

Abstract
NF-κB transcription factors induce a host of genes involved in pro-inflammatory/stress-like responses; but the collateral effects and consequences of sustained NF-κB activation on other cellular gene expression programming remain less well understood. Here enforced expression of a constitutively active IKKβ T-loop mutant (IKKβca) drove murine fibroblasts into transient growth arrest that subsided within 2-3 weeks of continuous culture. Proliferation arrest was associated with a G1/S phase block in immortalized and primary early passage MEFs. Molecular analysis in immortalized MEFs revealed that inhibition of cell proliferation in the initial 1-2 weeks after their IKKβca retroviral infection was linked to the transient, concerted repression of essential cell cycle effectors that are known targets of either E2F or FoxM1. Co-expression of a phosphorylation resistant IκBα super repressor and IKKβca abrogated growth arrest and cell cycle effector repression, thereby linking IKKβca's effects to canonical NF-κB activation. Transient growth arrest of IKKβca cells was associated with enhanced p21 (cyclin-dependent kinase inhibitor 1A) protein expression, due in part to transcriptional activation by NF-κB and also likely due to strong repression of Skp2 and Csk1, both of which are FoxM1 direct targets mediating proteasomal dependent p21 turnover. Ablation of p21 in immortalized MEFs reduced their IKKβca mediated growth suppression. Moreover, trichostatin A inhibition of HDACs alleviated the repression of E2F and FoxM1 targets induced by IKKβca, suggesting chromatin mediated gene silencing in IKKβca's short term repressive effects on E2F and FoxM1 target gene expression.
Keywords: NF-κB, IKKβca mutant, FoxM1 and E2F targets, p21, cell cycle arrest, HDACs
INTRODUCTION
The NF-κB transcription factor family encodes essential regulators of innate and adaptive immunity. NF-κB becomes activated in pro-inflammatory stress-like responses initiated by a host of extracellular stimuli including bacterial and viral infections, cytokines and DNA damaging agents (Baldwin, 1996; Bonizzi and Karin, 2004; Hoffmann et al., 2006; Karin and Greten, 2005; Karin and Lin, 2002; May and Ghosh, 1998; Wu et al., 2006). The mammalian NF-κB transcription factor family consists of 5 members (RelA/p65, c-Rel, RelB, p105/p50 and 100/p52) each containing a Rel-homology domain (RHD) required for their DNA binding (Baldwin, 1996; May and Ghosh, 1998). NF-κB subunits hetero- or homo- dimerize with each other to bind to their bipartite consensus sequence (GGGRNWTYCC) in the promoters or enhancers of chromosomal target genes to induce their transcription (Baldwin, 1996; May and Ghosh, 1998). However, in most cells in the absence of an activating signal, prototypical NF-κB hetero-dimers are sequestered in a cytoplasmic inactive state by members of the inhibitory IκB family, which prevent NF-κB nuclear import and transcriptional activation (Baldwin, 1996; Basak et al., 2007; Derudder et al., 2003; Ghosh et al., 1998; May and Ghosh, 1998). Unlike p65/RelA, c-Rel and RelB, the p50 and p52 NF-κB subunits lack transcriptional activation domains (TADs); and in association with other regulatory factors DNA bound p50 homo-dimers have the capability to suppress the transcription of NF-κB targets (Grundstrom et al., 2004; Pan and McEver, 1995; Zhong et al., 2002). The transcriptional competence of DNA bound NF-κB subunits is also post-translationally regulated by site specific phosphorylations mediated by the IKKs as well as other kinases, which can have positive as well as negative effects on the transcription of NF-κB target genes (Barre and Perkins, 2007; Jiang et al., 2003; Perkins, 2006; Perkins, 2007; Sizemore et al., 2002).
The diverse intracellular signaling pathways of the majority of extracellular activators of NF-κBs all converge on the same cytoplasmic signaling complex, the IKK signalsome to activate NF-κBs. Three major proteins, IKKα and IKKβ, (two homologous serine-threonine kinases), and NEMO/IKKγ, (an adaptor protein which coordinates IKK assembly and activation), comprise the IKK signalsome complex (Ghosh and Karin, 2002; Karin, 1999; May and Ghosh, 1999; Yamamoto and Gaynor, 2004). Upstream signal induced IKK activating kinases are delivered to the IKK signalsome by protein-protein interactions facilitated by NEMO's polyubiquitination and oligomerization (Ea et al., 2006; Poyet et al., 2000; Tegethoff et al., 2003; Wuerzberger-Davis et al., 2007). IKKα and IKKβ are subsequently activated by the site specific phosphorylation of two conserved serine residues located within their T-loop activation domains. These activated IKKs in turn, phosphorylate IκBs on two conserved, amino-terminal serine residues, which flags them for ubiquitination, and proteasome dependent hydrolysis thereby liberating NF-κB dimers for nuclear translocation and subsequent target gene activation. In vivo, the IKKβ protein is almost always responsible for IκB phosphorylation, but IKKα can also provide this function in some unique circumstances (Cao et al., 2001; Hansberger et al., 2007). IKKα has also been reported to localize to the promoters of canonical NF-κB p65/p50 target genes, where it has been shown to either mediate histone H3 phosphorylation or release repressive factors to facilitate transcriptional activation (Anest et al., 2003; Hoberg et al., 2006; Hoberg et al., 2004; Li et al., 2002; Yamamoto and Gaynor, 2004; Yamamoto et al., 2003). Importantly the NEMO/IKKγ independent, alternative or non-canonical NF-κB activation pathway also solely depends on activated IKKα, which phosphorylates two serines in an IκB-like carboxy-proximal domain of the NF-κB2/p100 precursor protein thereby promoting its proteasomal processing to mature NF-κB p52 (Amir et al., 2004; Bonizzi and Karin, 2004; Pomerantz and Baltimore, 2002; Senftleben et al., 2001). The non-canonical pathway liberates RelB/p52 heterodimers and a subset of p50/p65 heterodimers residing in cytoplasmic complexes with p100 (Basak et al., 2007) to enter the nucleus and activate target genes involved in some innate and most adaptive immune responses. The NF-κB p52 subunit has also been reported to have functions outside of the NF-κB pathway by differentially regulating the activity of p53 target genes (Schumm et al., 2006). In addition IKKα-dependent phosphorylation of the CBP co-activator was recently shown to enhance its association with p52 while simultaneously displacing p53 (Huang et al., 2007).
Activated NF-κB has been reported to interfere with the transcriptional activities of p53 and c-Myb by the squelching of shared, limiting transcriptional co-activators (Nicot et al., 2001; Wadgaonkar et al., 1999; Webster and Perkins, 1999); but a general role for activated NF-κB in the interference mediated repression of other transcription factor networks and more importantly the short vs. long term effects this could have on cellular physiology remain unclear. In a prior study we employed DNA microarrays to reveal the global effects of TNFα, a strong archetypical stimulus of canonical NF-κB activation, on the induction of NF-κB and IKK dependent genes (Li et al., 2002). Although not reported in that prior study these DNA microarray screens also revealed a novel subset of TNFα and NF-κB dependent repressed genes, which were enriched in E2F and FoxM1 targets that control cell cycle progression. Herein we have gone on to show that IKKβca mediated sustained activation of canonical NF-κB signaling in murine fibroblasts induces the transient suppression of essential cell cycle effectors regulated by E2F and FoxM1, which occurs in conjunction with a short-term block to cellular proliferation.
MATERIALS AND METHODS
Tissue Culture and Retroviral Infections
Culture of IKKα (−/−), IKKβ (−/−), NEMO (−/−) and IκBαSR expressing mouse embryonic fibroblasts (MEFs) and their stimulation with TNF-α have been previously described (Li et al., 2002). Trichostatin-A (TSA) (Sigma) treatment was performed at a concentration of 100 nM for a total of 4 hours (Ashburner et al., 2001). For the latter 2 hours, the cells were either left untreated (US control) or were co-incubated with TNF-α (2Τ) at a concentration of 20 ng/ml. High titre amphotyped retroviruses were prepared by transient calcium phosphate transfection of Phoenix A packaging cells (kindly provided by Dr. Gary Nolan, Stanford Univ). For retroviral infections sixty thousand cells were seeded per well in six well plates one day prior to retroviral transduction. Retroviral infections were carried out by centrifuging viral supernatants onto cells @2000-2400 RPM for 45-60′ @30-32°C) in media supplemented with 8 μg/ml polybrine (Sigma) followed by continued incubation for 5 hrs at 32°C in a 5% CO2 incubator prior to changing to fresh growth media. Stable populations of cells expressing puromycin resistance retroviruses were obtained by commencing puromycin selections (1 μg/ml) 48 hrs post-infection with three changes of media over the next 4-5 days. Populations of cells transduced with retroviruses conferring neomycin resistance were selected in 1 mg/ml neomycin over 1-2 weeks (Li et al., 2001; Li et al., 2002).
Retroviral vectors
Retroviral expression vectors BIP and MP9 and IBIN {containing an IκBα super-repressor (SR) and a neomycin selection marker in an IκBαSR-Ires-Neo bicistronic cassette} have been previously described (Facchini et al., 2005; Li et al., 2001; Li et al., 2002; Palumbo et al., 2007; Zhang et al., 2005). A constitutively activated Human Flag-IKKβ (IKKβca) mutant in pcDNA3.1 was generated by changing the T-loop activation serines (177 & 181) to glutamic acids with a QuickStart PCR mutagenesis kit (Stratagene). The IKKβca cassette was removed from pcDNA3.1 and placed under the control of a moloney 5′ LTR by insertion into the unique SnaB1 or BamHI sites of the BIP and MP9(GFP) moloney retroviral vectors respectively by standard sub-cloning procedures.
Microarray analysis
Affymetrix MG-U74Av2 chips were used for all experiments. Chips were stained with streptavidin-phycoerythrin (Molecular Probes) and scanned with a Hewlett-Packard Gene Array Scanner and DNA microarray chip data analysis was performed using MAS5.1 software (Affymetrix) as previously described (Li et al., 2002; Massa et al., 2005). To identify TNFα stimulus dependent repression targets, two independently derived stocks of wild type immortalized MEFs were used. The expression signal values from matched stimulated (2T) and unstimulated (US) Wt. MEF samples were compared with the following criteria: (a) a change call of Decrease or Marginal Decrease, and a fold change value of −1.5 or lower with each Wt. MEF 2T versus Wt. MEF US comparison; (b) an average fold change value of −2.0 or lower in the Wt MEF comparisons. The NF-κB dependency of repression targets was determined by a change call of Decrease or Marginal Decrease and a fold change value of −2.0 or lower in both comparisons of each Wt. MEF 2T experimental screen file versus the Wt. MEF + IκBαSR 2T baseline file. This class of NF-κB/TNFα dependent repressed genes were next screened for their IKKα, IKKβ and NEMO requirements by comparing their expression signal values in each Wt. MEF 2T screen file vs. screens of IKKα(−/−) 2T, IKKβ(−/−) 2T and NEMO(−/−) 2T cell samples (as previously described for TNFα/NF-κB dependent induced genes) (Li et al., 2002; Massa et al., 2005). Because the Wt. TNFα sample appears in the numerator of each screen comparison (for example Wt. 2T vs. Wt. US or Wt. 2T vs. Wt + IκBαSR 2T respectively represent Wt. 2T divided by Wt. US and Wt. 2T divided by Wt. + IκBαSR 2T) this repressed class of genes would have fractional fold change values. However to simplify data presentation and interpretation, fractional fold change values were converted to their corresponding negative integers (see Figure 1).
Figure 1. Genes repressed in response to TNFα with a dependency on NF-κΒ signaling.

Twenty-five genes, enriched in mediators of cell cycle progression, that were suppressed in response to TNFα stimulation dependent on canonical NF-κB signaling are shown. Cell lines, stimulation conditions and gene selection criteria are described in Materials and Methods and also in more detail in prior work (Li et al., 2002). Data for each gene is presented as fold change values from duplicate microarray screens with two independently derived stocks of immortalized MEFs. As elaborated in Materials and Methods, to simplify the presentation of this data negative integer fold change values for repressed genes were derived from their fractional fold change values,. Accession numbers and gene names are shown in columns one and two. TNFα dependency for gene repression is shown in Column 3 (Wt. MEF 2T vs. Wt. MEF US). Column 4 shows the NF-κB dependency of each repressed gene by comparing their expression in Wt. MEF 2T vs. Wt. MEF + IκBαSR 2T (i.e., Wt. MEF 2T : Wt. MEF + IκBαSR 2T). IKK signalsome subunit dependencies are shown in an analogous way in columns 5-7 displaying Wt. MEFs 2T MEFs vs. IKKα(−/−) 2T, vs. IKKβ(−/−) 2T and vs. NEMO(−/−) 2T MEFs respectively. Genes with 2 hits were identified by multiple Affymetrix oligo probes corresponding to distinct regions within the same gene with the data for one hit shown. In the two far right columns genes which have been reported to be direct targets of the FoxM1 and E2F transcription factors are indicated by + signs (Bindra and Glazer, 2006; Costa, 2005; Ishida et al., 2001; Wang et al., 2005; Yang et al., 2007), and those marked by a # sign were found to have E2F DNA binding sites in their promoters.
RNA preparation and cDNA synthesis
Total cell RNAs were extracted from lysates of wild type and populations of stably retrotransduced cells (seven or twenty-one days post retroviral infection) with RNeasy spin column extraction and purification kits (Qiagen). Extracted RNAs were quantified with a Nanodrop spectrophotometer and 2 μg of total RNA were reverse transcribed with Superscript II Reverse transcriptase (InVitrogen). cDNAs were routinely diluted 12.5 fold in nuclease-free water (Qiagen) prior to SYBR Green real time PCR analysis.
SYBR Green Real-Time PCR
Semi- quantitative SYBR Green real time PCR reactions were assembled by mixing iQ™SYBR Green Supermix (BioRad), forward and reverse primers at final concentrations of 200 nM, 1.6 μl of diluted cDNA and nuclease-free water in 25 μl. All real time PCR quantifications were carried out in a BioRad iCycler machine. The PCR amplification protocol was as follows: Cycle 1: 95°C for 90″; Cycle 2 (50 times): 95°C for 15″, 60°C for 30″ (with SYBR green emitted fluorescence collection). The specificities of all PCR reactions were routinely inspected by analysis of their melting curves carried out as follows: 55°C for 10″, with an increase of 0.5°C at each cycle for 80 cycles (Dussault and Pouliot, 2006). By the latter method PCR products for each primer pair in all reactions produced a single species melting curve verifying their specificity. The relative yields of PCR products were quantified on the basis of their threshold cycle (Ct) values. PCR reactions were done in duplicate, averaged and fold differences were calculated by the ΔΔCt method (Dussault and Pouliot, 2006). Variations between PCR duplicates were not more than 0.5 Ct and results were confirmed by one or more independent experiments also performed in duplicate. All cDNAs were normalized versus one another by comparing their individual glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression levels against the median expression level for the cDNA set. GAPDH correction was then used as a normalization factor for the experimental primer data. All PCR oligonucleotide primer pairs were designed using BioRad Beacon Designer 2.0 software and were purchased from InVitrogen. Forward (F) and reverse (R) PCR primer sequences for each mRNA were as follows: p21 F(CAGACATTCAGAGCCACAGG), p21 R(AGTGTGCCGTTGTCTCTTCG); Cdc25b F(TGAGTGCTCCCTGTCATCTGA), Cdc25b R(TCACTGGTAAGGATCGGAAGC); Skp2 F(CCTCCAACACCTCTCGCTCAG), Skp2 R(GGTTCCCTCTGGCACGATTCC); Csk1 F(TTGAGCCACCAGTGCCACAG), Csk1 R(ACGTCAGCAAATTCACACCATCC); IL6 F(TGGGAAATCGTGGAAATGAG), IL-6 R(CTCTGAAGGACTCTGGCTTTG); Plk1 F(AGCTGCACAAGAGGAGGAAG), Plk1 R(GCTTGAGGTCCCTGTGAATG); Kif20a F(TGAAGGAGATGGTGAAGGATG), Kif20a R(CAGGTCAGGTGTCGGATG); CcnA2 F(CAGGCGGTGCTGAAGG), CcnA2 R(TTTCTTGCTGCGGGTAAAG); FoxM1: F(AAGATTATCAACCACCCCACCAC), FoxM1 R(CCAGAGCTGATGAGGATGAACC); Ect2 F(GTGTTTGAGAAGGATAAGCGAGGA), Ect2 R(TTCCCGAATCCCTTCCCGTC); Casp8 F(CGGAATCGGTAGCAAACCTCTG) Casp8: R(GGTCACAACTCCAGCTCGGG); AurkB F(GGAAGGAGAGGTGCAGGAGTA), AurkB R(AACAACAGCGAAGGCAAGAGA); Brca2 F(CCGAGATGAAGAAGCACGCA), Brca2 R(TCACTGTCAATCACTGAGGGAAC; Saa3 F(ATGAGTGGGGCCGGAGTG), Saa3 R(GACTGGGAACAACAGGAAGAGAA); ISG15 F(CGCAGACTGTAGACACGCTTAAG), ISG15 R(CCCTCGAAGCTCAGCCAG); Cdc6 F(TTATCTCCCTGTTCTCCACCAAAGC), Cdc6 R(ATCTGCTTTGCTCTCTGGACTTCTT); Rantes F (CTGCCCTCACCATCATCC), Rantes R(ACACTTGGCGGTTCCTTC); Ezh2 F(ACCCGAAAGGGCAACAAAATTC), Ezh2 R(AAGGATCCTATCCTGTGGTCACC); CcnE2 F(GCTGCTGCCGCCTTATGTC), CcnE2 R(GCACCATCCAGTCTACACATTCC); IκBα F(TACCCGAGAGCGAGGATG), IκBα R(GCTGGCCTCAAACACAC); Gapdh F(GACCCGCTTCATGCCTGG), Gapdh R (GGTGATGGTGTCCATCTGGAC).
Nuclear Extracts
Cell pellets were lysed in Hypotonic buffer (10 mM Hepes pH 7.9, 1.5 mM MgCl2, 10 mM KCl) supplemented with protease inhibitors, and centrifuged to separate the cytoplasmic fraction from the nuclear pellet. Nuclei were lysed in a 1:1 mixture of Low Salt Buffer (20 mM Hepes pH 7.9, 25% glycerol, 0.2 mM EDTA, 20 mM KCl, 1.5 mM MgCl2) and High Salt Buffer (20 mM Hepes pH 7.9, 25% glycerol, 0.2 mM EDTA, 1.2 M KCl, 1.5 mM MgCl2) (plus protease inhibitors) with nuclear extracts obtained by high speed centrifugation at 12,000 RPM at 4°C for 30 minutes.
Immunoblotting
Cells were lysed in Triton-X buffer supplemented with a cocktail of protease inhibitors (1% Triton-X100, 100mM Tris-cl pH 7.6, 150 mM NaCl, 10 μg/ml Leupeptin, 1 μg/ml Pepstatin, 100 ng/ml PMSF and 2 μg/ml Aprotinin); and proteins were quantified by a colorimetric based assay in comparison to BSA standard (BioRad) (Harlow and Lane, 1988). Total cellular (30 μg) and nuclear proteins (8 μg) were diluted in Laemmli Sample Buffer (BioRad) and boiled for 10′ in 400 mM β-mercaptoethanol prior to loading on a 12% SDS-PAGE polyacrylamide gel (Harlow and Lane, 1988). Proteins were transferred to either PVDF or cellulose nitrate membranes (Amersham Biosciences) using a Mini Trans Blot apparatus according to the manufacturer's specifications (BioRad). After electroblotting, membranes were blocked in 5% milk solution (PVDF filters) or 2% blocking buffer (GE Healthcare) for cellulose nitrate filters in TN buffer (25 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween 20) and subsequently probed overnight at 4°C with either a 1:4000 dilution of rabbit anti-mouse p21 primary antibody (BD Pharmingen) in TN+5% milk overnight at 4°C, a 1:1000 dilution of rabbit anti-Phospho-NF-κB p65 (Ser536) polyclonal antibody (Cell Signaling Technology) in TN or with a 1: 2 × 105 dilution of monoclonal anti-mouse α-tubulin antibody (Sigma) in TN +5% milk. Anti-p21 and anti-Tubulin blots were then washed and incubated for 1 hr @RT with either secondary anti-mouse IgG HRP or anti-rabbit IgG HRP antibodies (DakoCytomation) both diluted 1:200 in TN+5% milk; and phospho- NF-κB p65 (Ser536) blots were probed with a 1:5000 fold dilution of anti-rabbit IgG HRP in TN + 2% blocking buffer (GE Healthcare). Phospho-NF-κB p65 (Ser536) blots were also stripped and re-probed at 4°C overnight with rabbit polyclonal anti-p65 primary antibody (Santa Cruz Biotechnology) diluted 1:1000 in TN followed by signal detection with anti-rabbit IgG HRP secondary antibody. After detection of the latter signals membranes were stripped again and re-probed for 1 hour at RT with a primary goat polyclonal anti-Lamin B antibody (Santa Cruz Biotechnology) diluted 1:1000 in TN+3% BSA, followed by secondary bovine anti-goat antibody (Jackson) diluted 1:2 × 105 in TN+3% BSA for 1 hour at RT. After washing, HRP conjugated antibodies were detected with an enhanced chemiluminescent detection kit according to the manufacturer's specifications (GE Healthcare). Autoradiography was performed for 30 seconds to 10 minutes (depending on the antigen) before development (Kodak). Images were acquired with a Fluor-S Multimager (BioRad).
Cell proliferation analysis
Cell proliferation rates were determined essentially as previously described with minor modifications (Facchini et al., 2005). On day “0” cells were seeded at ∼1000 cells per well in quadruplicate in 96 well plates and their growth monitored every 24 hr over a 1-14 day time span. At this initial seeding density actively asynchronously growing wild type MEFs generally reached confluence by ∼7-8 days. Relative cell proliferation rates were compared by quantifying the amounts of DNA per well by means of the PicoGreen dsDNA (Molecular Probes, Eugene, OR) quantitation reagent on a daily basis over 5-7 days. Minor differences in initial cell seedings were corrected for based on DNA quantities at “Day 0”. At each time point, media was removed; and wells were washed with PBS. Plates were immediately sealed in Parafilm™ and stored frozen at −20°C until analysis. Cell lysis buffer (100 μl) (Molecular Probes) was added to each well followed by an equal volume of TE working solution containing a 1:40 dilution of PicoGreen dsDNA quantification reagent. After 5 min of incubation at RT, sample fluorescence was measured with a Spectra Max Gemini plate fluorometer (Molecular Devices Sunnyvale, CA). The instrument was set in the well scan mode with 480 excitation and 540 emission-cut off 515. Means of quadruplicate samples were compared by the Wilcoxon matched pairs test. Statistical analysis was performed using CSS statistical software (StatSoft, Tulsa, OK).
Cell cycle analysis
Cell cycle phases were visualized and quantified by FACS analysis of propidium iodide (PI) stained asynchronously growing cells. Briefly 1 × 106 exponentially growing cells were fixed in 70% ethanol, washed and then resuspended in 500 μl PBS. Cells were incubated at 37°C for 20′ in the presence of PI (50 μg/ml) and RNAse A (10 μg/ml), washed 3× with PBS and then kept on ice in the dark. PI stained cells were submitted to flow cytometry on a FACSAria instrument (Becton Dickinson, Mountain View, CA). FACSDiva software (Becton-Dickinson) was used to prepare graphs of PI content and for gating of different phases of the cell cycle.
Lactate dehydrogenase (LDH) assays
LDH catalyzes the reversible conversion of lactate to pyruvate in which the coenzyme NAD is reduced to NADH, which can be measured by a variety of quantitative photometric techniques including the stoichiometric conversion of a tetrazoilum dye (Decker and Lohmann-Matthes, 1988; Legrand C, 1992). Release of LDH by cultured cells was quantified spectrophotometically as described by the manufacturer (Roche Inc.).
Microscopy
Phase contrast images of cells were taken at 200× magnification with an Olympus CKX41 microscope fitted with an Olympus C-5060 wide zoom camera.
Promoter sequence analysis
The analysis of selected gene promoter sequences for NF-κB and E2F transcription factor (TF) binding sites was performed with the TRED database (http://rulai.cshl.edu/cgi-bin/TRED/ tred.cgi?process=home) (Zhao et al., 2005). The matrix search function was used to scan a continuous sequence string from 1000 base pairs upstream to 200 base pairs downstream with respect to each transcription start site. To derive a minimum permissible score (MPS) for each TF matrix, a threshold was determined by scanning the promoter regions of 5 genes known to be direct targets. For NF-κB, the MPS corresponded to a score of 5.0 by all three JASPAR (Lenhard and Wasserman, 2002; Sandelin et al., 2004) matrices for NF-κB (NF-κB, p50 and p65), and E2F was determined to have an MPS of 4.0.
RESULTS
Microarray screens reveal that part of the genomic response of MEFs to TNFα stimulation involves the IKK and NF-κB dependent repression of a subset of essential cell cycle effectors
In an earlier study we showed by microarray analysis that mouse embryonic fibroblasts (MEFs) required each of the three IKK signalsome subunits (IKKα, IKKβ, or NEMO/IKKγ) for the global induction of NF-κB dependent genes in response to TNFα (Li et al., 2002); but we did not elaborate repressive effects on the expression profile of other cellular genes. Figure 1 shows twenty-five TNFα and NF-κB dependent repressed genes that are enriched in positive effectors of cell cycle progression. To simplify the presentation of the microarray screening data, the fractional gene expression values for repressed genes, (obtained in Wt. MEF (2T) vs. other cell comparisons or conditions), were converted into their corresponding negative integer fold change values (see Materials and Methods and results in Figure 1). Blocking canonical NF-κB activity by transducing Wt. MEFs with an IκBα super-repressor (IκBαSR) retrovirus prevented the TNFα induced repression effect, which also required each of the three major subunits of the NF-κB activating IKK signalsome (IKKβ, IKKα and NEMO/IKKγ). Interestingly, the majority of these genes are known targets of either the FoxM1 or E2F transcription factors (Bindra and Glazer, 2006; Costa, 2005; Ishida et al., 2001; Wang et al., 2005; Yang et al., 2007) and included essential cell cycle checkpoint regulators of the G1/S (Cdc6, p107, RFC4, RFC5, multiple MCM proteins, FoxM1, Ect2 and Pole2) and G2/M transitions (Cdc6, RFC4, FoxM1, INCEP proteins, Bub1, AurkB, Cdcd20, Kif20a, CcnA2 and Plk1), effectors of tumor suppression (Brca1, Brca2 and p107) and apoptosis (Casp8) (see Figure 1). Moreover, scanning 1000 bp upstream and 200 bp downstream of the transcription start sites of a selected subset of these repressed genes (including FoxM1, AurkB, Brca1, Brca2 and Bub1) revealed E2F DNA binding sites in their promoters (see Figure 1) but did not identify canonical NF-κB consensus binding sites. Using the duplicate microarray data in Figure 1 as a preliminary gene screening tool, for the remainder of this report we have gone on to explore if some of the same positive effectors of cell cycle progression are suppressed in the context of NF-κB activation alone and then to elaborate some of the resultant effects on cell cycle progression.
Sustained NF-κB activation is sufficient to repress the expression of a number of essential cell cycle effectors
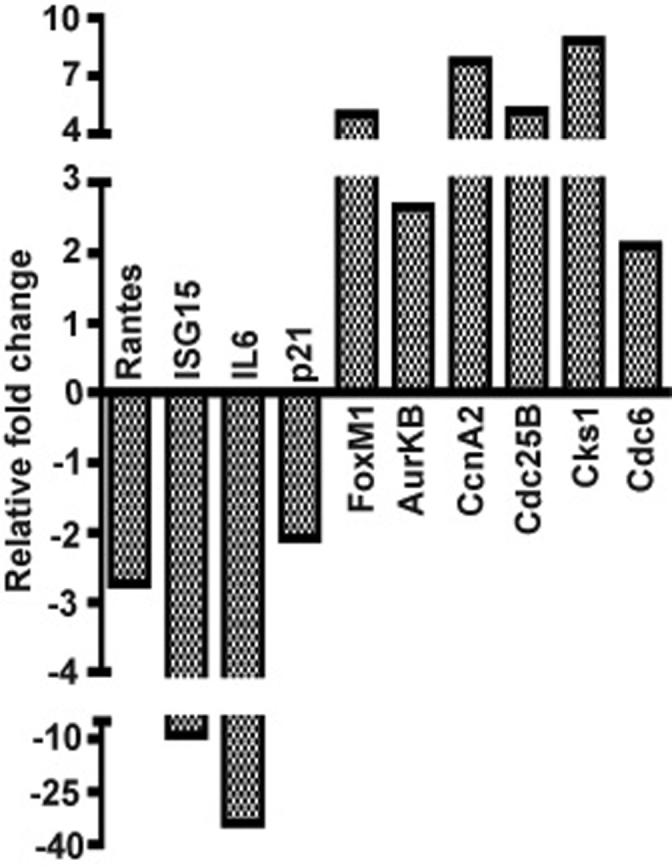
To determine whether NF-κB activation could alone repress the expression of the gene subset identified by the above microarray screens, we enforced the expression of a constitutively active T-loop mutant of IKKβ (IKKβca) in immortalized MEFs by retroviral transduction to selectively drive the activation of the canonical NF-κB pathway. A constitutively active mutant of IKKβ (IKKβca) was generated by site directed mutagenesis of serines 177 & 181 in its T activation loop to glutamic acids and was subcloned into moloney retroviral vectors co-expressing either a puromycin resistance gene (BIP) or GFP (MP9) (Li et al., 2001; Li et al., 2002; Palumbo et al., 2007; Zhang et al., 2005). Two days after infection with either the BIP empty vector or BIP co-expressing IKKβca populations of puromycin resistant cells were efficiently selected after 3 changes of growth media over the next 3-4 days. Little evidence of cell death occurred during this rapid course of puromycin selection, which produced puromycin resistant cells in 5 days post infection (5 dpi), indicative of a retroviral transduction efficiency of at least 90%. Similar high efficiency retrotransduction was also achieved with a retrovirus co-expressing IKKβca and GFP. Seven days post infection quantitative real time PCR assays were performed to assess the expression of twenty-one genes, {including 10 genes repressed in the TNFα/NF-κB microarray screens (FoxM1, AurKB, KIF20A, CcnA2, Plk1, Ect2, Cdc6, Casp8, Brca1 and Brca2), 2 additional well accepted direct targets of E2F transcription factors (CcnE2 and Ezh2), 3 other direct targets of FoxM1 (Cdc25B, Skp2 and Csk1) and six known direct targets of canonical NF-κB signaling (RANTES, IκBα, ISG15, IL-6, SAA3 and p21)}, (Costa, 2005; Hellin et al., 2000; Li et al., 2002; Pahl, 1999; Wang et al., 2005; Wuerzberger-Davis et al., 2005). Repression of the 15 above genes, (which aside from Brca2 have been described as downstream targets of either FoxM1 or E2F), was observed along with the simultaneous induction of each of the six NF-κB dependent targets in IKKβca cells in comparison to MEFs harboring an empty vector puromycin control virus (Figure 2). Similar experiments employing a GFP retrovirus co-expressing IKKβca also showed evidence of comparable gene suppression effects at earlier times of 2-5 days post infection (data not shown). To demonstrate that IKKβca caused this repression effect by canonical NF-κB activation and not by an NF-κB independent mechanism, the same experiment was carried out in IBIN-MEFs that constitutively express an IκBα super-repressor (SR), which is resistant to IKKβca activating phosphorylation. As shown in Figure 3, a comparison of 7 dpi IKKβca IBIN-MEFs vs. 7 dpi IKKβca MEFs shows that inhibition of canonical NF-κB by IκBαSR in the IBIN-MEFs abrogated both the IKKβca mediated induction of direct NF-κB targets and the repression effect on the FoxM1 and E2F targets in these cells.
Figure 2. Sustained canonical NF-κB activation induces a cell cycle effector suppression response in conjunction with the activation of known NF-κB targets.

The expression of twenty-one genes (six induced and 15 repressed) are examined by real time PCR in MEFs stably expressing a constitutively activated IKKβ mutant in a puromcyin resistant retroviral vector (IKKβca-BIP) vs. MEFs harboring a BIP empty retroviral vector. All RNAs were prepared from populations of MEFs 7 days post-infection (dpi) with either IKKβca-BIP or empty BIP. Fold change values for 7 dpi IKKβca-BIP vs. 7 dpi BIP MEFs were derived by the ΔΔCt method (with duplicate samples differing by no more than 0.5Ct) as described in Materials and Methods (Dussault and Pouliot, 2006). Relative fold change values were the averages of duplicate samples from one representative experiment out of two independent experiments producing similar results.
Figure 3. Gene repression induced by IKKβca requires canonical NF-κB signaling.
The canonical NF-κB requirement for gene repression by IKKβca were evaluated in a population of MEFs stably expressing an IκBα super-repressor (SR) (IBIN-MEFs). Fold change values of four induced and six repressed genes were determine by real time PCR on total cell RNAs prepared from 7 dpi IKKβca-BIP in IBIN-MEFs vs. 7 dpi IKKβca-BIP in wild type MEFs. Results were quantified as described in Materials and Methods and in Figure 2's legend and are averages of duplicate samples from one representative experiment out of two independent experiments producing similar results.
Canonical NF-κB activation causes a strong, short-term inhibition of cell proliferation linked to the concerted suppression of cell cycle progression factors
MEFs transduced with IKKβca virus began to display a cell proliferation block within two days post-infection as evidenced by only a very modest increase in cell number, while cells infected with an empty control vector continued to grow during the course of puromycin selection with an 18-24 hr doubling time. In Figure 4A, rates of cell proliferation were compared with high sensitivity by quantifying the amounts of cellular DNA accumulating as a function of time in days in cultures of control vs. IKKβca cells (500-1000 cells seeded on Day 0 per well in quadruplicate in 96 well plates). The rate of proliferation of IKKβca cells was severely compromised compared to cells infected with an empty vector control virus (Fig. 4A). In agreement with the results in Fig. 4A, other independent experiments in which 50-100,000 5 dpi IKKβca cells were seeded per well of six well plates showed little if any change in cell numbers over 2-3 days, while 5 dpi BIP infected control MEFs doubled each 18-24hr (data not shown). IKKβca's pronounced growth inhibitory effect was mediated by canonical NF-κB activation and not by other potential IKKβ targets, because MEFs expressing a phosphorylation resistant IκBα-SR mutant were refractory to the growth suppressive effects of enforced IKKβca expression (Fig. 4A). Comparative cell cycle analyses by FACS profiling of BIP empty vector vs. IKKβca-BIP cells at 7 dpi revealed that IKKβca cells at this early time point accumulate in G1 phase with a sharp reduction of cells in S phase in keeping with their proliferation block (Fig. 5). IKKβca cells generally had an abnormal, elongated morphology compared to their control empty BIP vector counterparts (Fig. 5). Similar experiments were performed with an IKKβca-IRES-GFP virus revealing comparable results (data not shown).
Figure 4. Chronic canonical NF-κB activation by IKKβca induces a pronounced, short-term cell proliferation block, whose relief correlates with the dissipation of cell cycle effector repression.

A: Comparisons of proliferation rates of 7 and 14 dpi IKKβca MEFs, 7 dpi IKKβca IBIN-MEFs and 7 dpi BIP control MEFs. Cells were seeded at low density (∼1000 cells per well) in quadruplicate in 96 well plates and the relative amounts of cell growth were determined by quantifying the increase in DNA (in ng) every 24 hr over a 5 day period as described in materials and methods. All DNA values are expressed as the mean ± s.e.m. Proliferation rates for 7 dpi IKKβca vs. 7 dpi BIP control cells were statistically significant on days 2, 3, 4 and 5 (p values of 0.009 and 0.005 on days 2-3 and 4-5 respectively) as were the results for 14 dpi IKKβca vs. 7 dpi BIP control cells on days 2, 3 and 5 (p values of 0.020). B: Sybr green real time PCR results for 4 induced and 8 repressed genes are shown as fold change values obtained with total cell RNAs prepared from 21 dpi IKKβca vs. 21 dpi BIP control MEFs. Results were quantified as described in Materials and Methods and in the above legends of Figures 2 and 3 and are averages of duplicate samples from one representative experiment out of two independent experiments producing similar results. C: Immunoblot analysis of nuclear extracts of control vs. IKKβca-BIP immortalized MEFs (5 dpi and 15 dpi) for Phospho-NF-κB p65 (Ser536), total p65 protein and lamin B as a protein loading reference control.
Figure 5. Enforced IKKβca expression in immortalized MEFs produces a G1/S cell cycle block during the 1st week of culture.

Flow cytometry was performed on propidium iodide (PI) stained cells to reveal their cell cycle distribution profiles (as described in Materials and Methods). Data was analyzed with FACSDiva software (Becton-Dickinson). The percentages of cells in G1, S and G2 phases are indicated. Results are representative of several independent experiments. Images of 7 dpi IKKβca-BIP and 7 dpi Wt. MEF-BIP control cells (200X magnification phase contrast microscopy) are shown next to each FACS profile.
The canonical NF-κB induced block of cellular growth in conjunction with the concerted suppression of cell cycle effectors was a transient phenomenon. IKKβca MEFs began to show significant improvement in their growth rate by ~10-12 days post-infection (see Figure 4A); and their proliferation rate began to parallel that of BIP control MEFs during the 3rd week post-infection (see growth curve in Figure 4A initiated with 14 dpi IKKβca MEFs). Multiple experiments consistently showed that the majority (not a minor subset) of the initial population of IKKβca cells improved their growth rate by ∼12 dpi. The increases in DNA quantities in Figure 4A visually corresponded to an increase in cell numbers over the 1-6 day time course with wells seeded with either control or 14 dpi IKKβca cells approaching confluence in 6-7 days. Moreover, to rule out the possibility that the initial reduced growth rate of IKKβca cells was due to non-apoptotic cell death, we assayed for lactate dehydrogenase (LDH) released by IKKβca and control cells. LDH, a stable cytoplasmic enzyme, is released by dying or damaged cells upon rupture of the plasma membrane and quantifying its release by cells is a standard method for assessing cell death and cytotoxicity (Decker and Lohmann-Matthes, 1988; Legrand C, 1992). Briefly a sensitive spectrophotometric based assay (Roche Inc.) was employed to quantify the levels of lactate dehydrogenase (LDH) released after 24 and 48 hrs by 6 dpi IKKβca-BIP and BIP empty vector control cells seeded in triplicate in six well plates. Because the IKKβca cells are growth arrested at 6 dpi, we seeded 7.5 × 105 and 1.5 × 105 control and IKKβca cells respectively. After 24 hrs IKKβca-BIP cells had slightly (∼1.2×) more LDH in their tissue culture media than BIP control cells, and after 48 hrs there was no apparent difference in LDH released between the two cell populations (data not shown). Thus growth curves, FACS analysis and LDH assays indicated that IKKβca cells initially fail to proliferate due to a transient cell cycle arrest, from which cells recover at a high frequency by ∼12 days post infection.
Importantly, in conjunction with the resumption in cellular proliferation the concerted repression of cell cycle progression factors in 21 dpi IKKβca MEFs was relieved albeit with some genes remaining modestly repressed, whilst direct canonical NF-κB targets (including IκBα, the archetypical canonical NF-κB target) remained activated (Figure 4B). Moreover the timing of alleviation of gene repression correlated very well with the onset of improved cell growth rate. These observations were consistently reproduced in multiple experiments with either puromycin or GFP co-expressing viruses. In addition canonical NF-κB activation by IKKβca was also visualized by the accumulation of activated phospho-NF-κB p65 (Ser536) subunit in cell nuclei in 5 dpi growth arrested cells and at a longer time point (15 dpi), when their cell proliferation rate had improved to nearly the control cell growth rate (Figure 4C). Although the fraction of p65 in the nucleus was predominantly its activated phospho-Ser536 form the total level of p65 subunit in the nuclei of IKKβca cells was similar to that observed in the control cells. Since IκBα expression was induced by IKKβca signaling this would establish a continuous cycle of breakdown and re-synthesis of IκBα resulting in cycles of NF-κB activation over real time (Hoffmann et al., 2002; Hoffmann et al., 2006; Werner et al., 2005). The latter intrinsic, cyclical characteristic of NF-κB activation could have contributed to establishing a more balanced level of p65 in the cytoplasmic and nuclear compartments over time but with a fraction of the nuclear NF-κB p65 remaining in a post-translationally modified, activated state. Moreover evidence has also been reported for an IKKβ signaling dependent pathway driving phospho-NF-κB p65 (Ser536) (Mattioli et al., 2004; Perkins, 2006) into the nucleus and other evidence for the signaling induced nuclear import of phospho-NF-κB p65 (Ser536) by an IκBα independent pathway has also been documented (Sasaki et al., 2005).
IKKβca experiments with primary MEFs (early passage 4) revealed a similar short-term growth inhibitory effect as observed above for immortalized MEFs. As shown in Figure 6A, during the initial 12-14 days of cell culture the proliferation rate of IKKβca expressing primary MEFs was greatly retarded compared to control primary MEFs harboring an empty BIP retroviral vector. Akin to immortalized MEFs, most 7 dpi IKKβca primary MEFs presented an abnormal, elongated cell morphology (Fig. 6A); and FACS analysis also revealed a G1 cell cycle arrest in IKKβca cells during their 1st week in culture (Fig. 6B) (similar to that observed with immortalized MEFs in Figure 5). LDH release assays revealed no difference between IKKβca 7 dpi primary MEFs and their controls (data not shown). In addition, IKKβca induced suppression of positive cell cycle effectors was also observed in the context of primary MEFs (data not shown). However since a similar transient IKKβca induced cell proliferation arrest was observed in immortalized (Fig. 4A and 5) and primary MEFs (Fig. 6), we undertook the molecular analysis of IKKβca's effects in immortalized MEFs. Moreover because the growth rate of primary MEFs reduced by 3rd weeks in culture as they approached replicative senescence, IKKβca's NF-κB dependency (as shown in Figure 3) and analysis of the temporal nature of its repressive effects on E2F and FoxM1 targets were more feasible to execute and less subject to other collateral growth associated gene expression effects in immortalized cells.
Figure 6. Enforced IKKβca expression in primary early passage MEFs produces a transient cell proliferation arrest in association with a G1/S phase cell cycle block during the 1st week of culture.

A: Wild type primary MEFs (passage 4) were stably infected with IKKβca-BIP or BIP empty control vector and maintained under puromycin selection for 5 days post infection. At 7 dpi cells were seeded at low density (∼1000 cells per well) in quadruplicate in 96 well plates and the relative amounts of cell growth were determined by quantifying the increase in DNA (in ng) every 24 hr over a 14 day period (equivalent to 8-21 dpi) as described in Materials and Methods (and also in Fig. 4A legend). All DNA values are expressed as the mean ± s.e.m. Phase contrast microscopy (20X magnification) images of 1° 7 dpi IKKβca-BIP and 1° BIP control MEFs are shown adjacent to the growth curves. B: Vector control and IKKβca 1°MEFs 7 dpi were stained with propidium iodide and submitted to flow cytometry to reveal their cell cycle distribution profiles (as described in Materials and Methods). Data was analyzed with FACSDiva software (Becton-Dickinson). The percentages of cells in G1, S and G2 phases are indicated. Results are representative of several independent experiments.
The IKKβca/NF-κB mediated cell proliferation arrest in immortalized MEFs is partially dependent on p21
Since evidence exists that the p21 cyclin dependent kinase inhibitor is a potential downstream target of NF-κB in several other cell types (Bash et al., 1997; Basile et al., 2003; Chang and Miyamoto, 2006; Hinata et al., 2003; Pennington et al., 2001; Wuerzberger-Davis et al., 2005) and because p21 RNA levels were increased in IKKβca cells (see Figure 2), we investigated the contribution of p21 to IKKβca induced growth suppression. As shown in Figure 7A, the highest level of p21 protein was found in 7 dpi IKKβca immortalized MEFs, whilst p21 protein was reduced by 21 dpi in the same cells and was negligibly expressed by 7 dpi IBIN-IKKβca cells. Next we stably expressed IKKβca in p21 null immortalized MEFs by retroviral transduction and their proliferation rates were assessed in Figure 7B. During their initial two weeks post infection IKKβca p21 KO MEFs had ∼50% of the proliferation rate of control p21 KO MEFs transduced with a empty BIP retroviral vector. However after 2 weeks post infection IKKβca p21 KO MEFs had attained the growth rate of the control cells (Figure 7B). The latter initial reduced growth rate of IKKβca p21 KO MEFs could be correlated with the repression of cell cycle effectors controlling the G1>S and G2>M transition in these cells (see Figure 7C). Interestingly IKKβca dependent induction of p21 mRNA levels was not the sole reason for the very poor growth rate of 7 dpi IKKβca vs. 7 dpi IKKβca p21 null MEFs. Surprisingly higher levels of p21 mRNA were observed in 21 dpi IKKβca cells, which had substantially recovered from growth suppression (compare real time PCR results for p21 in Figures 2 and 4B). As indicated above this enigma was resolved by western blot analysis, which revealed the highest levels of p21 protein in the growth inhibited 7 dpi IKKβca cells (Figure 7A). Taken together these observations suggest that the pronounced growth arrest phenotype of 7 dpi IKKβca Wt. MEFs was due at least in part to the enhanced accumulation of p21 protein by a combination of transcriptional and post-transcriptional mechanisms requiring sustained, canonical NF-κB activation. Moreover the post-transcriptional enhancing effect on p21 protein levels in 7 dpi IKKβca cells was well correlated with the repression of positive cell cycle effectors at this early time point (compare real time gene expression profiles of the repressed gene set in Figures 2 and 4B).
Figure 7. Short-term IKKβca induced cell proliferation arrest is partially p21 dependent.

A: Comparisons of p21 protein levels in 7 dpi IKKβca MEFS, 7 dpi IKKβca IBIN-MEFs, 21 dpi IKKβca MEFs and 7 dpi BIP control MEFs. Western blots of whole cell lysates derived from the indicated cell populations were probed with anti-p21 Ab and with an anti-tubulin Ab as a protein reference control. B: Cell proliferation rates over a 1-5 day time period of 7 and 14 dpi IKKβca p21 (−/−) MEFs are compared to p21 (−/−) control cells infected with an empty BIP vector 7 dpi. Cells were seeded in quadruplicate and DNAs were quantified and shown as the mean ± s.e.m. Results on days 3 and 5 of 7 dpi IKKβca p21 (−/−) vs. 7 dpi BIP p21 (−/−) control cells were statistically significant (p values of 0.009). C: Sybr green real time PCR results for 2 induced and 5 repressed genes are shown as fold change values obtained with total cell RNAs prepared from 10 dpi IKKβca p21 (−/−) vs. p21 (−/−) control MEFs. Results were quantified as described in Materials and Methods and in the legend for Figures 2 and are averages of duplicate samples from one representative experiment out of two independent experiments producing similar results.
Repressive effects of enforced IKKβca expression on E2F and FoxM1 target genes involves HDACs
Next we began to investigate aspects of how IKKβca mediated sustained NF-κB activation invoked the concerted, short-term repression of cell cycle progression factors. Because many of the repressed genes are direct targets of either E2F or FoxM1 and their NF-κB dependent repression subsided after 2-3 weeks, it seemed likely to us that this repression phenomenon was caused by epigenetic changes in chromatin structure. The ability of E2F transcription factors to repress their target genes is well known to involve their recruitment of transcriptional co-repressors of the Rb family, which act in part by targeting HDACs to E2F responsive gene promoters {reviewed in (Rowland and Bernards, 2006)}. Thus we explored if histone deacetylases (HDACs) were involved in the transient repressive effects of enforced IKKβca on E2F and FoxM1 target genes. Gene expression profiling by real time PCR revealed that 4 hrs of treatment with trichostatin A (TSA), an HDAC inhibitor, was sufficient to rescue the expression of the repressed genes but was without effect on the NF-κB induced genes (Figure 8A). In keeping with these results, TSA mediated HDAC inhibition also reversed TNFα induced, canonical NF-κB dependent gene repression, having essentially the same effect as blocking canonical NF-κB activation with an IκBαSR (Fig. 8B). Although the latter observations could not be correlated with effects on cellular growth, (due to the prolonged adverse effects of TSA treatment on cellular physiology), these results suggest that the recruitment of HDACs to E2F and FoxM1 target gene promoters may contribute to IKKβca's transient, repressive effects on their activity.
Figure 8. NF-κB mediated gene repression is an active mechanism involving histone deacetylases.
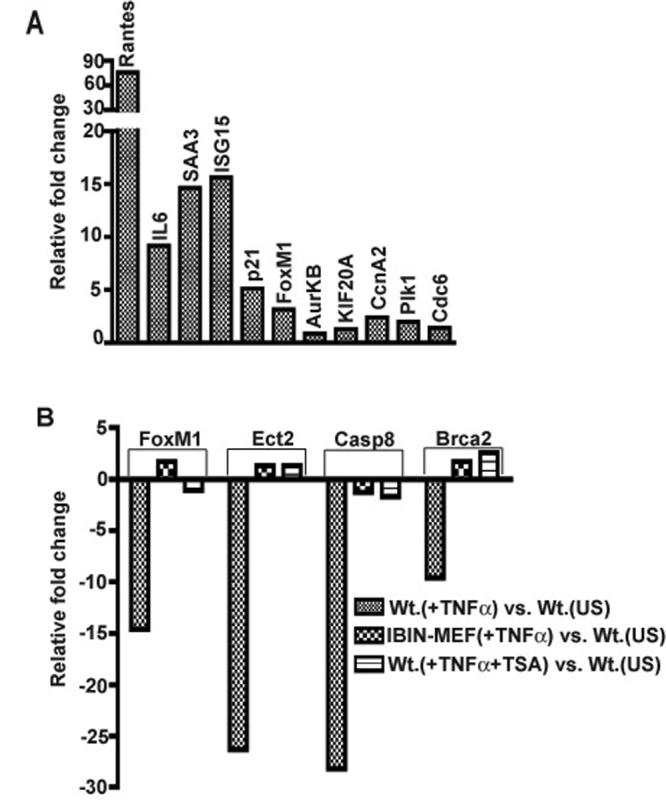
A: HDAC inhibition prevents IKKβca induced gene repression. 7 dpi IKKβca MEFs were treated with 100 nM trichostatin A (TSA) for 4 hours. The expressions of 5 induced and 6 repressed target gene are shown as fold change values in 7 dpi IKKβca MEFs +TSA vs. 7 dpi BIP control MEFs. B: HDAC inhibition or constitutive expression of IκBαSR blocks TNFα induced gene repression. Expression results for four repressed genes (FoxM1, Ect2, Casp8 and Brca2) that were identified by our microarray screens are shown as fold change values in Wt MEFs (+TNFα), IBIN-MEFs and Wt. MEFs (+TNFα & TSA) vs. Wt. unstimulated MEFs in each case as indicated. For the TSA experiment wild type MEFs were pretreated with 100 nM Trichostatin-A (TSA) for two hours followed by TNFα + TSA for an additional two hours. Results were quantified as described in Materials and Methods and also in Fig. 2 legend and are expressed as averages of duplicate samples from one representative experiment out of two independent experiments producing similar results.
DISCUSSION
Roles of NF-κB in cell proliferation: mediator of cell cycle progression or arrest?
Early work identified the cyclin D1 promoter as a direct target of canonical NF-κB in transient, co-transfection experiments (Hinz et al., 1999). Inhibition of basal canonical NF-κB activity in several cell types with an IκBα super repressor (IκBαSR) reduced cyclin D1 expression and associated Cdk4 activity, which also prevented cell cycle re-entry of G0 synchronized serum starved or quiescent serum deprived cells (Guttridge et al., 1999; Hinz et al., 1999). Moreover IκBαSR mediated inhibition of endogenous NF-κB also facilitated the differentiation of C2C12 myeloblast cells by reducing their proliferation and enhancing their cell cycle exit upon differentiation, which were also correlated with NF-κB dependent activation of cyclin D1 (Guttridge et al., 1999). Subsequent work showed that a complex of p52 homodimers and Bcl3 co-activator activate cyclin D1 transcription to facilitate Rb hyperphosphorylation and more rapid transit from G1 to S phase (Westerheide et al., 2001). In addition cyclin D1 was previously reported to be a downstream target of IKKα and RANKL dependent signaling (Cao et al., 2001); and an NF-κB independent requirement for IKKα in TCF mediated induction of cyclin D1 expression has also been noted (Albanese et al., 2003). However IKKα's role in maintaining cyclin D1 levels would appear to be a more complex phenomenon, because other experiments have surprisingly revealed elevated levels of nuclear cyclin D1 protein in IKKα compromised cells due to its IKKα mediated phosphorylation, nuclear export and subsequent proteolysis (Kwak et al., 2005). Other more recent work in either immortalized or transformed cellular contexts has convincingly shown that the IKKα and p52 dependent non-canonical NF-κB pathway is necessary for maintaining the cell cycle specific expression of important cell cycle progression factors (including cyclin D1, c-Myc and Skp2) (Barre and Perkins, 2007; Schneider et al., 2006).
DNA damage responses strongly activate canonical NF-κB via NEMO dependent IKKβ signaling to induce anti-apoptotic factors thereby giving cells time to repair their damaged DNA before an irreversible cell death response can become invoked (Janssens et al., 2005; Wu et al., 2005; Wu et al., 2006). Moreover studies in several cell types (including transformed T lymphocytes, monocytes, epithelial cells, and keratinocytes) have also shown that either genotoxic stress responses (Berchtold et al., 2007; Wuerzberger-Davis et al., 2005), enforced expression of canonical NF-κB subunits (Bash et al., 1997; Hinata et al., 2003; Seitz et al., 2000), or persistent activation of endogenous NF-κB (Basile et al., 2003; Pennington et al., 2001) can lead to increased p21 expression resulting in enhanced cellular survival (Gartel and Tyner, 2002) or inhibition of cell growth (Bash et al., 1997; Basile et al., 2003; Chang and Miyamoto, 2006; Hinata et al., 2003; Seitz et al., 2000; Wuerzberger-Davis et al., 2005). However, in contrast to the above reports two other studies in which IκBαSR was over-expressed in transformed clones of MEFs or epithelial cells showed elevated p21 levels in association with retarded cell growth due to a partial G1 arrest (Kaltschmidt et al., 1999 ; Sakaida et al., 2003). Taken collectively these prior studies have indicated that NF-κB can have either a positive or negative impact on cell cycle progression depending on the NF-κB subunit, the presence or absence of a specific activating stimulus, and the cellular physiological context.
In an earlier study we employed DNA microarrays to analyze the global response to TNFα, a potent activator of canonical NF-κB signaling, to reveal that each IKK signalsome subunit was required for the induction of NF-κB target genes (Li et al., 2002); and these screens also uncovered a repressed subset of genes whose identification form the basis of this report. Unlike the induced class of genes, which were mostly direct targets of NF-κB associated with stress-like inflammatory responses, transcriptional repression in this context appears to be an indirect, concerted phenomenon effecting the expression of target genes of either the E2F or FoxM1 transcription factors (Costa, 2005; Ishida et al., 2001; Laoukili et al., 2005; Wang et al., 2005). Here we show that IKKβca mediated sustained NF-κB activation in MEFs causes a short-term cell proliferation block, which requires p21 along with the simultaneous, concerted repression of positive cell cycle effectors regulated by E2F and FoxM1.
Constitutive NF-κB activation causes a short-term cell growth arrest by transcriptional and post-transcriptional mechanisms
To determine if canonical NF-κB activation was sufficient to repress critical FoxM1 and E2F regulated cell cycle progression factors and to determine the immediate and long term consequences for cell growth, MEFs were stably transduced with a constitutively active mutant of IKKβ (IKKβca). Enforced IKKβca expression simultaneously induced NF-κB targets while repressing effectors of cell cycle progression, as we observed in the NF-κB dependent response to TNFα stimulation. Moreover the specific sustained activation of canonical NF-κB signaling by IKKβca in wild type MEFs lead to a strong but short-term cell proliferation block lasting up to 2 weeks. Enforced IKKβca expression transiently arrested the proliferation of either immortalized or early passage primary MEFs with similar kinetics in association with a G1/S phase cell cycle block (see Figs. 4-6). Interestingly the NF-κB dependent suppression of cell cycle effectors was well correlated with the growth rate of established MEFs, because cell cycle effectors became de-repressed within 3 weeks of IKKβca retroviral transducton in conjunction with the resumption in their proliferation.
Growth suppression of immortalized IKKβca MEFs during their first 1-2 weeks of culture was associated with the enhanced accumulation of p21 protein due to its NF-κB dependent transcriptional induction and post-transcriptional stabilization. Our data point to the strong repression of Skp2 and Csk1 in 7 dpi IKKβca MEFs as a likely reason for the high levels of p21 protein in these growth arrested cells (Figure 2). Skp2 and Csk1 are direct targets of FoxM1 and essential subunits of the Skp1-Cullin 1-F-box (SCF) ubiquitin ligase complex that targets p21 for degradation between the G1 and S phases of the cell cycle (Wang et al., 2005). By 2-3 weeks of culture even though IKKβca induced p21 transcription remained quite high, p21 protein levels had significantly abated (see Figures 4B and 7A respectively). The latter reduction in p21 protein could at least in part be explained by the substantial recovery of Csk1 expression in 21 dpi IKKβca MEFs (Figure 4B). Cell proliferation experiments with immortalized p21 null MEFs confirmed that a portion (but not all) of the pronounced growth suppression of 7 dpi IKKβca MEFs was most likely mediated by the super-induction of p21 protein. Although much improved over their Wt. IKKβca counterparts the still reduced growth rate of 1-2 week IKKβca expressing p21 null MEFs was correlated with the repression of FoxM1 or E2F targets, which are necessary for the timely transit of cells from G1 to S (Cdc6 and Cdc25B) and G2 to M (Ccna2, AurkB and Kif20a) (Figure 7C).
Growth and cell cycle effector gene suppressive effects of sustained NF-κB activation are transient and appear to involve chromatin silencing
Cell cycle effector expression substantially recovered in 21 dpi IKKβca cells, even though the expression of direct NF-κB targets (including p21) remained high (see Figure 4B). We believe that the recovery of cell cycle effector expression probably occurs due to the strong selective pressure for cells to find a means of escaping from the severe growth suppression instigated by IKKβca in conjunction with sustained canonical NF-κB activation. The short-term gene repression effect could involve transcriptional interference or cross-talk between IKKβca and NF-κB and other transcription factors at the level of chromatin modification. In this regard, it has previously been documented that activated NF-κB can interfere with the transcriptional activities of p53 and c-Myb at the level of limiting transcriptional co-activators (Nicot et al., 2001; Wadgaonkar et al., 1999; Webster and Perkins, 1999) and perhaps E2F and FoxM1 target genes are also somewhat subject to such a phenomenon, because the same limiting p300/CBP co-activators are necessary for their activity and ability to activate the transcription of their target genes (Bandyopadhyay et al., 2002; Major et al., 2004; Martinez-Balbas et al., 2000; Morris et al., 2000; Taubert et al., 2004).
We hypothesize that IKKβca's transient cell growth and gene repression effects could involve alterations in the activities of either transcriptional co-activators or co-repressors recruited to the promoters of cell cycle effectors that are subject to regulation by either E2F or FoxM1 transcription factors. In support of this notion, gene repression in 7 dpi IKKβca MEFs was relieved by inhibiting histone deacetylases (Fig. 8A), in agreement with similar results obtained in the context of TNFα induced, NF-κB dependent gene repression (Figure 8B). Moreover, evidence has accumulated pointing to E2Fs predominantly acting as repressors (instead of activators) of cell cycle progression in asynchronously growing cells (Krek et al., 1995; Rowland and Bernards, 2006; Rowland et al., 2002; Zhang HS, 1999) and sustained NF-κB activation mediated by IKKβca could conceivably impact on degree of E2F mediated gene repression. Although our observations are suggestive of an epigenetic alteration in the chromatin activity status of batteries of E2F and FoxM1 target genes the detailed molecular basis for the initiation and subsequent relief of this effect after prolonged IKKβca exposure will be the focus of future work.
The same IKKβca mutant employed in our study produced physiologically relevant results in other in vitro and in vivo experiments, wherein it has been shown to specifically activate targets of the canonical NF-κB pathway (Denk et al., 2001; Sasaki et al., 2006). IKKβca expression in the T lymphoid lineage allowed for survival of CD4+/CD8+ double positive cells in the absence of T cell receptor formation which is important for NF-κB signaling for the survival of differentiating T cells (Voll et al., 2000). In addition targeted expression of IKKβca in the B lymphoid cell compartment did not elicit an abnormal cell proliferation response but instead allowed for the enhanced survival of resting mature B cells (Sasaki et al., 2006), indicating that the long term dominant effect of chronically activated canonical NF-κB does not necessarily lead to enhanced cell cycling. However, neither of these prior studies investigated the immediate vs. long term effects of IKKβca mediated chronic canonical NF-κB activation on cellular physiology, which we have addressed here.
In contrast to our observations herein, two prior studies in fibroblastic cells either showed no evidence of NF-κB dependent p21 induction or increased p21 expression upon inhibition of canonical NF-κB signaling respectively (Hinata et al., 2003; Sakaida et al., 2003). The contrasting outcomes of these earlier studies compared to ours could be due to differences in experimental conditions and/or the physiological behavior of the cells under study. Sakaida et al. observed that inhibiting endogenous NF-κB activity in Ras transformed NIH3T3 by over-expression of an IκBαSR or a dominant negative mutant of IKKβ resulted in enhanced p21 expression, which may have been caused by elevated levels of p53 (Sakaida et al., 2003). Hinata and colleagues reported that over-expression of NF-κB subunits by retroviral transduction induced p21 expression in primary human kerotincytes but not in dermal fibroblasts (Hinata et al., 2003). However, in addition to enhancing p21 expression, direct NF-κB targets (including Rantes, IL1RA, IL-1a) were also elevated by over-expression of NF-κB subunits in keratinocytes that were unaltered in fibroblasts, suggesting that NF-κB subunit over-expression without an NF-κB activating signal was insufficient to induce the same spectrum of NF-κB targets in these two cell types (Hinata et al., 2003). It is important to consider in this context that depending on their states of post-translational modification canonical NF-κB subunits have been found to act in either activating or repressing modes, thereby potentially provoking other complicating collateral effects in different cellular contexts (Ashburner et al., 2001; Campbell et al., 2004; Perkins, 2006; Zhong et al., 2002). To examine the short-term effects of sustained NF-κB activation in fibroblasts, we induced a persistent state of canonical NF-κB activation with a constitutively active mutant of IKKβ by efficient retroviral transduction. Importantly IKKβ(ca) not only drives canonical NF-κB subunits into the nucleus but by site specific phosphorylations can also contribute to the activation of their transcriptional competency{reviewed in (Perkins, 2006)},{exemplified in Figure 4C by the accumulation of phospho-NF-κB p65 (Ser536) in the nuclei of IKKβca cells}, which could directly or indirectly influence the activation status of NF-κB targets or other batteries of genes that cross-talk with activated, nuclear NF-κB p65.
Taken together our data reveal that the initial effects of sustained NF-κB activation can result in the repression of genes encoding effectors of cell cycle progression, which contributes to a short-term (1-2 week) inhibition of cellular growth. Importantly due to the nature of this short-term growth suppressive response, our results clearly show that this effect would not have been observed with other cell culture selection protocols in which cell populations harboring constitutively activated IKKβca/NF-κB are maintained for 2 weeks or more prior to their physiological or molecular analysis. Gene repression mediated by activated NF-κB under our experimental conditions largely appears to be a concerted phenomenon, because it negatively impacts on many targets of either the E2F or FoxM1 transcription factors. Because the downstream targets of either E2F or FoxM1 respectively include the Brca1 and Brca2 tumor suppressors, effectors of the homologous recombination DNA repair pathway {reviewed in (Yoshida and Miki, 2004)}, DNA repair in this state of cell cycle arrest could be shifted towards other pathways including the non-homologous end joining (NHEJ) route. We envision that this unanticipated short-term growth suppressive effect of persistent NF-κB activation could become important in the context of cellular responses to genotoxic insults, which are known to strongly activate canonical NF-κB signaling, thereby giving cells the time to attempt to repair their damaged DNA. In addition, because the transient growth suppression effects of IKKβca were observed in both immortalized and primary MEFs, wild type functionality of the p53 axis would not appear to be essential for IKKβca induced growth arrest (given that immortalization of MEFs by the 3T3 protocol commonly selects for cells with a compromised p53 activity) (Harvey and Levine, 1991). Similar persistent states of NF-κB activation in either normal senescence prone or immortalized cells in response to DNA damage provoked by the activation of specific transforming oncoproteins could potentially contribute to NF-κB's role as a tumor promoter.
ACKNOWLEDGEMENTS
We thank Drs Michael Karin for immortalized IKKα (−/−), IKKβ (−/−), and NEMO/IKKγ (−/−) MEFs and Dr. Luisa LanFrancone for immortalized p21 (−/−) MEFs. We also gratefully acknowledge Dr. Vicenzo Cerreta (CRBA) for performing LDH assays and Dr. Evangelos Kolettas (University of Ioannina Medical School, Ioannina Greece) for critical reading and suggestions on the manuscript. This work was supported in part by USA NIH grant GM-066882 (awarded to KBM), the CRBA laboratory, the Institute of Advanced Studies of the University of Bologna, the Rizzoli Research Institute, the Cassa di Risparmo di Bologna and the MAIN EU FP6 Network of Excellence. PEM and KBM were junior and senior scholars respectively of the Institute of Advanced Studies during part of these studies.
REFERENCES
- Albanese C, Wu K, D'Amico M, Jarrett C, Joyce D, Hughes J, Hulit J, Sakamaki T, Fu M, Ben-Ze'ev A, Bromberg JF, Lamberti C, Verma U, Gaynor RB, Byers SW, Pestell RG. IKKalpha Regulates Mitogenic Signaling through Transcriptional Induction of Cyclin D1 via Tcf. Mol Biol Cell. 2003;14(2):585–599. doi: 10.1091/mbc.02-06-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Haecker H, Karin M, Ciechanover A. Mechanism of processing of the NF-kappa B2 p100 precursor: identification of the specific polyubiquitin chain-anchoring lysine residue and analysis of the role of NEDD8-modification on the SCF(beta-TrCP) ubiquitin ligase. Oncogene. 2004;23(14):2540–2547. doi: 10.1038/sj.onc.1207366. [DOI] [PubMed] [Google Scholar]
- Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21(20):7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin A., Jr The NF-kappaB and IkappaB proteins: new discoveries and insights. Annu Rev Immunol. 1996;14:649–683. doi: 10.1146/annurev.immunol.14.1.649. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay D, Okan NA, Bales E, Nascimento L, Cole PA, Medrano EE. Down-Regulation of p300/CBP Histone Acetyltransferase Activates a Senescence Checkpoint in Human Melanocytes. Cancer Res. 2002;62(21):6231–6239. [PubMed] [Google Scholar]
- Barre B, Perkins ND. A cell cycle regulatory network controlling NF-kappaB subunit activity and function. EMBO J. 2007;26:4841–4855. doi: 10.1038/sj.emboj.7601899. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Basak S, Kim H, Kearns JD, Tergaonkar V, O'Dea B, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A. A Fourth IκB protein within the NF-κB Signaling Module. Cell. 2007;128:369–381. doi: 10.1016/j.cell.2006.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bash J, Zong WX, Gelinas C. c-Rel arrests the proliferation of HeLa cells and affects critical regulators of the G1/S-phase transition. Mol Cell Biol. 1997;17(11):6526–6536. doi: 10.1128/mcb.17.11.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basile JR, Eichten A, Zacny V, Munger K. NF-{kappa}B-Mediated Induction of p21Cip1/Waf1 by Tumor Necrosis Factor {alpha} Induces Growth Arrest and Cytoprotection in Normal Human Keratinocytes. Mol Cancer Res. 2003;1(4):262–270. [PubMed] [Google Scholar]
- Berchtold CM, Wu Z, Huang TT, Miyamoto S. Calcium-dependent regulation of NEMO nuclear export in response to genotoxic stimuli. Mol Cell Biol. 2007;27:497–509. doi: 10.1128/MCB.01772-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindra RS, Glazer PM. Basal expression of Brca1 by multiple E2Fs and pocket proteins at adjacent E2F sites. Cancer Biology and Therapy. 2006;5:1400–1407. doi: 10.4161/cbt.5.10.3454. [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Karin M. The two NF-kappaB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25(6):280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Rocha S, Perkins ND. Active repression of antiapoptotic gene expression by RelA(p65) NF-kappa B. Mol Cell. 2004;13(6):853–865. doi: 10.1016/s1097-2765(04)00131-5. [DOI] [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M. IKKalpha Provides an Essential Link between RANK Signaling and Cyclin D1 Expression during Mammary Gland Development. Cell. 2001;107(6):763–775. doi: 10.1016/s0092-8674(01)00599-2. [DOI] [PubMed] [Google Scholar]
- Chang PY, Miyamoto S. Nuclear factor-kappaB dimer exchange promotes a p21(waf1/cip1) superinduction response in human T leukemic cells. Mol Cancer Res. 2006;4(2):101–112. doi: 10.1158/1541-7786.MCR-05-0259. [DOI] [PubMed] [Google Scholar]
- Costa RH. FoxM1 dances with mitosis. Nat Cell Biol. 2005;7(2):108–110. doi: 10.1038/ncb0205-108. [DOI] [PubMed] [Google Scholar]
- Decker T, Lohmann-Matthes ML. A quick and simple method for the quantification of lactate dehydrogenase release in measurements of cellular cytotoxicity and tumor necrosis factor (TNF) activity. J Immunol Methods. 1988;15:61–69. doi: 10.1016/0022-1759(88)90310-9. [DOI] [PubMed] [Google Scholar]
- Denk A, Goebeler M, Schmid S, Berberich I, Ritz O, Lindemann D, Ludwig S, Wirth T. Activation of NF-kappa B via the Ikappa B kinase complex is both essential and sufficient for proinflammatory gene expression in primary endothelial cells. J Biol Chem. 2001;276(30):28451–28458. doi: 10.1074/jbc.M102698200. [DOI] [PubMed] [Google Scholar]
- Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 Dimers Are Differentially Regulated by Tumor Necrosis Factor-{alpha} and Lymphotoxin-{beta} Receptor Activation: Critical roles for p100. J Biol Chem. 2003;278(26):23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- Dussault A-A, Pouliot M. Rapid and simple comparison of messenger RNA levels using real-time PCR. Biol Proced Online. 2006;8:1–10. doi: 10.1251/bpo114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22(2):245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- Facchini A, Borzi RM, Marcu KB, Stefanelli C, Olivotto E, Goldring MB, Facchini A, Flamigni F. Polyamine depletion inhibits NF-kappaB binding to DNA and interleukin-8 production in human chondrocytes stimulated by tumor necrosis factor-alpha. J Cell Physiol. 2005;204(3):956–963. doi: 10.1002/jcp.20368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. The Role of the Cyclin-dependent Kinase Inhibitor p21 in Apoptosis. Mol Cancer Ther. 2002;1(8):639–649. [PubMed] [Google Scholar]
- Ghosh S, Karin M. Missing pieces in the NF-kappaB puzzle. Cell. 2002;109:S81–S96. doi: 10.1016/s0092-8674(02)00703-1. [DOI] [PubMed] [Google Scholar]
- Ghosh S, May MJ, Kopp EB. NF-kappaB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu Rev Immunol. 1998;16:225–260. doi: 10.1146/annurev.immunol.16.1.225. [DOI] [PubMed] [Google Scholar]
- Grundstrom S, Anderson P, Scheipers P, Sundstedt A. Bcl-3 and NF-kappaB p50-p50 homodimers act as transcriptional repressors in tolerant CD4+ T cells. J Biol Chem. 2004;279(9):8460–8468. doi: 10.1074/jbc.M312398200. [DOI] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19(8):5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansberger MW, Campbell JA, Danthi P, Arrate P, Pennington KN, Marcu KB, Ballard DW, Dermody TS. IkappaB kinase subunits alpha and gamma are required for activation of NF-kappaB and induction of apoptosis by mammalian reovirus. J Virol. 2007;81(3):1360–1371. doi: 10.1128/JVI.01860-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor Laboratory Press; 1988. pp. 483–509. [Google Scholar]
- Harvey DM, Levine AJ. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev. 1991;5(12b):2375–2385. doi: 10.1101/gad.5.12b.2375. [DOI] [PubMed] [Google Scholar]
- Hellin A-C, Bentires-Alj M, Verlaet M, Benoit V, Gielen J, Bours V, Merville M-P. Roles of Nuclear Factor-kappa B, p53, and p21/WAF1 in Daunomycin-Induced Cell Cycle Arrest and Apoptosis. J Pharmacol Exp Ther. 2000;295(3):870–878. [PubMed] [Google Scholar]
- Hinata K, Gervin AM, Jennifer Zhang Y, Khavari PA. Divergent gene regulation and growth effects by NF-[kappa]B in epithelial and mesenchymal cells of human skin. Oncogene. 2003;22(13):1955–1964. doi: 10.1038/sj.onc.1206198. [DOI] [PubMed] [Google Scholar]
- Hinz M, Krappmann D, Eichten A, Heder A, Scheidereit C, Strauss M. NF-kappaB function in growth control: regulation of cyclin D1 expression and G0/G1-to-S-phase transition. Mol Cell Biol. 1999;19(4):2690–2698. doi: 10.1128/mcb.19.4.2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg JE, Popko AE, Ramsey CS, Mayo MW. I{kappa}B Kinase {alpha}-Mediated Derepression of SMRT Potentiates Acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26(2):457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoberg JE, Yeung F, Mayo MW. SMRT derepression by the IkappaB kinase alpha: a prerequisite to NF-kappaB transcription and survival. Mol Cell. 2004;16(2):245–255. doi: 10.1016/j.molcel.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D. The Ikappa B-NF-kappa B Signaling Module: Temporal Control and Selective Gene Activation. Science. 2002;298(5596):1241–1245. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Natoli G, Ghosh G. Transcriptional regulation via the NF-kappaB signaling module. Oncogene. 2006;25(51):6706–6716. doi: 10.1038/sj.onc.1209933. [DOI] [PubMed] [Google Scholar]
- Huang WC, Ju TK, Hung MC, Chen CC. Phosphorylation of CBP by IKKalpha promotes cell growth by switching the binding preference of CBP from p53 to NF-kappaB. Mol Cell. 2007;26(1):75–87. doi: 10.1016/j.molcel.2007.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida S, Huang E, Zuzan H, Spang R, Leone G, West M, Nevins JR. Role for E2F in control of both DNA replication and mitotic functions as revealed from DNA microarray analysis. Mol Cell Biol. 2001;21(14):4684–4699. doi: 10.1128/MCB.21.14.4684-4699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssens S, Tinel A, Lippens S, Tschopp J. PIDD mediates NF-kappaB activation in response to DNA damage. Cell. 2005;123(6):1079–1092. doi: 10.1016/j.cell.2005.09.036. [DOI] [PubMed] [Google Scholar]
- Jiang X, Takahashi N, Matsui N, Tetsuka T, Okamoto T. The NF-kappa B activation in lymphotoxin beta receptor signaling depends on the phosphorylation of p65 at serine 536. J Biol Chem. 2003;278(2):919–926. doi: 10.1074/jbc.M208696200. [DOI] [PubMed] [Google Scholar]
- Kaltschmidt B, Kaltschmidt C, Hehner SP, Droge W, Schmitz ML. Repression of NF-kappaB impairs HeLa cell proliferation by functional interference with cell cycle checkpoint regulators. Oncogene. 1999;18(21):3213–3225. doi: 10.1038/sj.onc.1202657. [DOI] [PubMed] [Google Scholar]
- Karin M. How NF-kappaB is activated: the role of the IkappaB kinase (IKK) complex. Oncogene. 1999;18:6867–6874. doi: 10.1038/sj.onc.1203219. [DOI] [PubMed] [Google Scholar]
- Karin M, Greten FR. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol. 2005;5(10):749–759. doi: 10.1038/nri1703. [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3(3):221–227. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- Krek W, Xu G, Livingston DM. Cyclin A-kinase regulation of E2F-1 DNA binding function underlies suppression of an S phase checkpoint. Cell. 1995;83:1149–1158. doi: 10.1016/0092-8674(95)90141-8. [DOI] [PubMed] [Google Scholar]
- Kwak Y-T, Li R, Becerra CR, Tripathy D, Frenkel EP, Verma UN. I{kappa}B Kinase {alpha} Regulates Subcellular Distribution and Turnover of Cyclin D1 by Phosphorylation. J Biol Chem. 2005;280(40):33945–33952. doi: 10.1074/jbc.M506206200. [DOI] [PubMed] [Google Scholar]
- Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7(2):126–136. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- Legrand CBJ, Jacob C, Capiaumont J, Martial A, Marc A, Wudtke M, Kretzmer G, Demangel C, Duval D, et al. Lactate dehydrogenase (LDH) activity of the number of dead celsl in the medium of cultured enukaryotic cells as marker. J Biotechnol. 1992;25:231–243. doi: 10.1016/0168-1656(92)90158-6. [DOI] [PubMed] [Google Scholar]
- Lenhard B, Wasserman WW. TFBS: Computational framework for transcription factor binding site analysis. Bioinformatics. 2002;18(8):1135–1136. doi: 10.1093/bioinformatics/18.8.1135. [DOI] [PubMed] [Google Scholar]
- Li J, Peet GW, Balzarano D, Li X, Massa P, Barton RW, Marcu KB. Novel NEMO/IκB kinase and NF-κB target genes at the pre-B to immature B cell transition. J Biol Chem. 2001;276:18579–18590. doi: 10.1074/jbc.M100846200. [DOI] [PubMed] [Google Scholar]
- Li X, Massa PE, Hanidu A, Peet GW, Aro P, Savitt A, Mische S, Li J, Marcu KB. IKKα, IKKβ, and NEMO/IKKγ are each required for the NF-κB-mediated Inflammatory response program. J Biol Chem. 2002;277(47):45129–45140. doi: 10.1074/jbc.M205165200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major ML, Lepe R, Costa RH. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol Cell Biol. 2004;24(7):2649–2661. doi: 10.1128/MCB.24.7.2649-2661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Balbas MM, Bauer U-M, Nielsen sJ, Brehm A, Kouzarides T. Regulation of E2F1 activity by acetylation. Embo J. 2000;19:662–671. doi: 10.1093/emboj/19.4.662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa PE, Li X, Hanidu A, Siamas J, Pariali M, Pareja J, Savitt AG, Catron KM, Li J, Marcu KB. Gene expression profiling in conjunction with physiological rescues of IKKα-null cells with wild type or mutant IKKα reveals distinct classes of IKKα/NF-κB-dependent genes. J Biol Chem. 2005;280(14):14057–14069. doi: 10.1074/jbc.M414401200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattioli I, Sebald A, Bucher C, Charles RP, Nakano H, Doi T, Kracht M, Schmitz ML. Transient and Selective NF-κB p65 Serine 536 Phosphorylation Induced by T Cell Costimulation Is Mediated by IκB Kinase {beta} and Controls the Kinetics of p65 Nuclear Import. J Immunol. 2004;172(10):6336–6344. doi: 10.4049/jimmunol.172.10.6336. [DOI] [PubMed] [Google Scholar]
- May MJ, Ghosh S. Signal transduction through NF-kappa B. Immunol Today. 1998;19(2):80–88. doi: 10.1016/s0167-5699(97)01197-3. [DOI] [PubMed] [Google Scholar]
- May MJ, Ghosh S. IkappaB kinases: kinsmen with different crafts. Science. 1999;284:271–273. doi: 10.1126/science.284.5412.271. [DOI] [PubMed] [Google Scholar]
- Morris L, Allen KE, La Thangue NB. Regulation of E2F transcription by cyclin E-Cdk2 kinase mediated through p300/CBP co-activators. Nat Cell Biol. 2000;2(4):232–239. doi: 10.1038/35008660. [DOI] [PubMed] [Google Scholar]
- Nicot C, Mahieux R, Pise-Masison C, Brady J, Gessain A, Yamaoka S, Franchini G. Human T-cell lymphotropic virus type 1 Tax represses c-Myb-dependent transcription through activation of the NF-kappaB pathway and modulation of coactivator usage. Mol Cell Biol. 2001;21(21):7391–7402. doi: 10.1128/MCB.21.21.7391-7402.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6853–6866. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- Palumbo R, Galvez BG, Pusterla T, De Marchis F, Cossu G, Marcu KB, Bianchi ME. Cells migrating to sites of tissue damage in response to the danger signal HMGB1 require NF-{kappa}B activation. J Cell Biol. 2007;179(1):33–40. doi: 10.1083/jcb.200704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, McEver RP. Regulation of the human P-selectin promoter by Bcl-3 and specific homodimeric members of the NF-kappa B/Rel family. J Biol Chem. 1995;270(39):23077–23083. doi: 10.1074/jbc.270.39.23077. [DOI] [PubMed] [Google Scholar]
- Pennington KN, Taylor JA, Bren GD, Paya CV. IκB Kinase-Dependent Chronic Activation of NF-{kappa}B Is Necessary for p21WAF1/Cip1 Inhibition of Differentiation-Induced Apoptosis of Monocytes. Mol Cell Biol. 2001;21(6):1930–1941. doi: 10.1128/MCB.21.6.1930-1941.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25(51):6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49–62. doi: 10.1038/nrm2083. [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Baltimore D. Two pathways to NF-kappaB. Mol Cell. 2002;10:693–694. doi: 10.1016/s1097-2765(02)00697-4. [DOI] [PubMed] [Google Scholar]
- Poyet JL, Srinivasula SM, Lin JH, Fernandes-Alnemri T, Yamaoka S, Tsichlis PN, Alnemri ES. Activation of the Ikappa B kinases by RIP via IKKgamma /NEMO-mediated oligomerization. J Biol Chem. 2000;275(48):37966–37977. doi: 10.1074/jbc.M006643200. [DOI] [PubMed] [Google Scholar]
- Rowland BD, Bernards R. Re-evaluating cell-cycle regulation by E2Fs. Cell. 2006;127:871–874. doi: 10.1016/j.cell.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Rowland BD, Denissov SG, Douma S, Stunnenberg HG, Bernards R, Peeper DS. E2F transcriptional repressor complexes are critical downstream targets of p19ARF/p53-induced proliferative arrest. Cancer Cell. 2002;2(1):55–65. doi: 10.1016/s1535-6108(02)00085-5. [DOI] [PubMed] [Google Scholar]
- Sakaida T, Iwadate Y, Yamaji S, Uehara M, Noda A, Takiguchi M, Yamaura A, Hiwasa T. Acquisition of drug resistance by constitutive suppression of NF-kappaB in ras-transformed NIH3T3 mouse fibroblasts. Int J Oncol. 2003;23:1071–1077. [PubMed] [Google Scholar]
- Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki CY, Barberi TJ, Ghosh P, Longo DL. Phosphorylation of RelA/p65 on Serine 536 Defines an I{kappa}B{alpha}-independent NF-{kappa}B Pathway. J Biol Chem. 2005;280(41):34538–34547. doi: 10.1074/jbc.M504943200. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Derudder E, Hobelka E, Pelanda R, Reth M, Rajewsky K, Schmidt-Supprian M. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity. 2006;24:729–739. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Schneider G, Saur D, Siveke JT, Fritsch R, Greten FR, Schmid RM. IKKalpha controls p52/RelB at the skp2 gene promoter to regulate G1- to S-phase progression. Embo J. 2006;25(16):3801–3812. doi: 10.1038/sj.emboj.7601259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumm K, Rocha S, Caamano J, Perkins ND. Regulation of p53 tumour suppressor target gene expression by the p52 NF-kappaB subunit. Embo J. 2006;25(20):4820–4832. doi: 10.1038/sj.emboj.7601343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz CS, Deng H, Hinata K, Lin Q, Khavari PA. Nuclear factor kappaB subunits induce epithelial cell growth arrest. Cancer Res. 2000;60(15):4085–4092. [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science. 2001;293(5534):1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Sizemore N, Lerner N, Dombrowski N, Sakurai H, Stark GR. Distinct roles of the Ikappa B kinase alpha and beta subunits in liberating nuclear factor kappa B (NF-kappa B) from Ikappa B and in phosphorylating the p65 subunit of NF-kappa B. J Biol Chem. 2002;277(6):3863–3869. doi: 10.1074/jbc.M110572200. [DOI] [PubMed] [Google Scholar]
- Taubert S, Gorrini C, Frank SR, Parisi T, Fuchs M, Chan H-M, Livingston DM, Amati B. E2F-Dependent Histone Acetylation and Recruitment of the Tip60 Acetyltransferase Complex to Chromatin in Late G1. Mol Cell Biol. 2004;24(10):4546–4556. doi: 10.1128/MCB.24.10.4546-4556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegethoff S, Behlke J, Scheidereit C. Tetrameric Oligomerization of I{kappa}B Kinase {gamma} (IKK{gamma}) Is Obligatory for IKK Complex Activity and NF-{kappa}B Activation. Mol Cell Biol. 2003;23(6):2029–2041. doi: 10.1128/MCB.23.6.2029-2041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voll RE, Jimi E, Phillips RJ, Barber DF, Rincon M, Hayday AC, Flavell RA, Ghosh S. NF-κB activation by the pre-T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity. 2000;13(5):677–689. doi: 10.1016/s1074-7613(00)00067-4. [DOI] [PubMed] [Google Scholar]
- Wadgaonkar R, Phelps KM, Haque Z, Williams AJ, Silverman ES, Collins T. CREB-binding protein is a nuclear integrator of nuclear factor-kappaB and p53 signaling. J Biol Chem. 1999;274(4):1879–1882. doi: 10.1074/jbc.274.4.1879. [DOI] [PubMed] [Google Scholar]
- Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH. Forkhead Box M1 Regulates the Transcriptional Network of Genes Essential for Mitotic Progression and Genes Encoding the SCF (Skp2-Cks1) Ubiquitin Ligase. Mol Cell Biol. 2005;25(24):10875–10894. doi: 10.1128/MCB.25.24.10875-10894.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster GA, Perkins ND. Transcriptional cross talk between NF-kappaB and p53. Mol Cell Biol. 1999;19(5):3485–3495. doi: 10.1128/mcb.19.5.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner SL, Barken D, Hoffmann A. Stimulus Specificity of Gene Expression Programs Determined by Temporal Control of IKK Activity. Science. 2005;309(5742):1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Mayo MW, Anest V, Hanson JL, Baldwin AS., Jr The Putative Oncoprotein Bcl-3 Induces Cyclin D1 To Stimulate G1 Transition. Mol Cell Biol. 2001;21(24):8428–8436. doi: 10.1128/MCB.21.24.8428-8436.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZH, Mabb A, Miyamoto S. PIDD: a switch hitter. Cell. 2005;123(6):980–982. doi: 10.1016/j.cell.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular Linkage Between the Kinase ATM and NF-{kappa}B Signaling in Response to Genotoxic Stimuli. Science. 2006;311(5764):1141–1146. doi: 10.1126/science.1121513. [DOI] [PubMed] [Google Scholar]
- Wuerzberger-Davis SM, Chang PY, Berchtold C, Miyamoto S. Enhanced G2-M arrest by nuclear factor-{kappa}B-dependent p21waf1/cip1 induction. Mol Cancer Res. 2005;3(6):345–353. doi: 10.1158/1541-7786.MCR-05-0028. [DOI] [PubMed] [Google Scholar]
- Wuerzberger-Davis SM, Nakamura Y, Seufzer BJ, Miyamoto S. NF-kappaB activation by combinations of NEMO SUMOylation and ATM activation stresses in the absence of DNA damage. Oncogene. 2007;26:641–651. doi: 10.1038/sj.onc.1209815. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Gaynor RB. IkappaB kinases: key regulators of the NF-kappaB pathway. Trends Biochem Sci. 2004;29(2):72–79. doi: 10.1016/j.tibs.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423(6940):655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- Yang WW, Wang ZH, Yang HT. E2F6 negatively regulates ultraviolet-induced apoptosis via modulation of Brca1. Cell Death and Differentiation. 2007;14:807–817. doi: 10.1038/sj.cdd.4402062. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Miki Y. Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Science. 2004;95:866–871. doi: 10.1111/j.1349-7006.2004.tb02195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HSPA, Dean DC. Active transcriptional repression by the Rb-E2F complex mediates G1 arrest triggered by p16INK4a, TGFbeta, and contact inhibition. Cell. 1999;97:53–61. doi: 10.1016/s0092-8674(00)80714-x. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Ting AT, Marcu KB, Bliska JB. Inhibition of MAPK and NF-κB Pathways Is Necessary for Rapid Apoptosis in Macrophages Infected with Yersinia. J Immunol. 2005;174(12):7939–7949. doi: 10.4049/jimmunol.174.12.7939. [DOI] [PubMed] [Google Scholar]
- Zhao F, Xuan Z, Liu L, Zhang MQ. TRED: a Transcriptional Regulatory Element Database and a platform for in silico gene regulation studies. Nucleic Acids Res. 2005;33:D103–D107. doi: 10.1093/nar/gki004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S. The Phosphorylation Status of Nuclear NF-kappaB Determines Its Association with CBP/p300 or HDAC-1. Mol Cell. 2002;9(3):625–636. doi: 10.1016/s1097-2765(02)00477-x. [DOI] [PubMed] [Google Scholar]