

Summary
The two-protein complex between putidaredoxin (Pdx) and cytochrome P450cam (CYP101) is the catalytically competent species for camphor hydroxylation by CYP101. We detected a conformational change in CYP101 upon binding of Pdx that re-orients bound camphor appropriately for hydroxylation. Experimental evidence shows that binding of Pdx converts a single X-proline amide bond in CYP101 from trans or distorted trans to cis. Mutation of proline 89 to isoleucine yields a mixture of both bound camphor orientations, that seen in Pdx-free and that seen in Pdx-bound CYP101. A mutation in CYP101 that destabilizes the cis conformer of the Ile 88-Pro 89 amide bond results in weaker binding of Pdx. This work provides direct experimental evidence for involvement of X-proline isomerization in enzyme function.
Introduction
Enzyme catalysis requires enzyme motion, a concept that is now generally appreciated. The structural requirements for selective binding of substrates, stabilization of transition states and product release by an enzyme appear to be mutually exclusive. However, they can be reconciled by assuming that each step in the catalytic cycle takes place from an appropriate enzyme conformation, with sequentially occupied conformations linked to one another by motions with frequencies low enough so that the individual states are discrete on the time scale of catalysis. Alternatively, conformational changes can be driven by binding of a co-factor or an effector. A complete catalytic cycle would start in the resting state (which may or may not be the enzyme conformation receptive to substrate binding), pass through conformations that sequentially stabilize substrate binding, reaction transition state(s), and finally product release.
We are using nuclear magnetic resonance (NMR) methods to investigate such conformational changes in cytochrome P450cam (CYP101) from Pseudomonas putida. CYP101 catalyzes the hydroxylation of camphor at the unactivated 5-exo C-H bond by molecular oxygen, the first step in camphor catabolism by P. putida. Such selective oxidations are among the most mechanistically complex chemical reactions that occur in living organisms. Nevertheless, CYP101 carries out the stereo- and regioselective oxidation of camphor with better than 99% efficiency at room temperature, and at a rate sufficient to supply all of the carbon and energy requirements of Pseudomonas growth and reproduction in the absence of other reduced carbon sources. Much of our current understanding of structure and function in the P450 superfamily comes from in-depth investigations of CYP101 that span over forty years, and have involved hundreds of researchers (Mueller et al., 1995; Poulos, 2003; Katagiri, 2005). Despite this, there remain some crucial unresolved mechanistic questions regarding CYP101 that have relevance for other P450 enzymes as well. One outstanding question concerns the role of conformational changes induced by effector binding in the stimulation of catalytic activity in CYP101. It has long been known that the physiological reductant of CYP101, the Cys4Fe2S2 ferredoxin putidaredoxin (Pdx), is an effector and required component of the catalytically competent CYP101 enzyme system (Lipscomb et al., 1976). In the absence of Pdx, no product formation is observed under standard assay conditions, even if a reductant of appropriate potential is present to provide the required electrons. As other P450s have become better characterized, it appears that the requirement for an effector is the norm rather than the exception (Urushino et al., 2006; Zhang et al. 2005; Zhang et al. 2006). The origin of effector activity in all cases remains unclear. Electronic changes that occur in the heme of CYP101 upon Pdx binding have been proposed to be important in facilitating the second electron transfer (Unno et al. 2002; Sjodin et al., 2001). However, effector activity has been observed with molecules that do not induce such electronic changes, suggesting that the origin of such activity lies elsewhere.
With ∼80% of the backbone 1H, 15N and 13C NMR resonances in the reduced (Fe+2) CO- and camphor (S)-bound form of the 414-residue monomeric CYP101 (CYP-S-CO) now assigned (Pochapsky et al., 2003; Rui et al., 2006; OuYang et al., 2006) we can obtain detailed and localized information on conformational changes in this enzyme. CYP-S-CO is iso-electronic with the physiologically relevant reduced camphor-bound O2 complex of CYP101 (CYP-S-O2) upon which effectors act, but is more convenient than CYP-S-O2 for equilibrium NMR studies because it does not lead to product formation. Titration of CYP-S-CO with reduced Pdx (Pdxr), causes conformational perturbations in CYP101 remote from the Pdx binding site (Pochapsky et al., 2003). Many of these perturbations are located in regions of CYP101 that have been implicated in substrate access to and orientation within the active site. Based on these observations, we proposed a model for effector activity in which the binding of Pdx forces selection of the active conformation of CYP101 and prevents loss of substrate and/or intermediate during the reaction cycle (Pochapsky et al., 2003). Support for this model was provided by the detection of a conformational shift that takes place in the active site of CYP101 upon Pdx binding and results in a re-orientation of bound substrate in the active site (Wei et al., 2005). We found that 1H chemical shifts of the methyl groups of bound camphor at saturating Pdx concentrations matched those calculated from the crystallographic structure of CYP-S-CO better than the shifts observed in the absence of Pdx, indicating that the crystallographic conformation of CYP-S-CO most resembles the Pdx-bound form in solution. A molecular dynamics simulation restrained by the 1H chemical shifts of camphor observed in Pdx-free CYP-S-CO suggested the active site cavity of CYP101 in the absence of Pdxr expands relative to the crystallographically-determined cavity in order to accommodate the alternate camphor orientation (Wei et al., 2005). Repositioning of bound substrate in response to changing conditions has been observed in other P450 enzymes as well (Jovanovic et al., 2005; Ravindranathan et al., 2007).
The time scale of the conformational change induced in CYP-S-CO by Pdxr is intriguing. Line widths of NH correlations in TROSY-HSQC spectra of CYP-S-CO that shift upon titration with Pdxr as well as the time scales of observed chemical shift changes indicate that the conformational change takes place with a kex between 150 and 200 s-1 at half-saturation at 25 °C (Figure 1) (Pochapsky et al., 2003; Wei et al., 2005). Recently, Glascock et al. described a corresponding conformational change with a rate constant of 150 s-1 at 25 °C upon titration of the active intermediate complex CYP-S-O2 with Pdxr using time-resolved optical spectroscopy (Glascock et al., 2005). The time scale of the conformational change suggested to us that a cis-trans isomerization of an X-proline (X-Pro) amide might be involved. While uncatalyzed X-Pro cis-trans isomerizations are considerably slower than this (∼0.01 s-1 in isolated peptides, (Stein, 1993)), enzymes that catalyze such isomerizations (peptide prolyl isomerases, or PPIases), e.g., cyclophylin A (CyA) and FK506 binding protein (FKBP), can increase isomerization rates by up to six orders of magnitude (Hur & Bruice, 2002). X-Pro peptide bond isomerization has been implicated in a number of biological switches involved in ion transport (Lummis et al., 2005; Andreotti, 2006), gene expression (Nelson et al., 2006) and signal transduction (Sarkar et al., 2007) among others (Grochulski et al., 1994; Andreotti, 2003). We therefore decided to investigate the possibility that the conformational change induced by Pdxr binding to CYP101 involves a X-Pro isomerization.
Figure 1.
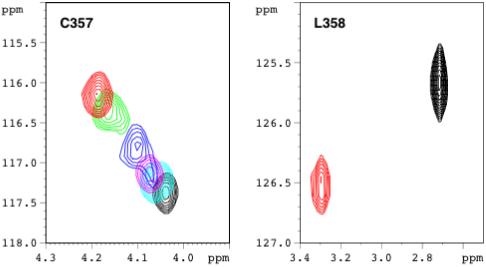
The time scale of the Pdxr-induced conformational change in WT CYP-S-CO at 298 K is bracketed by resonances showing slow vs. fast exchange behavior in the course of titration by Pdxr. The left hand figure shows the titration of the amide HN correlation of Cys 357 (which provides the heme iron axial thiolate ligand) as followed in 800 MHz 1H, 15N TROSY-HSQC spectra. A discrete signal is observed at each titration point, indicating fast-exchange behavior. The total 1H chemical shift change upon saturation with Pdxr relative to the Pdx-free form (δsat - δfree) for Cys 357 HN is 0.17 ppm (136 Hz). The right hand panel shows the HN correlation for the adjacent residue, Leu 358, from the same series of spectra. Discrete signals are seen only at the extrema of the titration (no Pdxr and saturating Pdxr), indicating slow exchange behavior. (δsat -δfree) for Leu 358 is 0.58 ppm (464 Hz). The rate for Pdx-induced conformational exchange at 298 K and half-saturation was estimated to be between 150 s-1 and 300 s-1 from line width and chemical shift measurements (Pochapsky et al., 2003; Wei et al., 2005). Current data further restricts this range to between 150 s-1 and 200 s-1 (see Supplementary Material). Spectrum colors correspond to relative concentrations of CYP-S-CO and Pdxr: black, no Pdx; cyan, 5/1 CYP/Pdx; magenta, 2/1; blue, 1/1; green, 1/2; red, 1/4 (saturating Pdxr). All spectra were obtained at 298 K.
One residue in particular, Pro 89, seemed a likely candidate for isomerization. Pro 89 is one of three prolines identified in all crystal structures of CYP101 as being in a cis conformation (ωCα88-C88-N89-Cα89 = 0°) around the peptide bond with the previous residue (Ile 88), and the only one of the three not involved in a regular type VI turn (Poulos et al., 1987). The carbonyl oxygen of Pro 89 provides the N-terminal hydrogen bond acceptor stabilizing the B’ helix (residues 90-96), which is the secondary structural element in CYP101 most uniformly perturbed by binding of Pdx. Tyr 96, the last residue in the B’ helix, provides a hydrogen bond to the carbonyl of bound camphor, and the B’ helix has also been implicated in gating access to the CYP101 active site (Poulos et al., 1986; Wade et al., 2004; Yao et al., 2007). As such, the conformation of the Ile 88-Pro 89 amide bond is important in determining the shape and accessibility of the active site of CYP101. We now report the results of spectroscopic and site-directed mutagenesis experiments indicating that isomerization of the Ile 88-Pro 89 amide is responsible for the high-barrier conformational change that takes place in CYP101 upon Pdxr binding, and leads to the catalytically competent conformation of CYP-S-O2 prior to the second electron transfer and subsequent hydroxylation.
Results
Population of a cis X-Pro conformation in WT CYP-S-CO driven by Pdx binding
A sample of WT CYP-S-CO selectively labeled with 1H,13C-proline and isoleucine but otherwise uniformly labeled with 2H and 15N was titrated with perdeuterated Pdxr. Due to spectral overlap (there are thirty prolines in CYP101), we were unable to definitively assign the proline ring 1H and 13C resonances of Pro 89, even in the selectively labeled sample. However, a difference spectrum obtained by subtracting the 1H,13C HSQC spectrum of Pdx-free WT CYP-S-CO from that of the same sample saturated with 4 equivalents of Pdxr shows a correlation that appears or is greatly intensified in the Pdx-bound form in a spectral region considered diagnostic for a cis-proline Cγ (Figure 2) (Dorman and Bovey, 1973; Schubert et al., 2002). This is evidence for population of a cis X-Pro conformation in the Pdx-bound CYP-S-CO that is absent or only lightly populated in the absence of Pdx. Furthermore, weak NOEs appearing in the region expected for proline CδH protons to the NH of Arg 90 in the 15N-edited NOESY spectrum of Pdx-free CYP-S-CO are consistent with a largely trans conformation of the 88-89 bond in the absence of Pdxr (see Supplementary material).
Figure 2.
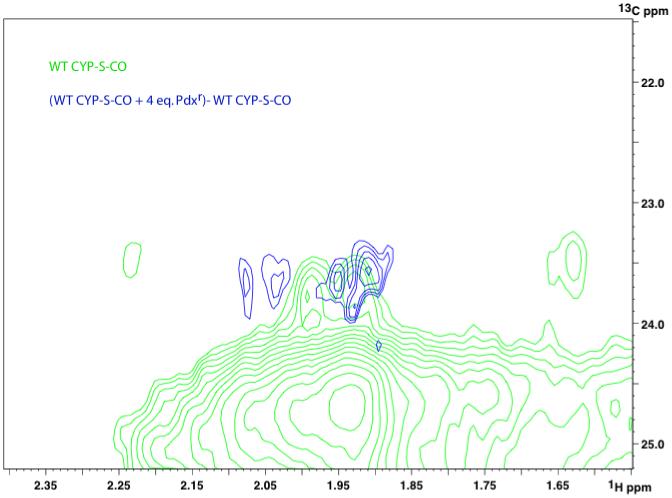
Expansion of the 800 MHz 1H,13C HSQC spectrum (green) of 1H,13C-Pro, Ile-u-2H, 15N-labeled WT CYP-S-CO, showing details of the region corresponding to CγH2 proline correlations. Overlaid in blue is the difference spectrum obtained by subtracting the spectrum obtained in the absence of Pdx from one obtained with 4 eq. (saturating) Pdxr present. Difference spectrum (blue) shows increased peak intensity for Pdxr-saturated CYP-S-CO in the region (between 23 and 24 ppm in the 13C dimension) where Cγ chemical shifts are observed for cis X-Pro.
Site directed mutagenesis
Three proline residues in CYP101 were tested for potential involvement in the conformational change that accompanies Pdxr binding to CYP-S-CO: Pro 86, Pro 89 and Pro 106. These were chosen based on their locations in regions affected by Pdx binding as determined by NMR titration and inspection of likely structural consequences of isomerization. Pro 86, in the B-B’ loop, stabilizes the positions of Phe 87 (which is in van der Waals contact with substrate) and the B’ helix. Mutations at Pro 86 result in marked loss of binding affinity for Pdxr (Kd = 450 μM for the P86V mutant CYP-S-CO vs. 32 μM for WT at 298 K as measured by NMR titration), but also multiple conformations of camphor in the active site that are not observed in WT, as determined by 1H NMR. Mutations at Pro 106, which provides the N-terminal cap for the C-helix in the Pdx binding site (Figure 3), resulted in low expression levels and poor enzyme stability. However, mutations at Pro 89, the carbonyl of which provides the N-terminal hydrogen bond acceptor of the B’ helix (Fig. 3), unambiguously give rise to the same two camphor orientations in the active site that are observed to interconvert in WT CYP-S-CO upon Pdxr binding. The proline to isoleucine mutant, P89I, yields the two conformations in approximately equal amounts (Figure 4), and so was chosen for further investigation.
Figure 3.
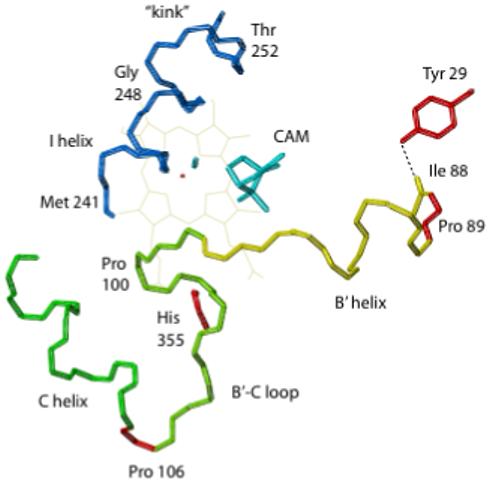
Peptide backbone structure of relevant portions of the CYP101 structure showing locations of mutations described in the text. Backbone positions of mutated residues are shown in red. Others are color-coded according to secondary structural feature. The position of Pro 86 (not shown) is almost directly in front of the first turn of the B’ helix. The heme porphyrin is shown in light lines, with the iron as a small red polygon. Substrate camphor (CAM) and carbon monoxide ligand are shown in cyan. The hydrogen bond between the carbonyl oxygen of Ile 88 and phenolic OH of Tyr 29 that stabilizes the cis conformation of the Ile 88-Pro 89 amide is indicated by a dotted line. Figure was generated using MOLMOL (Koradi et al., 1996).
Figure 4.
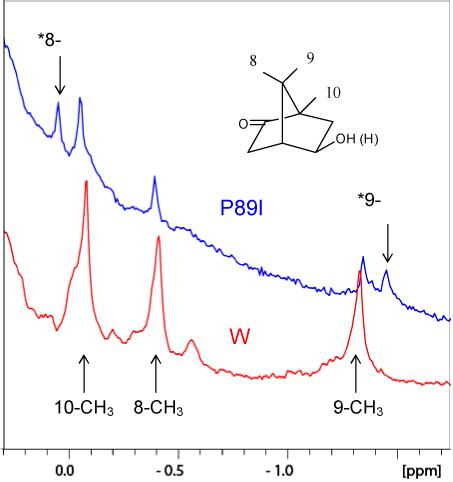
Upfield regions of the 800 MHz 1H NMR spectra of perdeuterated WT (red) and P89I (blue) CYP-S-CO showing the 8-, 9- and 10-CH3 resonances of camphor (structure at upper right) bound in the CYP101 active site. Arrows above the P89I spectrum with assignments labeled * indicate the positions of the 8- and 9-CH3 resonances observed in Pdxr-saturated WT CYP-S-CO (Wei et al., 2005). These correspond to the positions of one set of signals assigned to the 8- and 9-CH3 groups in P89I. The two camphor orientations observed at slow exchange in P89I thus correspond to the different camphor orientations seen in the Pdxr-free and Pdxr-bound forms of WT CYP-S-CO. The position of the 10-CH3 resonance in WT CYP-S-CO is not significantly perturbed by Pdxr binding (Wei et al., 2005).
A related mutation, Tyr 29 to Phe, was also made. The phenolic OH group of Tyr 29 stabilizes the cis conformer of the Ile 88-Pro 89 amide by hydrogen bonding with the carbonyl oxygen of Ile 88 (Fig. 3). While the Y29F mutation introduces only minor perturbations in the TROSY-HSQC spectrum of CYP-S-CO, the mutation increases the Kd for Pdxr binding to Y29F CYP-S-CO by an order of magnitude relative to WT (32 ± 10 μM for WT vs. 441 ± 202 μM for Y29F at 25 °C).
Both the P89I and Y29F mutants produce hydroxycamphor in the standard reconstituted camphor hydroxylase assay, although at rates slower than WT. Y29F catalyzed the oxidation of NADH with a Vmax ∼75% that of WT, while P89I exhibited a Vmax ∼25% of WT. No significant uncoupling of camphor hydroxylation from NADH consumption was observed with either the P89I or Y29F mutants; ratios of hydroxycamphor product to reducing equivalents (NADH) consumed were the same as WT within experimental error. However, the P89I mutant does not produce any detectable hydroxycamphor using sodium dithionite as a reductant in the absence of Pdx under single turnover conditions at 4 °C, although some product formation was observed with both WT and the L358P mutant, in which camphor is found primarily in the orientation induced by Pdx binding to WT (Tosha et al., 2004; OuYang et al., 2006). We speculate that the lack of product from P89I in single-turnover mode is due to the high barrier to interconversion between conformers, making side reactions more competitive, but more detailed kinetic experiments are required to confirm this.
Two other prolines are located in the Pdx-perturbed regions of CYP101, Pro 100 and Pro 105. Isomerization of Pro 105 seems unlikely in that it precedes Pro 106 (vide supra), and should be too sterically restrained to readily undergo isomerization. Furthermore the destabilization and low expression levels observed upon mutation of Pro 106 suggests that a well-defined conformation in this region of the polypeptide is required for correct folding and/or heme incorporation. The proximity of Pro 100 to the heme and the number of critical tertiary contacts in this region suggested to us that mutation of Pro 100 would also be seriously destabilizing, so instead we chose to mutate His 355, which could be in a position to catalyze a cis-trans isomerization of Pro 100 via a general acid mechanism. However, all three His 355 mutants that we made, H355F, H355D and H355N, showed loss of heme upon exposure to air, suggesting that heme is oxidatively degraded in the His 355 mutants.
Localization of perturbations due to the P89I mutation and selective Pdx binding to one conformer of P89I
1H,15N correlations in TROSY-HSQC of P89I CYP-S-CO spectra allow us to identify two conformations at slow exchange on the 1H chemical shift time scale in ∼50/50 mixture as indicated by doubling of NMR resonances in the mutant (Figures 4 and 5). Besides the aforementioned doubling of the 1H methyl resonances of bound camphor (Fig. 4), doubling is confined to residues spatially adjacent to the P89I mutation (β3 and β5 strands), residues in the active site that interact with bound camphor, residues in the B-B’ loop, B’ and I helices, and residues that are mechanically linked to the B-B’ loop, B’ and I helices (see Supplementary material). The 1:1 ratio of the two forms indicates that the cis conformation of the Ile 88-Ile 89 bond is stabilized and/or the trans conformation destabilized by the local protein environment, since in isolated non-prolyl peptide bonds, the trans conformation is favored by 2.5 kcal/mol, corresponding to only 1.5% cis conformation at ambient temperature (Pal and Chakrabarti, 1999). Molecular modeling indicates that the hydrophobic side chain of Ile 89 is solvent exposed in the trans conformation, but not in the cis, which would destabilize the trans conformation. The cis conformation is also stabilized by hydrogen bonding from Tyr 29 (vide supra), an interaction that molecular modeling indicates is not available to the trans form. Both of these factors are likely to contribute to the observed cis-trans ratio in P89I.
Figure 5.

Corresponding resonances in the 800 MHz 1H, 15N TROSY-HSQC spectra of WT (red) and P89I (black) CYP-S-CO showing splitting of resonances due to two conformations at slow exchange in the P89I mutant. Ile 88 immediately precedes Pro 89, and the sidechain of Ile 395 (adjacent to Gly 394) is in van der Waals contact with Ile 88 NH.
For most NH correlations split by the P89I mutation, only one of the pair of resonances is perturbed by the addition of Pdxr (Figure 6). Because of spectral complexity, it was not possible to accurately measure Kd of Pdxr for the affected conformer. However, it is striking that in many cases, the first addition of Pdxr to the sample resulted in the disappearance of one of a pair of correlations associated with a single residue, while the other member of the pair was unaffected or only weakly perturbed (see Supplementary material). This indicates that either one of the two conformers interacts preferentially with Pdxr, or that Pdx drives a conformational change in only one of the two conformers.
Figure 6.
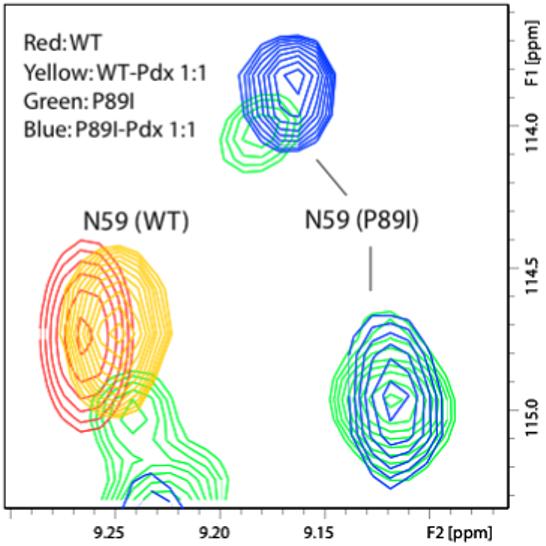
Pdxr interacts preferentially with one conformer of P89I CYP-S-CO, as detected by TROSY-1H, 15N HSQC. Only one of the split resonances of Asn 59 (the nearest non-sequential contact to Ile 89) titrates upon addition of Pdxr, while the other is unperturbed. Scale on the P89I spectrum has been increased relative to WT for clarity. All spectra were obtained at 800 MHz 1H, 298 K with sample conditions as described in the Experimental section.
Discussion
The hypothesis driving the current work is that binding of Pdxr to CYP-S-CO drives isomerization of the Ile 88-Pro 89 amide bond from a trans or distorted trans conformation in the absence of Pdxr to one that is cis in the Pdxr-bound form. Furthermore, we propose that the structural perturbations detected by NMR in the active site and other distal locations of CYP-S-CO upon Pdxr binding at the proximal face are due primarily to this isomerization. Mutation of Pro 89 to Ile results in population of both cis and trans conformations around the Ile 88-Ile 89 amide bond in P89I CYP-S-CO, as reflected by the doubling of NMR signals from residues affected by the mutation. The two conformations are at slow exchange on the chemical shift time scale in the P89I mutant, which is expected since the barrier to cis-trans isomerization is raised considerably in X-Y peptides when Y is not proline (Schulz and Schirmer, 1979). Based on the ratios of split signal intensities, the two conformers are roughly equal in energy in the P89I mutant, although line width differences between the split signals for many residues suggest that the two conformers have different dynamics on the ms timescale in some regions of the enzyme (see Figs. 5, 6 and Supplementary material). The 1H resonances of the 8- and 9-methyl groups of bound camphor indicate that camphor occupies two equally populated orientations in the active site of P89I (Fig. 4), and that these orientations are identical to the two camphor orientations found to interconvert in WT CYP-S-CO as a function of Pdxr binding (Wei et al., 2005). Furthermore, those residues affected by the P89I mutation (as determined by doubling of NMR resonances) are for the most part also those perturbed by Pdxr binding in WT CYP-S-CO. In particular, the NH correlations of residues Gly 248-Val 253 in the I helix, all of which are strongly affected by the P89I mutation, show the largest perturbations of any distal residues in WT CYP-S-CO upon Pdxr binding (Rui et al., 2006). The NH correlations of Gly 248, Gly 249, Asp 251, Thr 252 and Val 253 in WT CYP-S-CO all respond to Pdxr binding in the slow-exchange regime, that is, with 1H chemical shift changes greater than 200 Hz (Figure 7). Poulos and co-workers noted that the I helix is distorted in (cis-Ile 88-Pro 89 conformer) crystallographic structures between Gly 248 and Thr 252 with the Thr 252 OγH side chain hydroxyl acting as the proton donor to the carbonyl of Gly 248, replacing the i, i+4 hydrogen bond from the Gly 248 carbonyl to the NH of Thr 252 expected in α-helical structures. This distortion opens a gap in the I helix that provides a pocket to accommodate the Fe-bound O2 in the appropriate orientation for chemistry (Poulos, 2007). Clearly, both the binding of Pdx to WT CYP101 and perturbations introduced by the mutation of Pro 89 result in changes in the O2 binding pocket. It is likely that in both cases, displacements of the B’ helix are coupled to conformational changes in the I helix by interactions between the side chains of Phe 98 and Ile 99 in the B’-C loop with Met Leu 244 and Met 241 in the I helix (Fig. 3).
Figure 7.
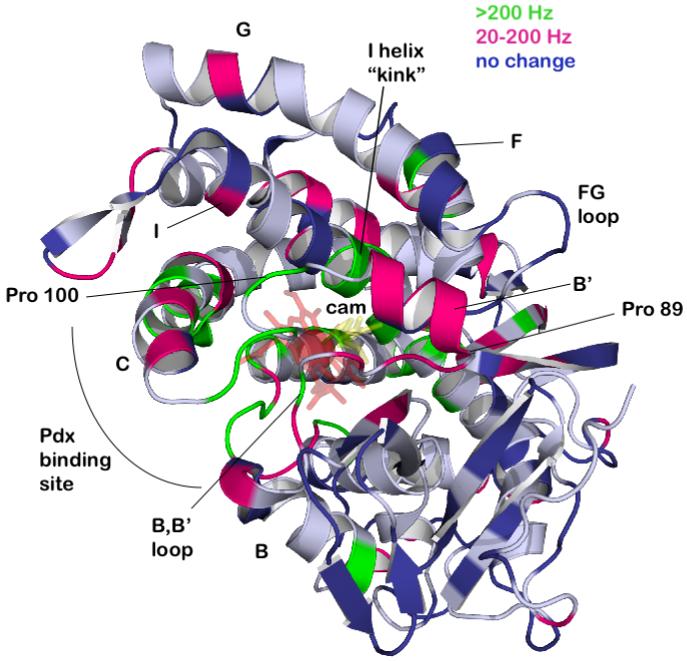
Distribution of residues in CYP-S-CO affected by Pdxr titration, color-coded to match residue labels in Figures S1 and S2 in supplementary material. Slow exchange residues (Δδmax > 200 Hz) are in green, titrating residues in crimson and non-perturbed residues are in dark blue. Residues in gray are either unassigned or titration behavior is undetermined due to spectral overlap. Structural features discussed in the text are labeled.
Evidence for Pdxr-enforced population of the cis conformation of the Ile 88-Pro 89 bond in CYP-S-CO
There are several reasons to conclude that the crystallographically observed cis conformer of the Ile 88-Pro 89 bond is also that favored upon binding of Pdxr. First, we see the appearance of an upfield proline 13Cγ signal in the Pdxr-bound WT CYP-S-CO that is either absent or much less intense in the Pdxr-free form (Fig. 2). An upfield-shifted proline 13Cγ is usually diagnostic for a cis X-Pro conformation. Secondly, we found that 1H ring-current shifts calculated for the methyl resonances of bound camphor from the (cis) 3CPP crystal structure are in better agreement with the shifts observed for the Pdxr-bound CYP-S-CO than are those seen for the Pdx-free form (Wei et al., 2005).
Results from the Y29F mutant of CYP101 also support the idea that the cis conformer is favored upon binding of Pdx, if, as expected, the cis conformer is destabilized by the absence of the hydrogen bond between the carbonyl oxygen of Ile 88 and the Tyr 29 hydroxyl group. The change in Kd for Pdxr (by a factor of ∼10 in Y29F relative to WT) corresponds to destabilization of the Pdx-bound form of Y29F by ∼6 kJ/mol, which is appropriate for the loss of a weak to medium hydrogen bond. Conversely, the lack of large spectral perturbations in the Y29F mutant relative to WT suggests that the cis conformer is not a significant contributor to the solution conformations present in the Pdx-free form of CYP101.
One might ask why, if it is the dominant form in solution in the absence of Pdx, the trans conformer has not been detected crystallographically. Although we cannot answer this question unequivocally, it is likely that crystal packing favors the cis form. Furthermore, the barrier to isomerization is not high (vide infra), and packing forces in the crystal may be sufficient to drive the isomerization in situ. It has been shown that crystalline CYP-S-O2 is capable of turning over substrate in the absence of effector using radiolytically generated electrons, confirming that the crystallographic conformer is catalytically competent (Schlichting et al., 2000).
Do local structural features in CYP101 catalyze Ile 88-Pro 89 isomerization?
The rate constant for the conformational exchange observed upon Pdxr binding to WT CYP-S-CO (between 150 s-1 and 200 s-1 at 298 K and half-saturation (Wei et al., 2005), see also Fig. 1) indicates an activation free energy of ∼60 kJ/mol, which lies between the barriers for an uncatalyzed X-Pro isomerization (∼80 kJ/mol) and those catalyzed by PPIases (∼50 kJ/mol for CyA) (Stein, 1993). We might therefore expect the structure of CYP101 near the Ile 88-Pro 89 dipeptide to have features in common with the active sites of known PPIases. PPIases are thought to catalyze X-Pro cis-trans isomerization by providing a hydrophobic environment for the amide carbonyl (thereby destabilizing the charge-separated O--C=N+ resonance structure that imparts double bond character to the C-N bond) and by providing a localized hydrogen bond donor to stabilize the lone pair on the amide nitrogen due to developing sp3 character in the transition state (Stein, 1993). The structure of CYP101 in the vicinity of the Ile 88-Pro 89 amide bond meets both of these requirements. The closest side chains to the Ile 88-Pro 89 amide (Met 28, Tyr 29, Phe 87, Ile 88, Pro 89 and Ile 395) are hydrophobic, and there are no water molecules within hydrogen bonding distance of the amide in the 3CPP crystal structure. The side chain hydroxyl of Tyr 29, the only hydrogen bond donor to the Ile 88-Pro 89 peptide, is positioned such that it stabilizes the cis conformation of the 88-89 amide, but not the trans. This hydroxyl group is also in a position to stabilize a developing electron lone pair on the Pro 89 amide nitrogen by hydrogen bonding in the transition state of the isomerization. A hydrogen bond from a tyrosine hydroxyl group has been proposed to stabilize the sp3 nitrogen electron lone pair in the transition state of X-Pro isomerization catalyzed by the PPIase FKBP (Stein, 1993).
One other comparison with PPIases is worth making. Ground state binding of the trans substrate conformer by CyA is thought to involve considerable distortion of the target peptide bond away from planarity by a combination of steric interactions and the destabilization of the charge-separated O--C=N+ resonance structure discussed above (Hur and Bruice, 2002; Zhou and Ke, 1996). If the same holds true for CYP101, one might expect that the trans conformation of the Ile 88-Pro 89 bond is likewise distorted in the ground state. (In the cis conformer, the Tyr 29 OH hydrogen bond is expected to stabilize the charge-separated resonance form and maintain bond planarity).
Generality of mechanism
Based on our conclusions regarding Pdx-enforced conformational selection in CYP101, we might expect to find similar mechanisms active in other P450 enzymes. BLAST multiple sequence alignment (Altschul et al., 1997) reveals that proline is conserved at the beginning of the B’ helix in a majority of known and putative bacterial P450 genes with homology to CYP101, and if present, is usually preceded by a hydrophobic residue (Ile, Val, Phe or Leu). The same motif is found in other P450s for which structures have been determined, and may correlate with enzymes that have specific substrates and give products with a single regio- or stereochemistry. For example, in CYP2C5 (progesterone 21-hydroxylase) Val-Pro precedes the B’ helix, but not in CYP2B4, which oxidizes a wide range of substrates, despite 72% sequence similarity between the two enzymes. There could also be a substrate size limitation for this mechanism: P450eryF, which is involved in biosynthesis of a 14-atom ring macrolide, the B’ helix is preceded by Phe-Pro (Cupp-Vickery and Poulos, 1997) but there is no Pro initiator for either the B1′ or B2′ helices in P450epoK, which acts on a 16-member macrocycle (Nagano et al., 2003).
In summary, our observations support the existence of a hitherto unsuspected and functionally relevant X-Pro isomerization in a well-characterized enzyme. X-Pro isomerizations have been implicated in a variety of biological switches, so it is likely that such isomerizations can control conformational changes necessary for efficient activity in enzymes as well.
Experimental Procedures
Site-directed mutagenesis
All mutants were generated using the CYP101 gene encoding the C334A mutant of CYP101 as template. The C334A mutant has been shown to be spectroscopically and enzymatically identical to wild-type CYP101 enzyme, but does not form dimers in solution, and so is more suitable for solution NMR studies than wild-type enzyme (Nickerson and Wong, 1997). For convenience, the abbreviation WT in this paper refers to the C334A mutant. A four-primer mutagenesis protocol was used to introduce mutations. Experimental methods used for four-primer mutagenesis were described previously (Pochapsky et al., 2001). The presence of the appropriate mutation was confirmed in each case by sequencing the entire CYP101 gene after isolation and purification of the mutant plasmid.
Mutant proteins were overexpressed, isolated, and purified from Escherichia coli strain NCM533 harboring modified pDNC334A plasmids that encoded the appropriate mutant of CYP101 under control of the lac promoter (Nickerson and Wong, 1997). Mutant proteins were expressed and purified using standard methods (Rui et al., 2006). The purity of WT CYP101 was determined spectroscopically. Fractions with an absorption ratio A391/A280 greater than 1.4 were used for experiments described below. For mutant proteins, purity was confirmed by gel electrophoresis.
Activity assays
Percent uncoupling was determined by gas chromatography from the ratio of hydroxycamphor to camphor peak areas for fixed amounts of camphor present and NADH consumed in a standard reconstituted camphor hydroxylase assay. Initial reaction rates were determined spectroscopically from the rate of change in NADH absorption under standard assay conditions. Details of the assay have been published previously (Rui et al., 2006). Each experiment was performed at least twice. Single-turnover assays in the absence of effector were performed as described by elsewhere (Tosha et al., 2004).
NMR Spectroscopy
The expression and purification of isotopically labeled CYP101 for NMR samples has been described previously (Rui et al., 2006). Samples of WT CYP101 were prepared with the following combinations of isotopic labels: uniform (u)-15N; u-2H,u-15N; u-2H,u-15N, (1H,13C)-Pro, and u-2H,u-15N, (1H,13C)-Pro, Ile. Mutant proteins were expressed and purified in either u-15N or u-2H,u-15N labeled form. CYP-S-CO samples for all NMR experiments were 0.2-0.4 mM in either 100% D2O or 90% H2O/10% D2O, pH 7.4, 50 mM deuterated TrisCl, 100 mM KCl and 1 mM (d)-camphor. Samples of Pdx and CYP101 were reduced and prepared for spectroscopy using previously described methods (Pochapsky et al., 2003). Pdx samples used for titration were concentrated to ∼ 6 mM prior to titration. NMR experiments were performed on a Bruker Avance 800 MHz spectrometer operating at 800.13, 201.2 and 81.08 MHz for 1H, 13C and 15N respectively. All NMR experiments were performed at 25 °C. Experiments used for sequential resonance assignments and structural analyses have been described previously (Pochapsky et al., 2003; Wei et al., 2005; OuYang et al., 2006; Rui et al., 2006). Dissociation constants for Pdxr-CYP-S-CO complexes were calculated from chemical shift perturbations induced by Pdxr titration as described previously (Pochapsky et al., 2003).
Supplementary Material
Acknowledgements
This work was supported by a grant from the US Public Health Service (RO1-GM44191, TCP). The 800 MHz NMR was purchased with a grant from the NCRR HEI program.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti AH. Native state proline isomerization: An intrinsic molecular switch. Biochemistry. 2003;42:9515–9524. doi: 10.1021/bi0350710. [DOI] [PubMed] [Google Scholar]
- Andreotti AH. Opening the pore hinges on proline. Nature Chem. Biol. 2006;2:13–14. doi: 10.1038/nchembio0106-13. [DOI] [PubMed] [Google Scholar]
- Cupp-Vickery JR, Poulos TL. Structure of cytochrome P450eryf: Substrate, inhibitors, and model compounds bound in the active site. Steroids. 1997;62:112–116. doi: 10.1016/s0039-128x(96)00168-7. [DOI] [PubMed] [Google Scholar]
- Dorman DE, Bovey FA. C-13 magnetic resonance spectroscopy - Spectrum of proline in oligopeptides. J. Org. Chem. 1973;38:2379–2383. [Google Scholar]
- Glascock MC, Ballou DP, Dawson JH. Direct observation of a novel perturbed oxyferrous catalytic intermediate during reduced putidaredoxin-initiated turnover of cytochrome P450cam - Probing the effector role of putidaredoxin in catalysis. J. Biol. Chem. 2005;280:42134–42141. doi: 10.1074/jbc.M505426200. [DOI] [PubMed] [Google Scholar]
- Grochulski P, Li Y, Schrag JD, Cygler M. Two conformational states of Candida rugosa lipase. Prot. Sci. 1994;3:82–91. doi: 10.1002/pro.5560030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur S, Bruice TC. The mechanism of cis-trans isomerization of prolyl peptides by cyclophilin. J. Am. Chem. Soc. 2002;124:7303–7313. doi: 10.1021/ja020222s. [DOI] [PubMed] [Google Scholar]
- Jovanovic T, Farid R, Friesner RA, McDermott AE. Thermal equilibrium of high- and low-spin forms of cytochrome P450BM-3: Repositioning of the substrate? J. Am. Chem. Soc. 2005;127:13548–13552. doi: 10.1021/ja0524604. [DOI] [PubMed] [Google Scholar]
- Katagiri M. Early years of oxygenase research in Bethesda, Osaka, Urbana, and Kanazawa. Biochem. Biophys. Res. Commun. 2005;338:285–289. doi: 10.1016/j.bbrc.2005.07.179. [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graphics. 1996;14:51. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- Lipscomb JD, Sligar SG, Namtvedt MJ, Gunsalus IC. Autooxidation and hydroxylation reactions of oxygenated cytochrome P450cam. J. Biol. Chem. 1976;251:1116–1124. [PubMed] [Google Scholar]
- Lummis SCR, Beene DL, Lee LW, Lester HA, Broadhurst RW, Dougherty DA. Cis-trans isomerization at a proline opens the pore of a neurotransmitter-gated ion channel. Nature. 2005;438:248–252. doi: 10.1038/nature04130. [DOI] [PubMed] [Google Scholar]
- Mueller EJ, Loida PJ, Sligar SG. Twenty five years of P450cam research. In: Ortiz de Montellano P, editor. Cytochrome P450: Structure, Function and Biochemistry. Plenum Press; New York: 1995. pp. 83–124. [Google Scholar]
- Nagano S, Li HY, Shimizu H, Nishida C, Ogura H, Ortiz de Montellano PR, Poulos TL. Crystal structures of epothilone D-bound, epothilone B-bound, and substrate-free forms of cytochrome P450epoK. J. Biol. Chem. 2003;278:44886–44893. doi: 10.1074/jbc.M308115200. [DOI] [PubMed] [Google Scholar]
- Nelson CJ, Santos-Rosa H, Kouzarides T. Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell. 2006;126:905–916. doi: 10.1016/j.cell.2006.07.026. [DOI] [PubMed] [Google Scholar]
- Nickerson DP, Wong LL. The dimerization of Pseudomonas putida cytochrome P450cam: Practical consequences and engineering of a monomeric enzyme. Protein Eng. 1997;10:1357–1361. doi: 10.1093/protein/10.12.1357. [DOI] [PubMed] [Google Scholar]
- OuYang B, Pochapsky SS, Pagani GM, Pochapsky TC. Specific effects of potassium ion binding on wild-type and L358P cytochrome P450cam. Biochemistry. 2006;45:14379–14388. doi: 10.1021/bi0617355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, Chakrabarti P. Cis peptide bonds in proteins: Residues involved, their conformations, interactions and locations. J. Mol. Biol. 1999;294:271–288. doi: 10.1006/jmbi.1999.3217. [DOI] [PubMed] [Google Scholar]
- Pochapsky TC, Kostic M, Jain N, Pejchal R. Redox-dependent conformational selection in a Cys4Fe2S2 ferredoxin. Biochemistry. 2001;40:5602–5614. doi: 10.1021/bi0028845. [DOI] [PubMed] [Google Scholar]
- Pochapsky SS, Pochapsky TC, Wei JW. A model for effector activity in a highly specific biological electron transfer complex: The cytochrome P450cam-putidaredoxin couple. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- Poulos TL, Finzel BC, Howard AJ. Crystal structure of substrate-free Pseudomonas putida cytochrome P450. Biochemistry. 1986;25:5314–5322. doi: 10.1021/bi00366a049. [DOI] [PubMed] [Google Scholar]
- Poulos TL, Finzel BC, Howard AJ. High resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- Poulos TL. The past and present of P450cam structural biology. Biochem. Biophys. Res. Commun. 2003;312:35–39. doi: 10.1016/j.bbrc.2003.09.239. [DOI] [PubMed] [Google Scholar]
- Poulos TL. Structural biology of P450-oxy complexes. Drug Metabol. Rev. 2007;39:557–566. doi: 10.1080/03602530701498240. [DOI] [PubMed] [Google Scholar]
- Raag R, Poulos TL. Crystal structure of the carbon monoxide substrate cytochrome P450cam ternary complex. Biochemistry. 1989;28:7586–7592. doi: 10.1021/bi00445a013. [DOI] [PubMed] [Google Scholar]
- Ravindranathan KP, Gallicchio E, McDermott AE, Levy RM. Conformational dynamics of substrate in the active site of cytochrome P450BM-3/NPG complex: Insights from NMR order parameters. J. Am. Chem. Soc. 2007;129:474–475. doi: 10.1021/ja0672371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui LY, Pochapsky SS, Pochapsky TC. Comparison of the complexes formed by cytochrome P450cam with cytochrome b5 and putidaredoxin, two effectors of camphor hydroxylase activity. Biochemistry. 2006;45:3887–3897. doi: 10.1021/bi052318f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar P, Reichman C, Saleh T, Birge RB, Kalodimos CG. Proline cis-trans isomerization controls autoinhibition of a signaling protein. Mol. Cell. 2007;25:413–426. doi: 10.1016/j.molcel.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert M, Labudde D, Oschkinat H, Schmieder P. A software tool for the prediction of Xaa-Pro peptide bond conformations in proteins based on C-13 chemical shift statistics. J. Biomol. NMR. 2002;24:149–154. doi: 10.1023/a:1020997118364. [DOI] [PubMed] [Google Scholar]
- Schlichting I, et al. The catalytic pathway of cytochrome P450cam at atomic resolution. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- Schulz GE, Schirmer RH. Principles of Protein Structure. Springer-Verlag; Berlin: 1979. p. 25. [Google Scholar]
- Sjodin T, Christian JF, Macdonald IDG, Davydov R, Unno M, Sligar SC, Hoffman BM, Champion PM. Resonance Raman and EPR Investigations of the D251N oxycytochrome P450cam/putidaredoxin complex. Biochemistry. 2001;40:6852–6859. doi: 10.1021/bi002510b. [DOI] [PubMed] [Google Scholar]
- Stein RL. Mechanism of enzymatic and nonenzymatic prolyl cis-trans isomerization. Adv. Prot. Chem. 1993;44:1–24. doi: 10.1016/s0065-3233(08)60562-8. [DOI] [PubMed] [Google Scholar]
- Tosha T, Yoshioka S, Ishimori K, Morishima I. L358P mutation on cytochrome P450cam simulates structural changes upon putidaredoxin binding - The structural changes trigger electron transfer to oxy-P450cam from electron donors. J. Biol. Chem. 2004;279:42836–42843. doi: 10.1074/jbc.M404216200. [DOI] [PubMed] [Google Scholar]
- Unno M, Christian JF, Sjodin T, Benson DE, Macdonald IDG, Sligar SG, Champion PM. Complex formation of cytochrome P450cam with putidaredoxin - Evidence for protein-specific interactions involving the proximal thiolate ligand. J. Biol. Chem. 2002;277:2547–2553. doi: 10.1074/jbc.M108917200. [DOI] [PubMed] [Google Scholar]
- Urushino N, Yamamoto K, Kagawa N, Ikushiro S, Kamakura M, Yamada S, Kato S, Inouye K, Sakaki T. Interaction between mitochondrial CYP27B1 and adrenodoxin: Role of arginine 458 of mouse CYP27B1. Biochemistry. 2006;45:4405–4412. doi: 10.1021/bi060072o. [DOI] [PubMed] [Google Scholar]
- Wade RC, Winn PJ, Schlichting I, Sudarko A. A survey of active site channels in cytochromes P450. J. Inorg. Biochem. 2004;98:1175–1182. doi: 10.1016/j.jinorgbio.2004.02.007. [DOI] [PubMed] [Google Scholar]
- Wei JY, Pochapsky TC, Pochapsky SS. Detection of a high-barrier conformational change in the active site of cytochrome P450cam upon binding of putidaredoxin. J. Am. Chem. Soc. 2005;127:6974–6976. doi: 10.1021/ja051195j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, McCullough CR, Costache AD, Pullela PK, Sem DS. Structural evidence for a functionally relevant second camphor binding site in P450cam: Model for substrate entry into a P450 active site. Proteins-Struct. Funct. Bioinf. 2007;69:125–138. doi: 10.1002/prot.21508. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Myshkin E, Waskell L. Role of cytochrome b5 in catalysis by cytochrome P4502B4. Biochem. Biophys. Res. Commun. 2005;338:499–506. doi: 10.1016/j.bbrc.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Zhang HM, Im SC, Waskell L. Cytochrome b5 increases the rate of catalysis by cytochrome P4502B4. Acta Pharm. Sinica. 2006;27:213–213. [Google Scholar]
- Zhao Y, Ke H. Crystal structure implies that cyclophilin predominantly catalyzes the trans to cis isomerization. Biochemistry. 1996;35:7356–7361. doi: 10.1021/bi9602775. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.