

Abstract
Double-strand breaks (DSBs) are highly deleterious DNA lesions as they lead to chromosome aberrations and/or apoptosis. The formation of nuclear DSBs triggers phosphorylation of histone H2AX on Ser-139 (defined as γH2AX), which participates in the repair of such DNA damage. Our aim was to compare the induction of γH2AX in relation to DSBs induced by topoisomerase II (TOPO II) poisons, etoposide (ETOP) and mitoxantrone (MXT), in V79 cells. DSBs were measured by the neutral comet assay, while γH2AX was quantified using immunocytochemistry and flow cytometry. Stabilized cleavage complexes (SCCs), lesions thought to be responsible for TOPO II poison-induced genotoxicity, were measured using a complex of enzyme–DNA assay. In the case of ETOP, a no observed adverse effect level (NOAEL) and lowest observed effect level (LOEL) for genotoxicity was determined; γH2AX levels paralleled DSBs at all concentrations but significant DNA damage was not detected below 0.5 μg/ml. Furthermore, DNA damage was dependent on the formation of SCCs. In contrast, at low MXT concentrations (0.0001–0.001 μg/ml), induction of γH2AX was not accompanied by increases in DSBs. Rather, DSBs were only significantly increased when SCCs were detected. These findings suggest MXT-induced genotoxicity occurred via at least two mechanisms, possibly related to DNA intercalation and/or redox cycling as well as TOPO II inhibition. Our findings also indicate that γH2AX can be induced by DNA lesions other than DSBs. In conclusion, γH2AX, when measured using immunocytochemical and flow cytometric methods, is a sensitive indicator of DNA damage and may be a useful tool in genetic toxicology screens. ETOP data are consistent with the threshold concept for TOPO II poison-induced genotoxicity and this should be considered in the safety assessment of chemicals displaying an affinity for TOPO II and genotoxic/clastogenic effects.
Keywords: DNA double-strand breaks, Topoisomerase II, Etoposide, Mitoxantrone, Neutral comet assay, γH2AX, Genotoxicity thresholds
1. Introduction
Double-strand breaks (DSBs) are highly deleterious lesions in genomic DNA [1,2]. They can be generated by ionizing irradiation and a variety of chemical agents, e.g. topoisomerase II (TOPO II) poisons, heavy metal ions and reactive oxygen species (ROS), and hence arise through diverse mechanisms [3-5]. DSBs, if not repaired efficiently, lead to chromosomal aberrations and apoptosis; in higher eukaryotes, even a single DSB in an essential gene can trigger the apoptosis signaling cascade [6,7]. Interestingly, key steps in meiosis, V(D)J and immunoglobulin class-switch recombination are mediated via formation of DSBs, thus also demonstrating their essentiality in organism development [8].
Several methods are available for the detection and quantification of DSBs in mammalian cells. The neutral single-cell gel electrophoresis or ‘comet’ assay is a sensitive technique that has been used in genotoxicity testing and in vitro DNA damage and repair investigations [9]. By using electrophoresis buffer at non-denaturing or neutral pH, DSBs are specifically revealed and can be quantified using specialized image analysis software [10,11]. The neutral comet assay allows the detection of DSBs in individual cells and an estimation of their distribution in cell populations.
Bioindicators frequently serve as surrogate endpoints in toxicological investigations [12]. They are often more accessible and assayed more rapidly, and, providing that they have been validated, can provide similar information to the biological effect of interest. Phosphorylated histone H2AX is a promising molecular marker for DSBs produced in nuclear chromatin [13]. Following the generation of a DSB, PI-3-like kinases, e.g. ATM, ATR and DNA-PK, are activated and phosphorylate Ser-139 of H2AX (defined as γH2AX) molecules in a megabase chromatin domain flanking the lesion [14,15]. γH2AX is thought to participate in DSB repair by holding broken DNA ends in close proximity and recruiting other DNA repair factors to the damaged area [16]. Radiation and chemotherapeutic agents known to cause DSBs, e.g. X-rays and TOPO II poisons, have been shown to induce γH2AX foci in various cell types [4,17,18]. Discrete γH2AX foci can be easily detected and quantified using an immunocytochemical and multiparameter flow cytometry approach [19]. Furthermore, this technique offers several advantages over traditional methods, e.g. Western blotting, as it allows high-throughput screening, rapid and accurate analysis of individual cells, and the ability to correlate γH2AX with cell cycle phase and apoptosis [4,20].
The neutral comet assay and detection of γH2AX have previously been utilized in parallel to assess the level of DSBs in γ-irradiated cells [21]. It has also been suggested that γH2AX expression can be used as a surrogate of cell killing by drugs that create DSBs [22]. TOPO II poisons are a class of antitumor drugs that induce cancer cell death via this mechanism [23]. TOPO II enzymes are responsible for modulating DNA topology in regulatory processes such as chromosome condensation, DNA replication, transcription and recombination [24]. Inhibition of TOPO II, particularly the α isoform (TOPO IIα), in rapidly dividing cells leads to the formation of stabilized cleavage complexes (SCCs), generation of DSBs and ultimately apoptosis [25]. To the best of our knowledge, no direct comparison of both methods in the assessment of DSBs generated as a result of TOPO II poisoning has been reported.
The aim of the present study was to compare the induction of γH2AX vis-à-vis DSBs detected by the neutral comet assay using two classical TOPO II poisons, etoposide (ETOP) and mitoxantrone (MXT). We studied the formation of intracellular SCCs with a sensitive complex of enzyme–DNA assay, which utilizes immunoblotting to measure DNA-bound TOPO IIα. These complementary assays were applied in concentration–response studies in V79 cells extending to very low concentrations in order to investigate the threshold concept for genotoxicity, which has been reported with various TOPO II poisons [26].
2. Materials and methods
2.1. Cell culture and treatment
Chinese hamster lung fibroblasts (V79 cells) were cultured at 37 °C (in a humidified, 5% CO2 atmosphere) in high-glucose DMEM (Gibco, USA) supplemented with 10% (v/v) fetal bovine serum, 2 mM l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. To identify TOPO IIα-DNA SCCs, V79 cells were treated with ETOP (0–10 μg/ml) and MXT (0–1 μg/ml) for 4 h in 6-well culture plates (Gibco) and processed as in Section 2.2. For neutral comet assay and γH2AX, cells were treated with the same ETOP and MXT concentration range for 4 h in 12-well culture plates (Gibco). Following treatment, cells were gently scraped and split into two aliquots for use in the two assays. Cells were processed as in Sections 2.4 and 2.5, respectively.
2.2. Identification of stabilized TOPO IIα
ETOP- and MXT-mediated SCCs were identified using the In Vivo Link Kit (TopoGEN, USA) as per the manufacturer's instructions. Briefly, treated V79 cells were lysed in TE buffer (10 mM Tris, pH 7.5 and 1 mM Na2EDTA) containing sarkosyl (1%) and layered on to a cushion of cesium chloride (density of 1.5 g/ml). Following centrifugation (170,000 × g for 12h, at 25°C) in a SW50.1 rotor (Beckman Coulter, USA), isolated DNA was precipitated with 1 ml ethanol (100%) and resolubilized in TE buffer. DNA (10 μg) was applied to a nitrocellulose membrane (Bio-Rad, USA) using a slot-blotting apparatus (Bio-Dot SF, Bio-Rad). DNA-bound TOPO IIα was identified by immunoblotting with an anti-TOPO IIα antibody (diluted 1:1500; TopoGEN) and enhanced chemiluminescence reagents (GE Healthcare, USA).
2.3. Assessment of cytotoxicity
Cytotoxicity was assessed by performing relative cell counts (RCC). Following treatment, V79 cells were washed in PBS and fresh drug-free DMEM added. Cells were incubated at 37 °C for a further 20 h before media were collected and cells harvested from culture plates by trypsinization. Aspirated media and trypsinized cells were subjected to centrifugation (200 × g, 5 min) and the resulting cell pellet resuspended in HBSS solution (1 ml). An aliquot of cells (20 μl) was added to trypan blue solution (20 μl; Sigma–Aldrich, USA) and viable cells (non-trypan blue-containing) counted using a hemacytometer (only viable cells were used in RCC calculations).
2.4. Neutral comet assay
Isolated V79 cells were centrifuged (200 × g, 5 min) and resulting pellets resuspended in PBS (150 μl). An aliquot of resuspended cells (50 μl) was placed into a tube containing low melting point agarose (120 μl) and this cell suspension transferred to glass microscope slides (Fisher, USA) pre-coated with 0.5% normal melting point agarose. Glass coverslips (Fisher) were added and slides placed on ice for 10 min. Coverslips were subsequently removed and slides placed in lysis buffer (2.5 M NaCl, 0.1 M Na2EDTA, 10 mM Tris base, 1% sarkosyl, 10% DMSO and 1% Triton X-100, adjusted to pH 10) for 1 h at 4 °C. Following lysis, slides were transferred to a horizontal electrophoresis tank containing electrophoresis buffer (0.089M Tris base, 0.089M boric acid and 2 mM Na2EDTA, adjusted to pH 8.3) and DNA allowed to unwind for 30 min. DNA was then subjected to electrophoresis (0.7 V/cm and 50 mA, 30 min). Slides were subsequently stained with ethidium bromide solution (20 μg/ml). Comet images (at least 100 random nucleiods per slide) were examined at 160× magnification using a fluorescence microscope (Nikon Optiphot, Japan) and digitized, before being analyzed with CometScore™ software (TriTek Corp., USA). Measurements of percent (%) tail DNA were determined to assess the extent of DNA damage and median values of three separate experiments were analyzed using ANOVA and post hoc Student's t-test, as recommended by Duez et al. [27]. Neutral comet data were plotted on logarithmic–logarithmic axes.
2.5. Immunofluorescence detection of γH2AX
Isolated V79 cells were fixed in p-formaldehyde (1%) for 15 min at 4 °C and postfixed in ice-cold ethanol (70%) for at least 1 h at −20 °C. Fixed cells were incubated with BSA (1%) containing anti-human γH2AX antibody (diluted 1:100; Millipore Corp., USA) for 1 h at 25 °C. Following washing in BSA (1%), cells were incubated with Alexa Fluor 488 antibody (diluted 1:100; Molecular Probes, USA) for 1 h at 25 °C. Cells were counterstained with propidium iodide containing RNase (both 10 μg/ml) and stored at 4 °C overnight. Cellular green (γH2AX) and red (nuclear DNA) fluorescence was measured using a FACScan flow cytometer (Becton Dickinson, USA) with the standard emission filters for green (FL1) and red (FL3) fluorescence as described by Halicka et al. [20]. At least 4000 cells were counted per sample. γH2AX data were plotted on logarithmic–logarithmic axes.
3. Results
3.1. Stabilization of TOPO IIα by ETOP and MXT
Untreated V79 cells showed no presence of SCCs (Fig. 1A and B). Cells treated with 0.001 and 0.01 μg/ml ETOP also had no detectable SCCs present. A concentration-dependent increase in SCCs was detected in cells treated with higher concentrations of ETOP; a very faint level was observed at 0.1 μg/ml, with clear levels detected at both 1 and 10 μg/ml. Cells treated with 0.0001 μg/ml MXT had no detectable SCCs present. A concentration-dependent increase in SCCs was detected in cells treated with higher concentrations of MXT; a definite level was observed at 0.001 μg/ml, with greater levels evident at both 0.01 and 0.1 μg/ml. Cells treated with 1 μg/ml MXT showed some abundant SCCs, but the level was slightly lower than in cells treated with 0.1 μg/ml.
Fig. 1.
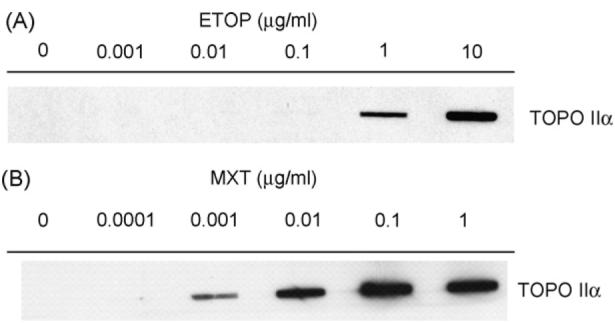
Formation of SCCs in V79 cells treated with TOPO II poisons: (A) ETOP and (B) MXT.
3.2. ETOP- and MXT-induced cytotoxicity
To investigate potential ETOP- and MXT-induced cytotoxicity in V79 cells, RCC were performed (Figs. 2 and 3). Treatment with 5 and 10 μg/ml ETOP produced 23% and 43% decreases in RCC compared to controls, respectively, whereas, all other concentrations were non-cytotoxic. MXT was more cytotoxic as treatment with 0.005, 0.01, 0.05, 0.1, 0.5 and 1 μg/ml produced 36%, 41%, 35%, 36%, 41% and 53% decreases in RCC compared to controls, respectively. Concentrations of 0.0001, 0.0005 and 0.001 μg/ml MXT were non-cytotoxic.
Fig. 2.

Induction of DSBs (measured as % tail DNA; solid line) by ETOP in V79 cells as assessed by the neutral comet assay (control; 2.7±0). * P < 0.05. RCC (dashed line) are also shown as a measure of cytotoxicity (control; 100%).
Fig. 3.
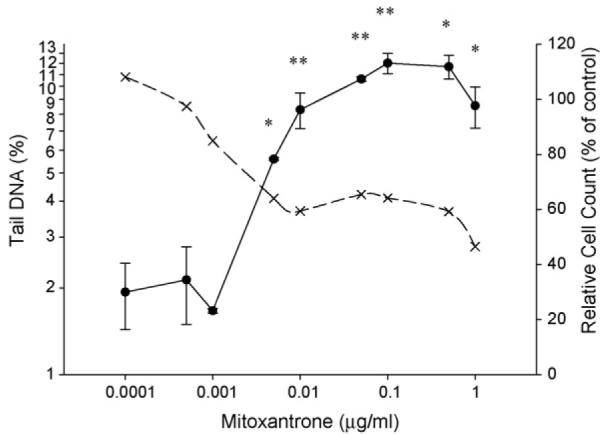
Induction of DSBs (measured as % tail DNA; solid line) by MXT in V79 cells as assessed by the neutral comet assay (control; 2.5±0.9). *P < 0.05, **P < 0.01. RCC (dashed line) are also shown as a measure of cytotoxicity (control; 100%).
3.3. Induction of DSBs by ETOP and MXT
Induction of DSBs by ETOP and MXT in V79 cells was assessed using the neutral comet assay. Results are expressed as mean of median values % DNA in the comet tail±standard error of the mean (S.E.M.) of three separate experiments and are shown in Figs. 2 and 3. No significant changes in comet tail DNA were detected following treatment with 0.001–0.1 μg/ml ETOP compared to controls, and concentration-dependent increases were observed with 0.5–10 μg/ml. No significant changes in comet tail DNA were detected following treatment with 0.0001–0.001 μg/ml MXT compared to controls. Treatment with 0.005–0.1 μg/ml MXT produced statistically significant increases in comet tail DNA in a concentration-dependent manner, whereas, it plateaued following treatment with 0.5–1 μg/ml.
3.4. Induction of H2AX by ETOP and MXT
Induction of γH2AX by ETOP and MXT in V79 cells was assessed using immunocytochemistry and flow cytometry. Results are expressed as mean green fluorescence (γH2AX) ±S.E.M. of three separate experiments and are shown in Fig. 4. γH2AX data were expressed in this manner as changes were pan-cell cycle without marked phase-specific effects. Small changes in γH2AX fluorescence were detected in cells treated with 0.001–0.05 μg/ml ETOP, whereas, marked concentration-dependent increases were observed with 0.1–10 μg/ml. Marked concentration-dependent increases in γH2AX levels were detected in cells treated with 0.0001–0.1 μg/ml MXT. γH2AX fluorescence plateaued in cells treated with 0.5 and 1 μg/ml MXT.
Fig. 4.

γH2AX mean fluorescence (±S.E.M.) in V79 cells treated with ETOP (control; 111±5) or MXT (control; 123±17).
4. Discussion
In the present study, ETOP- and MXT-treated V79 cells were assessed for the presence of DSBs using the neutral comet assay and γH2AX by immunocytochemistry and flow cytometry. Uniquely in this investigation, the same population of TOPO II poison-treated cells was used for both assays, thus enabling direct comparison of natural (DSBs) and surrogate (γH2AX) endpoints.
Treatment with ETOP produced similar concentration-response effects on DSBs and γH2AX induction. At low concentrations (0.001–0.1 μg/ml) no significant changes in DSBs were detected, although tail DNA was increased at 0.05 and 0.1 μg/ml ETOP compared to controls, and thus there was a no observed adverse effect level (NOAEL). Similarly, γH2AX levels were not distinct from controls at these concentrations. The lowest observed effect level (LOEL) occurred at ≥0.5 μg/ml ETOP as DSBs increased in a statistically significant, near-linear manner. Again, γH2AX levels reflected DSBs, with marked concentration-dependent increases detected at ≥0.1 μg/ml ETOP. In the case of ETOP, it appears that the level of γH2AX accurately reflects the level of genomic DSBs; accordingly, γH2AX could serve as a surrogate indicator of DSBs in future ETOP investigations. These findings also suggest the presence of a threshold for ETOP-mediated genotoxicity, which is in general agreement with the results of Lynch et al. [26]. In our model, the ‘pragmatic’ threshold for induction of both DSBs and γH2AX in V79 cells was determined to lie between 0.05 and 0.5 μg/ml. Lynch et al. using transformed L5178Y mouse lymphoma cells and a combination of hypothesis testing and mathematical modeling, reported the pragmatic threshold for ETOP-induced clastogenicity (as micronucleus induction) to be 0.00236 μg/ml [26]. In a dose–response study by Boos and Stopper, the lowest concentration (2 nM; 0.0012 μg/ml) for ETOP-induced micronuclei in L5178Y cells was also reported to be similar to that of Lynch et al. [26,28]. Taken together, these results suggest the existence of a threshold for ETOP-mediated genotoxicity. However, as stated by Lynch et al., we believe that differences in ‘threshold’ concentrations can arise from the use of different cell lines, i.e. genotoxic effects are cell type-specific and dependent on factors such as DNA damage response and repair capacity, which most likely vary between cell lines [26].
It is also of interest to note that ETOP-mediated SCCs are formed at a similar time to the significant induction of DSBs and γH2AX. SCCs are widely regarded as the lesion ultimately responsible for TOPO II poison-induced genotoxicity; they are thought to induce DSBs by disrupting the catalytic cycle of TOPO II and/or colliding with DNA replication and transcription machinery [23]. However, recent studies have shown that such covalent protein–DNA adducts can be degraded in a proteasome-mediated process and TOPO II poison-induced DSBs are substrates for repair by the non-homologous end-joining pathway [29,30]. In the light of our findings, it appears that a specific level of TOPO IIα inhibition is required for the induction of DNA damage, and below this level no significant genotoxic effects are manifested because of efficient repair. This is also consistent with a threshold concept and in support of the described genotoxicity results. Above the threshold, levels of ETOP-mediated SCCs are increased in a concentration-dependent manner, together with associated increases in both DSBs and γH2AX. Only at the top two ETOP concentrations (5 and 10 μg/ml) however, does the level of DNA damage overwhelm repair capacity, and manifest itself as significant cytotoxicity. Therefore, below a specific level, SCCs and DNA damage appear able to be processed and reversed but once the critical level is reached, cells are impelled into a quiescent state and/or apoptosis pathways are triggered (as determined by marked decreases in RCC at 5 and 10 μg/ml). Clearly, the temporal relationship between formation of SCCs and induction of DNA damage is important for the determination of thresholds and thus a temporal analysis of the various end-points may shed more light on this interesting phenomenon.
In the case of MXT, a marked induction of γH2AX by low concentrations (0.0001–0.001 μg/ml) was not accompanied by an induction of DSBs as measured by the neutral comet assay. Rather, DSBs were only significantly increased after treatment with ≥0.005 μg/ml. Although MXT is considered a TOPO II poison, its chemical structure and reported mode(s) of action are different to those of ETOP. ETOP is a semi-synthetic derivative of podophyllotoxin and binds stoichiometrically to TOPO II enzymes in the absence of DNA, whereas, MXT is an anthracenedione and can intercalate into duplex DNA, as well as interact with TOPO II [31-34]. Interestingly, several studies have shown that induction of γH2AX (and phosphorylation of its upstream mediator ATM) by ETOP and MXT can be partially abrogated by pre-treatment with the antioxidant N-acetyl-l-cysteine [35,36]. This suggests that ETOP and MXT can undergo redox cycling to generate ROS, which have the potential to act as DNA damaging agents [37]. As we detected no MXT-mediated SCCs at 0.0001 μg/ml, we speculate that at these concentrations some form of ROS- and/or intercalation-induced DNA damage, but not DSBs, is responsible for the increases in γH2AX in V79 cells. To this end, a number of recent studies have revealed that γH2AX can be induced by DNA excision repair intermediates, without the involvement of DSBs per se [38-40]. It is possible that excision repair-mediated removal of MXT-induced lesions yields intermediate moieties capable of activating ATM or other PI-3-related kinases, e.g. ATR, which subsequently phospho- rylate H2AX molecules.
At higher concentrations of MXT (>0.001 μg/ml), however, when SCCs were observed, significant levels of DSBs were also detected by the neutral comet assay (γH2AX also paralleled levels of DSBs over this concentration range). This suggests that DSBs produced in these cells were a direct consequence of TOPO II stabilization. Therefore, although our study did not detect a NOAEL for total MXT-induced DNA damage in V79 cells, it does provide useful information on its mechanisms of genotoxicity; at low concentrations it appears that MXT induces DNA damage via a TOPO II-independent mechanism(s), whereas, at higher concentrations, DNA damage is mainly in the form of DSBs and results from TOPO II inhibition. ETOP, on the other hand, appears to induce DSBs exclusively and these occur through the single mechanism of TOPO II poisoning. However, it should be noted that ETOP–DNA adducts (as determined by 32P-postlabeling) have been observed in vitro, so other mechanisms of ETOP-mediated DNA damage potentially exist (GM Williams, personal communication).
The approach used in this study also allowed us to assess the relative potencies of ETOP and MXT in terms of TOPO IIα inhibition, induction of DNA damage and cytotoxicity. Inhibition of TOPO IIα (measured as SCC formation) and significant induction of DSBs by MXT occurred at a concentration ∼90-fold (molar basis) lower than with ETOP. A recent study highlighted the importance of TOPO IIα inhibition in relation to cytotoxicity, so it was not surprising that MXT was significantly more cytotoxic than ETOP (as determined by RCC), thus confirming other reports [25,41]. However, it must be considered that MXT's TOPO II-independent mechanism of genotoxicity may also contribute to cytotoxicity, as cell counts were decreased with 0.001 μg/ml when SCCs and DSBs were not detected.
In conclusion, the present findings demonstrate that induction of γH2AX by TOPO II poisons, as with X- and γ-irradiation and other agents, is strongly associated with DSBs in nuclear chromatin. However, recent evidence suggests that γH2AX can also be induced by other types of DNA damage. Results should therefore be validated and interpreted carefully when using γH2AX as a marker of DSBs in future investigations. On the other hand, γH2AX, when measured using immunocytochemical and flow cytometric methods, appears to be a highly sensitive indicator of DNA damage and thus may prove to be a useful tool in genetic toxicology screens. The ETOP data are also in line with the current threshold concept for TOPO II poison-induced genotoxicity and this has implications in the safety assessment of chemicals displaying both an affinity for TOPO II and genotoxic/clastogenic effects.
Acknowledgments
DJS and GMW were supported by a grant from Bayer HealthCare AG, Germany. HDH, FT and ZD are supported in part by NCI RO1 28 704.
References
- 1.Scott SP, Pandita TK. The cellular control of DNA double-strand breaks. J. Cell. Biochem. 2006;99:1463–1475. doi: 10.1002/jcb.21067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mills KD, Ferguson DO, Alt FW. The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 2003;194:77–95. doi: 10.1034/j.1600-065x.2003.00060.x. [DOI] [PubMed] [Google Scholar]
- 3.Olive PL, Banath JP. Detection of DNA double-strand breaks through the cell cycle after exposure to X-rays, bleomycin, etoposide and 125IdUrd. Int. J. Radiat. Biol. 1993;64:349–358. doi: 10.1080/09553009314551531. [DOI] [PubMed] [Google Scholar]
- 4.Kurose A, Tanaka T, Huang X, Halicka HD, Traganos F, Dai W, Darzynkiewicz Z. Assessment of ATM phosphorylation on Ser-1981 induced by DNA topoisomerase I and II inhibitors in relation to Ser-139-histone H2AX phosphorylation, cell cycle phase, and apoptosis. Cytometry A. 2005;68:1–9. doi: 10.1002/cyto.a.20186. [DOI] [PubMed] [Google Scholar]
- 5.Ayene IS, Koch CJ, Krisch RE. DNA strand breakage by bivalent metal ions and ionizing radiation. Int. J. Radiat. Biol. 2007;83:195–210. doi: 10.1080/09553000601146956. [DOI] [PubMed] [Google Scholar]
- 6.Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature. 2000;407:777–783. doi: 10.1038/35037717. [DOI] [PubMed] [Google Scholar]
- 7.Lips J, Kaina B. DNA double-strand breaks trigger apoptosis in p53-deficient fibroblasts. Carcinogenesis. 2001;22:579–585. doi: 10.1093/carcin/22.4.579. [DOI] [PubMed] [Google Scholar]
- 8.Daboussi F, Dumay A, Delacote F, Lopez BS. DNA double-strand break repair signalling: the case of RAD51 post-translational regulation. Cell Signal. 2002;14:969–975. doi: 10.1016/s0898-6568(02)00052-9. [DOI] [PubMed] [Google Scholar]
- 9.Fairbairn DW, Olive PL, O'Neill KL. The comet assay: a comprehensive review. Mutat. Res. 1995;339:37–59. doi: 10.1016/0165-1110(94)00013-3. [DOI] [PubMed] [Google Scholar]
- 10.Olive PL, Wlodek D, Banath JP. DNA double-strand breaks measured in individual cells subjected to gel electrophoresis. Cancer Res. 1991;51:4671–4676. [PubMed] [Google Scholar]
- 11.Collins AR, Dobson VL, Dusinska M, Kennedy G, Stetina R. The comet assay: what can it really tell us? Mutat. Res. 1997;375:183–193. doi: 10.1016/s0027-5107(97)00013-4. [DOI] [PubMed] [Google Scholar]
- 12.Timbrell JA. Biomarkers in toxicology. Toxicology. 1998;129:1–12. doi: 10.1016/s0300-483x(98)00058-4. [DOI] [PubMed] [Google Scholar]
- 13.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 14.Sedelnikova OA, Pilch DR, Redon C, Bonner WM. Histone H2AX in DNA damage and repair. Cancer Biol. Ther. 2003;2:233–235. doi: 10.4161/cbt.2.3.373. [DOI] [PubMed] [Google Scholar]
- 15.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell. Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bassing CH, Alt FW. H2AX may function as an anchor to hold broken chromosomal DNA ends in close proximity. Cell Cycle. 2004;3:149–153. doi: 10.4161/cc.3.2.689. [DOI] [PubMed] [Google Scholar]
- 17.MacPhail SH, Banath JP, Yu TY, Chu EH, Lambur H, Olive PL. Expression of phosphorylated histone H2AX in cultured cell lines following exposure to X-rays. Int. J. Radiat. Biol. 2003;79:351–358. doi: 10.1080/0955300032000093128. [DOI] [PubMed] [Google Scholar]
- 18.Huang X, Traganos F, Darzynkiewicz Z. DNA damage induced by DNA topoisomerase I- and topoisomerase II-inhibitors detected by histone H2AX phosphorylation in relation to the cell cycle phase and apoptosis. Cell Cycle. 2003;2:614–619. [PubMed] [Google Scholar]
- 19.Olive PL. Detection of DNA damage in individual cells by analysis of histone H2AX phosphorylation. Methods Cell Biol. 2004;75:355–373. doi: 10.1016/s0091-679x(04)75014-1. [DOI] [PubMed] [Google Scholar]
- 20.Halicka HD, Huang X, Traganos F, King MA, Dai W, Darzynkiewicz Z. Histone H2AX phosphorylation after cell irradiation with UV-B: relationship to cell cycle phase and induction of apoptosis. Cell Cycle. 2005;4:339–345. [PubMed] [Google Scholar]
- 21.Mirzayans R, Severin D, Murray D. Relationship between DNA double-strand break rejoining and cell survival after exposure to ionizing radiation in human fibroblast strains with differing ATM/p53 status: implications for evaluation of clinical radiosensitivity. Int. J. Radiat. Oncol. Biol. Phys. 2006;66:1498–1505. doi: 10.1016/j.ijrobp.2006.08.064. [DOI] [PubMed] [Google Scholar]
- 22.Banath JP, Olive PL. Expression of phosphorylated histone H2AX as a surrogate of cell killing by drugs that create DNA double-strand breaks. Cancer Res. 2003;63:4347–4350. [PubMed] [Google Scholar]
- 23.Burden DA, Osheroff N. Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme. Biochim. Biophys. Acta. 1998;1400:139–154. doi: 10.1016/s0167-4781(98)00132-8. [DOI] [PubMed] [Google Scholar]
- 24.Berger JM. Structure of DNA topoisomerases. Biochim. Biophys. Acta. 1998;1400:3–18. doi: 10.1016/s0167-4781(98)00124-9. [DOI] [PubMed] [Google Scholar]
- 25.Azarova AM, Lyu YL, Lin CP, Tsai YC, Lau JY, Wang JC, Liu LF. From the cover: roles of DNA topoisomerase II isozymes in chemotherapy and secondary malignancies. Proc. Natl. Acad. Sci. U.S.A. 2007;104:11014–11019. doi: 10.1073/pnas.0704002104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lynch A, Harvey J, Aylott M, Nicholas E, Burman M, Siddiqui A, Walker S, Rees R. Investigations into the concept of a threshold for topoisomerase inhibitor-induced clastogenicity. Mutagenesis. 2003;18:345–353. doi: 10.1093/mutage/geg003. [DOI] [PubMed] [Google Scholar]
- 27.Duez P, Dehon G, Kumps A, Dubois J. Statistics of the comet assay: a key to discriminate between genotoxic effects. Mutagenesis. 2003;18:159–166. doi: 10.1093/mutage/18.2.159. [DOI] [PubMed] [Google Scholar]
- 28.Boos G, Stopper H. Genotoxicity of several clinically used topoisomerase II inhibitors. Toxicol. Lett. 2000;116:7–16. doi: 10.1016/s0378-4274(00)00192-2. [DOI] [PubMed] [Google Scholar]
- 29.Mao Y, Desai SD, Ting CY, Hwang J, Liu LF. 26S proteasome-mediated degradation of topoisomerase II cleavable complexes. J. Biol. Chem. 2001;276:40652–40658. doi: 10.1074/jbc.M104009200. [DOI] [PubMed] [Google Scholar]
- 30.Malik M, Nitiss KC, Enriquez-Rios V, Nitiss JL. Roles of nonhomologous end-joining pathways in surviving topoisomerase II-mediated DNA damage. Mol. Cancer Ther. 2006;5:1405–1414. doi: 10.1158/1535-7163.MCT-05-0263. [DOI] [PubMed] [Google Scholar]
- 31.Stahelin HF, von Wartburg A. The chemical and biological route from podophyllotoxin glucoside to etoposide: ninth Cain memorial Award lecture. Cancer Res. 1991;51:5–15. [PubMed] [Google Scholar]
- 32.Kingma PS, Burden DA, Osheroff N. Binding of etoposide to topoisomerase II in the absence of DNA: decreased affinity as a mechanism of drug resistance. Biochemistry. 1999;38:3457–3461. doi: 10.1021/bi982855i. [DOI] [PubMed] [Google Scholar]
- 33.Cheng CC, Zee-Cheng RK. The design, synthesis and development of a new class of potent antineoplastic anthraquinones. Prog. Med. Chem. 1983;20:83–118. doi: 10.1016/s0079-6468(08)70217-0. [DOI] [PubMed] [Google Scholar]
- 34.Kapuscinski J, Darzynkiewicz Z. Interactions of antitumor agents ametantrone and mitoxantrone (Novatrone) with double-stranded DNA. Biochem. Pharmacol. 1985;34:4203–4213. doi: 10.1016/0006-2952(85)90275-8. [DOI] [PubMed] [Google Scholar]
- 35.Tanaka T, Halicka HD, Traganos F, Seiter K, Darzynkiewicz Z. Induction of ATM activation, histone H2AX phosphorylation and apoptosis by etoposide: relation to cell cycle phase. Cell Cycle. 2007;6:371–376. doi: 10.4161/cc.6.3.3835. [DOI] [PubMed] [Google Scholar]
- 36.Huang X, Kurose A, Tanaka T, Traganos F, Dai W, Darzynkiewicz Z. Activation of ATM and histone H2AX phosphorylation induced by mitoxantrone but not by topotecan is prevented by the antioxidant N-acetyl-l-cysteine. Cancer Biol. Ther. 2006;5:959–964. doi: 10.4161/cbt.5.8.2878. [DOI] [PubMed] [Google Scholar]
- 37.Jeffrey AM, Williams GM. Oxidative DNA damage: endogenous and chemically induced. Regul. Toxicol. Pharmacol. 2000;32:283–292. doi: 10.1006/rtph.2000.1433. [DOI] [PubMed] [Google Scholar]
- 38.Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proc. Natl. Acad. Sci. U.S.A. 2006;103:9891–9896. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsumoto M, Yaginuma K, Igarashi A, Imura M, Hasegawa M, Iwabuchi K, Date T, Mori T, Ishizaki K, Yamashita K, Inobe M, Matsunaga T. Perturbed gap-filling synthesis in nucleotide excision repair causes histone H2AX phosphorylation in human quiescent cells. J. Cell Sci. 2007;120:1104–1112. doi: 10.1242/jcs.03391. [DOI] [PubMed] [Google Scholar]
- 40.Hanasoge S, Ljungman M. H2AX phosphorylation after UV-irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis. 2007;28:2298–2304. doi: 10.1093/carcin/bgm157. [DOI] [PubMed] [Google Scholar]
- 41.Stopper H, Boos G, Clark M, Gieseler F. Are topoisomerase II inhibitor-induced micronuclei in vitro a predictive marker for the compounds' ability to cause secondary leukemias after treatment? Toxicol. Lett. 1999;104:103–110. doi: 10.1016/s0378-4274(98)00353-1. [DOI] [PubMed] [Google Scholar]