioma growth and suggest therapeutic approaches that target either microglia or microglia-produced growth factors should be considered.
In addition to hyaluronidase, Nf1+/− microglia also produce increased levels of a cytokine termed CXCL12 or stromal-derived growth factor-1α (SDF-1α) that modulates both RAS activity and cyclic AMP levels.58 Previous studies have shown that another function of neurofibromin is regulation of intracellular cyclic AMP levels, such that loss of neurofibromin in astrocytes and other cell types leads to reduced cyclic AMP generation.59,60 Examination of both neurofibromatosis type 1–associated human and mouse optic gliomas revealed that CXCL12 was not only produced by microglia but also by endothelial cells and neurons.61 Interestingly, in these studies, CXCL12 expression was found to be highest along the optic pathway in young mice. This regional and spatial pattern of expression raises the possibility that glioma formation in the optic pathway in young children with neurofibromatosis type 1 may reflect the availability of a critical ligand, CXCL12, that dictates where and when tumors grow. Support for this hypothesis derives from the observation that CXCL12 promotes Nf1−/− astrocyte survival in a cyclic AMP-dependent fashion, whereas CXCL12 treatment leads to cell death (apoptosis) in wild-type astrocytes.
Lastly, the tumor microenvironment might not only influence glioma formation by providing critical developmentally regulated growth-promoting signals but also by generating cellular niches for specific cell types important for tumor maintenance. Recent studies have shown that a small proportion of cells present in human gliomas have stem cell-like properties and may be critical for the generation (or maintenance) of the tumor.62 In this regard, glioma stem cell-like cells can be isolated and shown to have properties attributed to normal neural stem cells (the ability to undergo self-renewal and multilineage differentiation).63,64 Moreover, transplantation of these cells into naïve mouse brains leads to the formation of a glioma that is histologically identical to the original tumor.65
Neural stem cells reside in specialized cellular compartments rich in blood vessels, suggesting that endothelial cells might provide unique signals important for stem cell maintenance. Support for this idea derives from elegant experiments by Calabrese and colleagues in which they demonstrate that glioma stem cell-like cell survival in vitro and in vivo is enhanced by endothelial cells.47 Because pilocytic astrocytomas are rather vascular tumors, it is possible that therapeutic agents that target the tumor vasculature will have the added effect of eliminating the cells in the tumor most responsible for tumor growth and maintenance.66
Genomic Influences
Epidemiologic studies focused on identifying environmental causes for brain tumors have largely been negative, with the notable exception of radiation exposure. While there may be no discernible connections between environmental exposure and glioma development, recent investigations have found that polymorphisms in the mouse genome may account for differences in glioma susceptibility. Seminal studies by Reilly and colleagues have shown that mice heterozygous for mutations in the Nf1 and p53 genes (NPCis mice) are prone to the development of malignant peripheral nerve sheath tumors or gliomas, depending on the genetic background of the mice.67 In this regard, NPCis mice maintained on a 129 inbred background do not develop gliomas, whereas NPCis mice on the C57BL/6 background develop gliomas (Figure 4). The investigators used advanced mouse genetic mapping techniques to localize a mouse glioma susceptibility gene to mouse chromosome 11.68 These exciting findings suggest that one important determinant influencing glioma formation is the genetic background, namely genetic polymorphisms in the genome that modify the effects of glioma-causing genetic changes. Future work in both mice and humans may lead to predictive testing for glioma susceptibility.
Figure 4.
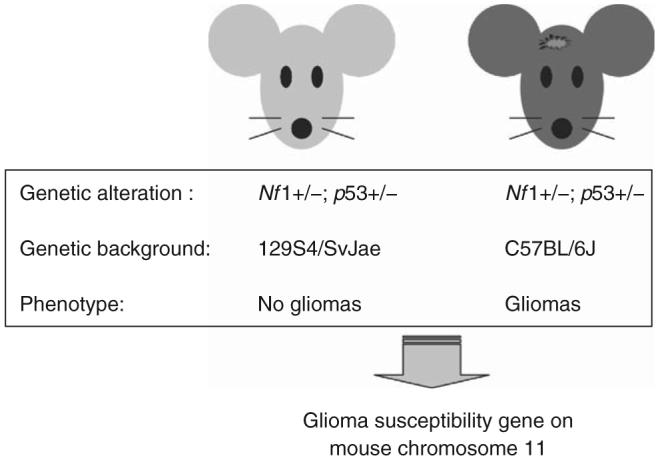
Glioma susceptibility in mice is determined by modifier genes in the mouse genome. Studies by Reilly and coworkers67 have shown that mice harboring the identical glioma-causing genetic changes (combined Nf1 and p53 heterozygosity; NPCis mice) exhibit different susceptibilities to glioma formation that reflects the presence of a glioma susceptibility locus on mouse chromosome 11.
Preclinical Mouse Glioma Models
The development of robust small-animal models of neurofibromatosis type 1–associated optic glioma provides unique opportunities not only to define the molecular and cellular pathogenesis of gliomagenesis but also to improve the treatment of individuals with these low-grade brain tumors (Figure 5). Investigations focused on identifying the critical molecular signals and participating cells in glioma formation and growth provide exceptional opportunities for discovery-based activities. As described above, future therapies may result from targeting different cell types within the tumor microcosm, including glioma stem cell-like cells, microglia, and endothelial cells as well as neutralizing or inhibiting stroma-derived signals that emanate from the tumor microenvironment (eg, hyaluronidase, CXCL12). These newly identified therapeutic targets can subsequently be evaluated in preclinical genetically engineered mouse treatment studies to determine their efficacy in the intact animal under conditions that most closely recapitulate glioma formation in humans.
Figure 5.
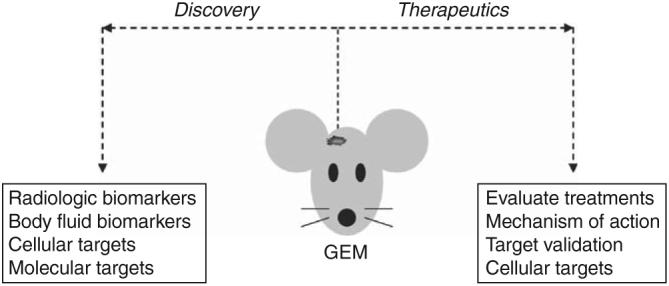
Genetically engineered mouse (GEM) glioma models provide unique opportunities to discover and evaluate new therapies for human low-grade gliomas (for details, refer to the text).
Importantly, small-animal low-grade glioma models may be used to identify surrogate markers of tumor progression or response to therapy. Current endpoints in human clinical trials of low-grade glioma employ changes in overall tumor size as measured by magnetic resonance imaging (MRI). Given the slow growth rate and infiltrative nature of these tumors, a reduction in overall tumor volume may not be an adequate endpoint measure. Advances in small-animal imaging now provide opportunities to exploit MRI to obtain information about the efficacy of chemotherapy using diffusion-based methods. While this has not been applied to low-grade glioma, experiments initially performed in rodent high-grade glioma models and later in human malignant gliomas have shown that changes in water diffusion significantly predate changes in tumor size.69,70 The ability to use MRI as a predictive measure of tumor response to therapy affords unprecedented opportunities to change therapy early during the course of treatment.
In addition to radiologic biomarkers, small-animal models allow investigators to obtain body fluids for analysis. The identification of serum or cerebrospinal fluid biomarkers of glioma disease activity would likewise provide surrogate measures of tumor growth and/or response to therapy. As a proof of concept exercise, we studied our Nf1 optic glioma mouse model to identify cerebrospinal fluid proteins overexpressed in tumor-bearing, but not in control, mice. One such marker, methionine aminopeptidase-2, was shown to be overexpressed in optic gliomas from both mice and children with neurofibromatosis type 1.71
Genetically engineered mouse models also can be used to evaluate candidate therapies.72 Using the Nf1 optic glioma mouse model, we have recently shown that temozolomide monotherapy results in decreased glioma proliferation, increased glioma apoptosis, and decreased tumor volume.73 Because optic gliomas can be detected in these mice using MRI,74 tumor-bearing mice can be randomly assigned to treatment or control arms, and the effect of therapy on tumor growth assessed. Current studies in our laboratory and others using biologically based therapies (eg, rapamycin) highlight some of the important information that can be derived from preclinical studies using Nf1 genetically engineered mouse models. First, one can determine whether the drug reaches its target and inhibits the molecule against which it is primarily designed (“target validation”). Second, the ability of the drug to inhibit other pathways against which the drug was not originally designed can be assessed (“off-target effects”). Third, the mechanism underlying reduced tumor growth can be determined. For example, the ability of the drug to cause cell death (apoptosis) versus reversible effects on cell proliferation can be measured. Similarly, the effect of the drug on stromal cells and glioma stem cell-like cells can be determined to identify the true cellular target of the drug. As such, it is possible that a drug could effectively inhibit differentiated glioma cell growth with little or no effect on cancer-generating cells. Similarly, a drug could target the endothelial cells without a demonstrable impact on neoplastic tumor cells.
Lastly, it is important to use small-animal models to determine why certain therapies might fail. In addition to the reasons outlined above, defining tumor escape mechanisms (eg, feedback loops, activation of other signaling pathways) are critical to the design of future anticancer drugs.75-77 As we move into an era of personalized medicine, it is important to begin to develop therapies that maximally inhibit tumor growth by disabling the cells most essential for continued glioma growth, blocking the intracellular and stroma-derived signals that drive glioma growth and preventing or minimizing the ability of the tumor cell to evade the effect of the anti-cancer drug.
Acknowledgments
This work was supported by grants from the Department of Defense (W81XWH061022) and National Institutes of Health (NS054629). This work also supported by National Institutes of Health grant 5R13NS040925-09. We also appreciate the generous support from Schnuck Markets, Inc. We apologize to investigators whose work we did not discuss in light of manuscript length restrictions.
Footnotes
There are no conflicts of interest.
Presented in part at the Neurobiology of Disease in Children Symposium on Central Nervous System Tumors in conjunction with the 36th annual meeting of the Child Neurology Society, Quebec City, Quebec, October 10, 2007.
References
- 1.CBTRUS 2005-2006 Primary Brain Tumors in the United States Statistical Report 1998-2002 Years Data Collected. Central Brain Tumor Registry of the United States; Available at: http://www.cbtrus.org. Accessed November 30, 2007. [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiester OD, et al., editors. WHO Classification of Tumours of the Central Nervous System. 4th ed. Vol. 1. WHO Press; Geneva: 2007. p. 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benesch M, Lackner H, Sovinz P, et al. Late sequela after treatment of childhood low-grade gliomas: a retrospective analysis of 69 long-term survivors treated between 1983 and 2003. J Neurooncol. 2006;78:199–205. doi: 10.1007/s11060-005-9091-z. [DOI] [PubMed] [Google Scholar]
- 4.Mulhern RK, Palmer SL. Neurocognitive late effects in pediatric cancer. Curr Probl Cancer. 2003;27:177–197. doi: 10.1016/s0147-0272(03)00026-6. [DOI] [PubMed] [Google Scholar]
- 5.Friedman JM, Gutmann DH, MacCollin M, et al. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Johns Hopkins Press; Baltimore, MD: 1999. [Google Scholar]
- 6.Listernick R, Charrow J, Greenwald M, et al. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125:63–66. doi: 10.1016/s0022-3476(94)70122-9. [DOI] [PubMed] [Google Scholar]
- 7.Guillamo JS, Creange A, Kalifa C, et al. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain. 2003;126:152–160. doi: 10.1093/brain/awg016. [DOI] [PubMed] [Google Scholar]
- 8.Gutmann DH, Donahoe J, Brown T, et al. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol. 2000;26:361–367. doi: 10.1046/j.1365-2990.2000.00258.x. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez FJ, Perry A, Gutmann DH, et al. Gliomas in neurofibromatosis type 1: a clinicopathologic study of 100 patients. J Neuropathol Exp Neurol. 2008;67:240–249. doi: 10.1097/NEN.0b013e318165eb75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schmandt SM, Packer RJ, Vezina LG, et al. Spontaneous regression of low-grade astrocytomas in childhood. Pediatr Neurosurg. 2000;32:132–136. doi: 10.1159/000028917. [DOI] [PubMed] [Google Scholar]
- 11.Wimmer K, Eckart M, Meyer-Puttlitz B, et al. Mutational and expression analysis of the NF1 gene argues against a role as tumor suppressor in sporadic pilocytic astrocytomas. J Neuropathol Exp Neurol. 2002;61:896–902. doi: 10.1093/jnen/61.10.896. [DOI] [PubMed] [Google Scholar]
- 12.Kluwe L, Hagel C, Tatagiba M, et al. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J Neuropathol Exp Neurol. 2001;60:917–920. doi: 10.1093/jnen/60.9.917. [DOI] [PubMed] [Google Scholar]
- 13.Bollag G, Clapp DW, Shih S, et al. Loss of NF1 results in activation of the RAS signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12:144–148. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- 14.Guha A, Lau N, Huvar I, et al. RAS-GTP levels are elevated in human NF1 peripheral nerve tumors. Oncogene. 1996;12:507–513. [PubMed] [Google Scholar]
- 15.Basu TN, Gutmann DH, Fletcher JA, et al. Aberrant regulation of RAS proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 16.Sharma MK, Zehnbauer BA, Watson MA, et al. RAS pathway activation and an oncogenic RAS mutation in sporadic pilocytic astrocytoma. Neurology. 2005;65:1335–1336. doi: 10.1212/01.wnl.0000180409.78098.d7. [DOI] [PubMed] [Google Scholar]
- 17.Janzarik WG, Kratz CP, Loges NT, et al. Further evidence for a somatic KRAS mutation in a pilocytic astrocytoma. Neuropediatrics. 2007;38:61–63. doi: 10.1055/s-2007-984451. [DOI] [PubMed] [Google Scholar]
- 18.Viskochil D, Buchberg AM, Xu G, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62:187–192. doi: 10.1016/0092-8674(90)90252-a. [DOI] [PubMed] [Google Scholar]
- 19.Wallace MR, Marchuk DA, Andersen LB, et al. Type 1 neurofibromatosis gene: identification of a large transcript disrupted in three NF1 patients. Science. 1990;249:181–186. doi: 10.1126/science.2134734. [DOI] [PubMed] [Google Scholar]
- 20.Xu GF, O'Connell P, Viskochil D, et al. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- 21.Bollag G, McCormick F. GTPase activating proteins. Semin Cancer Biol. 1992;3:199–208. [PubMed] [Google Scholar]
- 22.Sebti SM, Hamilton AD. Farnesyltransferase and geranylgeranyltransferase I inhibitors in cancer therapy: important mechanistic and bench to bedside issues. Expert Opin Investig Drugs. 2000;9:2767–2782. doi: 10.1517/13543784.9.12.2767. [DOI] [PubMed] [Google Scholar]
- 23.Karp JE, Lancet JE. Development of farnesyltransferase inhibitors for clinical cancer therapy: focus on hematologic malignancies. Cancer Invest. 2007;25:484–494. doi: 10.1080/07357900701359437. [DOI] [PubMed] [Google Scholar]
- 24.Widemann BC, Salzer WL, Arceci RJ, et al. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J Clin Oncol. 2006;24:507–516. doi: 10.1200/JCO.2005.03.8638. [DOI] [PubMed] [Google Scholar]
- 25.Mahgoub N, Taylor BR, Gratiot M, et al. In vitro and in vivo effects of a farnesyltransferase inhibitor on Nf1-deficient hematopoietic cells. Blood. 1999;94:2469–2476. [PubMed] [Google Scholar]
- 26.Dasgupta B, Li W, Perry A, et al. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 2005;65:236–245. [PubMed] [Google Scholar]
- 27.Khalaf WF, Yang FC, Chen S, et al. K-ras is critical for modulating multiple c-kit-mediated cellular functions in wild-type and Nf1+/− mast cells. J Immunol. 2007;178:2527–2534. doi: 10.4049/jimmunol.178.4.2527. [DOI] [PubMed] [Google Scholar]
- 28.Morgan KJ, Rowley MA, Wiesner SM, et al. The GAP-related domain of neurofibromin attenuates proliferation and downregulates N- and K-RAS activation in Nf1-negative AML cells. Leuk Res. 2007;31:1107–1113. doi: 10.1016/j.leukres.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dasgupta B, Yi Y, Chen DY, et al. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755–2760. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- 30.Sandsmark DK, Pelletier C, Weber JD, et al. Mammalian target of rapamycin: master regulator of cell growth in the nervous system. Histol Histopathol. 2007;22:895–903. doi: 10.14670/HH-22.895. [DOI] [PubMed] [Google Scholar]
- 31.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 32.Pelletier CL, Maggi LB, Jr, Brady SN, et al. TSC1 sets the rate of ribosome export and protein synthesis through nucleophosmin translation. Cancer Res. 2007;67:1609–1617. doi: 10.1158/0008-5472.CAN-06-2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandsmark DK, Zhang H, Hegedus B, et al. Nucleophosmin mediates mammalian target of rapamycin-dependent actin cytoskeleton dynamics and proliferation in neurofibromin-deficient astrocytes. Cancer Res. 2007;67:4790–4799. doi: 10.1158/0008-5472.CAN-06-4470. [DOI] [PubMed] [Google Scholar]
- 34.Yu Y, Maggi LB, Jr, Brady SN, et al. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol. 2006;26:3798–3809. doi: 10.1128/MCB.26.10.3798-3809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nellist M, Janssen B, Ward CJ, et al. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75:1305–1315. doi: 10.1016/0092-8674(93)90618-z. [DOI] [PubMed] [Google Scholar]
- 36.van Slegtenhorst M, de Hoogt R, Hermans C, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 37.Garami A, Zwartkruis FJ, Nobukuni T, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- 38.Inoki K, Li Y, Xu T, et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tee AR, Manning BD, Roux PP, et al. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 40.Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 41.Haas-Kogan D, Shalev N, Wong M, et al. Protein kinase B (PKB/Akt) activity is elevated in glioblastoma cells due to mutation of the tumor suppressor PTEN/MMAC. Curr Biol. 1998;8:1195–1198. doi: 10.1016/s0960-9822(07)00493-9. [DOI] [PubMed] [Google Scholar]
- 42.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 43.Wang L, Harris TE, Roth RA, et al. PRAS40 regulates mTORC1 kinase activity by functioning as a direct inhibitor of substrate binding. J Biol Chem. 2007;282:20036–20044. doi: 10.1074/jbc.M702376200. [DOI] [PubMed] [Google Scholar]
- 44.Johannessen CM, Reczek EE, James MF, et al. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tee AR, Anjum R, Blenis J. Inactivation of the tuberous sclerosis complex-1 and -2 gene products occurs by phosphoinositide 3-kinase/Akt-dependent and -independent phosphorylation of tuberin. J Biol Chem. 2003;278:37288–37296. doi: 10.1074/jbc.M303257200. [DOI] [PubMed] [Google Scholar]
- 46.Collins VP, James CD. Gene and chromosomal alterations associated with the development of human gliomas. Faseb J. 1993;7:926–930. doi: 10.1096/fasebj.7.10.8344489. [DOI] [PubMed] [Google Scholar]
- 47.Calabrese C, Poppleton H, Kocak M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82. doi: 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 48.Watters JJ, Schartner JM, Badie B. Microglia function in brain tumors. J Neurosci Res. 2005;81:447–455. doi: 10.1002/jnr.20485. [DOI] [PubMed] [Google Scholar]
- 49.Badie B, Schartner J. Role of microglia in glioma biology. Microsc Res Tech. 2001;54:106–113. doi: 10.1002/jemt.1125. [DOI] [PubMed] [Google Scholar]
- 50.Wesolowska A, Kwiatkowska A, Slomnicki L, et al. Microglia-derived TGF-beta as an important regulator of glioblastoma invasion—an inhibition of TGF-beta-dependent effects by shRNA against human TGF-beta type II receptor. Oncogene. 2008;27:918–930. doi: 10.1038/sj.onc.1210683. [DOI] [PubMed] [Google Scholar]
- 51.Weissenberger J, Loeffler S, Kappeler A, et al. IL-6 is required for glioma development in a mouse model. Oncogene. 2004;23:3308–3316. doi: 10.1038/sj.onc.1207455. [DOI] [PubMed] [Google Scholar]
- 52.Zhu Y, Ghosh P, Charnay P, et al. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bajenaru ML, Zhu Y, Hedrick NM, et al. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bajenaru ML, Hernandez MR, Perry A, et al. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–8577. [PubMed] [Google Scholar]
- 55.Graeber MB, Scheithauer BW, Kreutzberg GW. Microglia in brain tumors. Glia. 2002;40:252–259. doi: 10.1002/glia.10147. [DOI] [PubMed] [Google Scholar]
- 56.Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16:1098–1112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- 57.Adamia S, Maxwell CA, Pilarski LM. Hyaluronan and hyaluronan synthases: potential therapeutic targets in cancer. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:3–14. doi: 10.2174/1568006053005056. [DOI] [PubMed] [Google Scholar]
- 58.Yang L, Jackson E, Woerner BM, et al. Blocking CXCR4-mediated cyclic AMP suppression inhibits brain tumor growth in vivo. Cancer Res. 2007;67:651–658. doi: 10.1158/0008-5472.CAN-06-2762. [DOI] [PubMed] [Google Scholar]
- 59.Tong J, Hannan F, Zhu Y, et al. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci. 2002;5:95–96. doi: 10.1038/nn792. [DOI] [PubMed] [Google Scholar]
- 60.Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003;23:8949–8954. doi: 10.1523/JNEUROSCI.23-26-08949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warrington NM, Woerner BM, Daginakatte GC, et al. Spatio-temporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer Res. 2007;67:8588–8595. doi: 10.1158/0008-5472.CAN-06-2220. [DOI] [PubMed] [Google Scholar]
- 62.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 63.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 64.Yuan X, Curtin J, Xiong Y, et al. Isolation of cancer stem cells from adult glioblastoma multiforme. Oncogene. 2004;23:9392–9400. doi: 10.1038/sj.onc.1208311. [DOI] [PubMed] [Google Scholar]
- 65.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Folkins C, Man S, Xu P, et al. Anticancer therapies combining antiangiogenic and tumor cell cytotoxic effects reduce the tumor stem-like cell fraction in glioma xenograft tumors. Cancer Res. 2007;67:3560–3564. doi: 10.1158/0008-5472.CAN-06-4238. [DOI] [PubMed] [Google Scholar]
- 67.Reilly KM, Loisel DA, Bronson RT, et al. Nf1;Trp53 mutant mice develop glioblastoma with evidence of strain-specific effects. Nat Genet. 2000;26:109–113. doi: 10.1038/79075. [DOI] [PubMed] [Google Scholar]
- 68.Reilly KM, Tuskan RG, Christy E, et al. Susceptibility to astrocytoma in mice mutant for Nf1 and Trp53 is linked to chromosome 11 and subject to epigenetic effects. Proc Natl Acad Sci U S A. 2004;101:13008–13013. doi: 10.1073/pnas.0401236101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McConville P, Hambardzumyan D, Moody JB, et al. Magnetic resonance imaging determination of tumor grade and early response to temozolomide in a genetically engineered mouse model of glioma. Clin Cancer Res. 2007;13:2897–2904. doi: 10.1158/1078-0432.CCR-06-3058. [DOI] [PubMed] [Google Scholar]
- 70.Moffat BA, Chenevert TL, Lawrence TS, et al. Functional diffusion map: a noninvasive MRI biomarker for early stratification of clinical brain tumor response. Proc Natl Acad Sci U S A. 2005;102:5524–5529. doi: 10.1073/pnas.0501532102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dasgupta B, Yi Y, Hegedus B, et al. Cerebrospinal fluid proteomic analysis reveals dysregulation of methionine aminopeptidase-2 expression in human and mouse neurofibromatosis 1-associated glioma. Cancer Res. 2005;65:9843–9850. doi: 10.1158/0008-5472.CAN-05-1842. [DOI] [PubMed] [Google Scholar]
- 72.Gutmann DH, Hunter-Schaedle K, Shannon KM. Harnessing preclinical mouse models to inform human clinical cancer trials. J Clin Invest. 2006;116:847–852. doi: 10.1172/JCI28271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hegedus B, Banerjee D, Yeh TH, et al. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68:1520–1528. doi: 10.1158/0008-5472.CAN-07-5916. [DOI] [PubMed] [Google Scholar]
- 74.Banerjee D, Hegedus B, Gutmann DH, et al. Detection and measurement of neurofibromatosis-1 mouse optic glioma in vivo. Neuroimage. 2007;35:1434–1437. doi: 10.1016/j.neuroimage.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 76.Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2:117–125. doi: 10.1016/s1535-6108(02)00096-x. [DOI] [PubMed] [Google Scholar]
- 77.Giuriato S, Felsher DW. How cancers escape their oncogene habit. Cell Cycle. 2003;2:329–332. [PubMed] [Google Scholar]