

Abstract
The mitochondrial DNA hypervariable segment I (HVS-I) is widely used in studies of human evolutionary genetics, and therefore accurate estimates of mutation rates among nucleotide sites in this region are essential. We have developed a novel maximum-likelihood methodology for estimating site-specific mutation rates from partial phylogenetic information, such as haplogroup association. The resulting estimation problem is a generalized linear model, with a nonstandard link function. We develop inference and bias correction tools for our estimates and a hypothesis-testing approach for site independence. We demonstrate our methodology using 16,609 HVS-I samples from the Genographic Project. Our results suggest that mutation rates among nucleotide sites in HVS-I are highly variable. The 16,400–16,500 region exhibits significantly lower rates compared to other regions, suggesting potential functional constraints. Several loci identified in the literature as possible termination-associated sequences (TAS) do not yield statistically slower rates than the rest of HVS-I, casting doubt on their functional importance. Our tests do not reject the null hypothesis of independent mutation rates among nucleotide sites, supporting the use of site-independence assumption for analyzing HVS-I. Potential extensions of our methodology include its application to estimation of mutation rates in other genetic regions, like Y chromosome short tandem repeats.
IT has long been known that different regions in the genome mutate at vastly different rates (Tamura and Nei 1993). In particular, for the mitochondrial DNA (mtDNA) two hypervariable segments (HVS) have been identified and named HVS-I and HVS-II. Even within these segments, the mutation rates of the various sites are not fixed. Tamura and Nei (1993) showed that there is strong statistical support for a Gamma “prior” distribution of mutation rates across the mtDNA control region (which contains both HVS-I and HVS-II), with a shape parameter α = 0.1, implying many orders of magnitude difference in rates between the fastest and slowest mutating sites in these segments. Yang (1993, 1994) described methodologies for integrating this Gamma prior into maximum-likelihood (ML) phylogeny estimation.
Beyond the distribution of mutation rates, the next step is to estimate site-specific mutation and/or substitution rates. These are potentially important for understanding functionality of various genetic regions, as different functions are likely to impose selection or sequence constraints and these can be inferred through a good estimation methodology for site-specific rates. For example, in mtDNA HVS-I several termination-associated sequences (TAS) have been identified, on the basis of sequence properties and conservation indexes. These are suspected to play a central role in regulation between replication termination and elongation of the mtDNA (Falkenberg et al. 2007). If these suspicions are well founded, we would expect strong structural constraints to apply to these sequences and hence expect them to be subject to purifying selection. Although mutations might occur at a similar rate to the rest of HVS-I, the resulting variants would be selected against. In the presence of selection, neutral theory assumptions made by practically every estimation approach, including ours below, are violated, but the reduced diversity due to selection is still expected to lead to lower estimates. Thus, the task of identifying (or verifying) the functionality of such regions can be addressed in a hypothesis-testing framework for the “null” hypothesis of neutrality (under which the statistical model is valid and the rates should be “average”) against the alternative of slower rates.
Numerous approaches have been developed for estimating site-specific mutation rates. One flavor (e.g., Yang 1995; Siepel and Haussler 2005) is based on analyzing the mutation rates as a Markov process and hence identifying their sequential correlation. These approaches are theoretically attractive, but computationally intensive, and are usually applied to small sets of samples from different species, leading to a limited ability for high-quality estimation of individual rates, if the sequential correlation is not overwhelmingly high (as is the case in mtDNA, see our results below). Another family of methods is based on Bayesian or mixed-effects inference (Yang and Wang 1995; Mayrose et al. 2004; Mateiu and Rannala 2006), and these methods share both the favorable statistical properties and the computational difficulties in handling a large amount of data with the first group. The limitation on the amount of data that can be used is obviously of critical importance in determining the quality of estimates obtained. With a small number of samples (dozens or less), it is simply impossible to observe enough heterogeneity in the data to derive accurate estimates, however sound and theoretically appealing the methods are.
A third group of methods is based on phylogenetic reconstruction of potentially large samples of mtDNA HVS-I sequences, followed by estimation of the rates by counting the number of mutation events in each site. For a survey of these approaches, see Bandelt et al. (2006). As an example, we consider here the approach that Bandelt et al. (2006) develop and two previous approaches, by Excoffier and Yang (1999) and Meyer and von Haeseler (2003). Both of the latter are approximate ML methods, attempting to reconstruct the full distribution over possible tree topologies and estimate parameters simultaneously. Because of the extreme difficulty of this task, especially assuming rate variation, even for moderately sized data sets (up to several hundred samples), as used in these two articles, they develop different approximation approaches. Excoffier and Yang (1999) generate a limited set of parsimonious candidate trees and investigate the robustness of their estimates to their choice of topology from this set. Meyer and von Haeseler (2003), on the other hand, alternate between estimating phylogeny and mutation rates (where the phylogeny estimation step assumes known, but potentially variable, mutation rates). Bandelt et al. (2006) discuss these approaches and explore their limitations and shortcomings, which they consider to be critical. They therefore conclude that the best approach for mutation rate estimation is to manually construct a best tree (in their case, using parsimony considerations) and estimate the mutation rates by direct counting on this tree. They apply their methodology to ∼800 samples.
Our approach is motivated by the current availability of very large databases of HVS-I sequences, such as the genographic public participation database described in Behar et al. (2007), and by the realization that construction of reliable phylogenies for such large samples is a difficult, often impossible task. Instead we rely on partial, highly reliable phylogenetic information, in our case in the form of haplogroup (Hg) associations of the mtDNA samples we use. We develop a formal ML inference approach that allows us to find ML estimates of the site-specific rates without reconstructing the explicit phylogeny. We show that ML parameter estimation in our model is a binomial regression with complementary-log-log link function (a generalized linear model) for estimating the site-specific mutation rates and the size parameters for each Hg-specific phylogenetic tree. The main advantage of our approach is that it allows us to practically apply our method to large data sets and eliminate the difficulties resulting from uncertainty about the correct phylogeny. In our case, we apply it to a data set of 16,609 samples, collected in the Genographic Project (Behar et al. 2007) and classified into Hg's relying mostly on information from the slowly mutating coding region of mtDNA. We demonstrate the superiority of our estimates over the results of Bandelt et al. (2006) and others.
In addition to the search for functionality mentioned above, our methodology and the estimates it generates can be used to improve phylogeny estimation algorithms and sequence quality checking (Bandelt et al. 2002), as well as phylogenetic classification as we showed in Behar et al. (2007). Our likelihood-based approach also supports likelihood-ratio tests for the site independence hypothesis underlying much of the inference regularly performed on phylogenies. Below we perform these tests and demonstrate that this independence hypothesis is mostly reasonable for mtDNA HVS-I.
MATERIALS AND METHODS
Statistical estimation approach:
Assume we observe a large number of sequences of a nonrecombining DNA region. These samples are all located on a phylogenetic tree. We are not given their detailed phylogenetic relationship, but rather a haplogroup view of that relationship. That is, the samples are divided into groups that belong to the same haplogroup or paragroup, together abbreviated here as Hg, where each Hg can be thought of as a terminal subtree of the full phylogenetic tree, whose internal structure is not known. This situation is illustrated in Figure 1 (a paragroup may technically be a subforest of the full tree, but this has no bearing on our methodology).
Figure 1.—

(A) Schematic of the Hg view of a phylogenetic tree and (B) the full phylogenetic tree, including the internal Hg phylogenies, which we assume we do not observe.
We assume we have aligned sequences and concentrate on their differences through single-nucleotide polymorphisms (SNPs). We ignore insertions and deletions in our analysis. We do this because their mutation mechanisms are more difficult and less well understood than SNPs and because insertions and deletions in HVS-I appear to be unique events, not prone to homoplasy or back mutations (Behar et al. 2007). We also assume the following:
The haplogroup classification of all sequences is known and accurate.
The SNPs in each site of the considered DNA region are independent. It is important to differentiate this site-independence hypothesis from the rate-independence hypothesis tested and rejected by Yang (1995) and others. Our estimation approach is purely frequentist and we assume nothing about the “distribution” of the rates in our estimation methodology. We go beyond this only for inference on these estimates, as discussed in the next sections.
There is a global molecular clock; i.e., for every site considered, the rate of mutations per time unit is the same in every part of the phylogenetic tree.
Every site has a fixed Poisson rate with which the mutations occur. This assumption is exactly correct if we assume an appropriately simple substitution model, in particular one where the set of mutation rates is independent of the current nucleotide (and consequently all four nucleotides are equally likely to appear). This is true of the three simplest substitution models commonly used, including the Jukes–Cantor model (JC69) (Jukes and Cantor 1969), the Kimura two-parameter model (K80) (Kimura 1980), and the Kimura three-parameter model (K3ST) (Kimura 1981), which allows for different rates of transitions and two different types of transversions.
Assumption 1 is critical for our analysis and cannot be validated. The methodology we develop will allow us to do hypothesis testing to examine the validity of assumption 2. Assumption 3 can be relaxed as long as the clock changes uniformly for all sites in HVS-I. Assumption 4 is important to make our model formally correct, but slight violations of it (e.g., in substitution models that allow slightly different marginal rates for the different nucleotides) should not affect the practical validity of our methodology. Assumptions 2 and 4 clearly both depend on assuming neutrality of HVS-I. If the region is functional, it is very likely to create dependence between sites and violate standard substitution model assumptions. The selection it creates also clearly implies that our estimates would not correspond to true mutation rates. However, as we discuss below, we can still use our estimates in hypothesis tests for site independence and presence of selection.
Given a rooted phylogenetic tree T, let t(T) be the total time length of all branches on the tree. Subject to our assumptions, the number of mutations on this tree in a site i in total time t(T) is distributed Poisson(λi · t(T)), where λi is the rate parameter for this site (which is the same in all Hg's). In our case, we are not given the full tree T but a set of K Hg's, representing terminal subtrees T1, … , TK whose lengths t1, … , tK and internal structure are not known, with n samples sorted into n1, … , nK samples in each Hg, respectively.
Assume first we were able to observe the number of mutations mik in each site i in each Hg k; then the total log-likelihood of the data would be
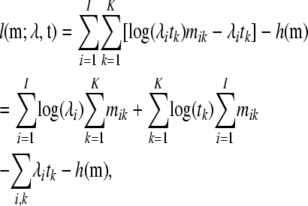 |
(1) |
where I is the number of sites in our genetic region and 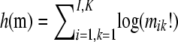 is of no consequence for ML estimation of the parameters
is of no consequence for ML estimation of the parameters 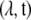 . This ML estimation problem is a straightforward Poisson regression with a (canonical) log link function. In fact, it is easy to show that the decomposition in Equation 1 implies that ML estimation of all λi's can be done by simple counting (up to multiplication by an overall constant factor).
. This ML estimation problem is a straightforward Poisson regression with a (canonical) log link function. In fact, it is easy to show that the decomposition in Equation 1 implies that ML estimation of all λi's can be done by simple counting (up to multiplication by an overall constant factor).
Given Hg-level classification only, however, we do not observe the mik's, but observe only the state of site i in all nk samples (leaves) in Hg k. If not all of these are identical, we know for certain that mik ≥ 1; i.e., site i has mutated at least once somewhere on the phylogenetic tree describing our haplogroup k samples. Without knowledge of the actual Hg-specific phylogeny we cannot make any further conclusions on mik in this case. If all of the nk samples have an identical nucleotide in position i, we conclude that this site has not mutated anywhere on the Hg's phylogenetic tree; i.e., mik = 0. This conclusion is not guaranteed to be correct; however, we can argue that with overwhelming probability it will be.
To demonstrate that our approach can properly capture whether a mutation did occur in a specific site, consider a simple phylogenetic tree like the one in Figure 2, where we assume a mutation from red triangle to black circle has occurred on the top right branch. The shapes at the bottom describe the states of the leaves (observed samples), if no other mutations have occurred at this site. Now assume we want all the leaves of the tree to have the same nucleotide (all triangle or all circle) at this site. This would clearly require that either the mutation reverted back from circle to triangle on a cut of the subtree below the original mutation (such as both branches marked with **) or the same exact mutation (triangle to circle) simultaneously happened on a set of branches completing a cut of the full tree (such as the branch marked with ×). If none of these highly unlikely events (requiring multiple “coordinated” mutations) occur, all leaves would not have the same nucleotide at this site, given the shown triangle to circle mutation.
Figure 2.—

Demonstration of our reasoning, that we know whether any mutations have occurred in a specific site.
We can illustrate the low probability of missing a mutation in our approach, by comparing it to another probability, that of not observing a mutation on a coalescent tree because it has mutated back on the same link and thus is completely unobservable. Assuming for simplicity that all polymorphisms are binary, consider, for example, the two links marked with ** in Figure 2, and assume they both have length t. It is easily seen that the probability that site i mutated and reverted on either one of them is 2 · exp(−2λit)(λit)2/2 + O((λi · t)3). The probability that the triangle to circle mutation reverted back on both of them simultaneously is similarly exp(−2λit)(λit)2 + O((λi · t)4), i.e., slightly smaller. If we do not assume both links have the same length, then the first probability is potentially much bigger than the second. Thus, under reasonable assumptions that reversion back is most likely on binary splits, our total chance of setting mik = 0 when the true value is mik > 0, is on the same order of magnitude as the chance that the coalescent tree contains mutations that reverted back on the same link, which are inherently unobservable. One caveat to keep in mind regarding our assertion that we likely know if mik = 0 and its illustration, is that gross violations of our model assumptions above (in particular, assumption 4) can make our chances of missing mutations much higher. For example, mutations creating a CpG dinucleotide may be likely to revert quickly, even multiple times. This logic also applies to the chance of multiple mutations occurring on the same link, of course.
It should be clarified that by setting mik = 0 we are not implying that the site i has never mutated in this haplgroup k anywhere in the world, but rather that it has not happened on the phylogenetic (coalescent) tree of the nk samples we observe in our data set. This is the tree whose total branch length tk is one of the parameters we will be estimating.
Thus, we are assuming that while we cannot observe our Poisson mutation counts mik, we can observe the binary variables 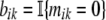 . It is easy to verify that these variables are distributed as bik ∼ Bernoulli(exp(−λi · tk)). If we now write the partial likelihood of the observed data b only, we get
. It is easy to verify that these variables are distributed as bik ∼ Bernoulli(exp(−λi · tk)). If we now write the partial likelihood of the observed data b only, we get
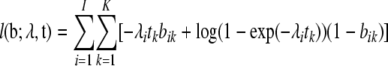 |
(2) |
and ML estimation of the parameters 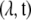 is now a binomial regression with a complementary log–log (CLL)-link function. In other words, the log of the negative log of the Bernoulli success probability is linearly related to the (log) parameters:
is now a binomial regression with a complementary log–log (CLL)-link function. In other words, the log of the negative log of the Bernoulli success probability is linearly related to the (log) parameters:
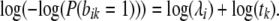 |
(3) |
This is still a generalized linear model (GLM) (McCullagh and Nelder 1989), although a less standard one than the Poisson regression we could use to estimate the parameters from the complete likelihood in Equation 1, if we could observe the actual counts.
This procedure yields ML estimates of both the Hg coalescent tree lengths 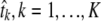 (without information about the actual phylogeny), and the site-specific instantaneous mutation rates
(without information about the actual phylogeny), and the site-specific instantaneous mutation rates 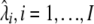 . However, note that this ML solution is defined only up to a multiplication of all the
. However, note that this ML solution is defined only up to a multiplication of all the 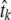 's by a constant and division of all the
's by a constant and division of all the 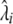 's by the same constant [the Bernoulli probabilities in (3) would not be affected]. Thus, to complete our estimation we need to resolve this remaining degree of freedom, for example, through calibration of the total mutation rate
's by the same constant [the Bernoulli probabilities in (3) would not be affected]. Thus, to complete our estimation we need to resolve this remaining degree of freedom, for example, through calibration of the total mutation rate 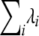 to an external accepted number. Following Forster et al. (1996) we use 1/20,180 mutations per year in the limited HVS-I (16,090–16,395) as our calibration number.
to an external accepted number. Following Forster et al. (1996) we use 1/20,180 mutations per year in the limited HVS-I (16,090–16,395) as our calibration number.
We summarize our modeling approach as follows:
We are given HVS-I sequences as data, and we assume that these sequences are correctly classified into Hg's and that we get the full, correct HVS-I sequence for every sample.
We make assumptions 1–4 above, under which the likelihood of the Hg-site-specific mutation counts mik is Poisson (1).
Since we do not know the intra-Hg phylogeny of our samples, we cannot observe mik; however, we can (with overwhelming probability) observe
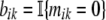 .
.ML estimation of the site-specific mutation rates and Hg-specific coalescent tree lengths is now a binomial regression with a CLL-link function.
Saturation and subsampling:
Since our method relies on high-quality Hg classification and then considers only the binary bik's, it can happen that a specific site i gives bik = 0 ∀k; i.e., it is polymorphic in all Hg's. This is especially likely if some of the λi's are much larger than others and if all Hg's contain a large number of samples. This is indeed the case for the genographic data set we use below for our experiments.
In the event that bik = 0 ∀k the rate λi is inestimable in our methodology (that is, the maximum-likelihood estimate is not finite). Even if bik ≠ 0 for a small number of Hg's, the estimate of λi may still suffer from stability problems. Ideally, we would like a balance between Hg's for which bik = 1 and ones for which bik = 0, especially for our fastest mutating sites.
In this situation, we propose to counter this problem by subsampling the large database multiple times and generating a distribution of estimates generated by applying our estimation approach to subsamples from the original larger sample. In fact, we advocate using a bootstrap-based subsampling approach, known as the m out of n bootstrap (Bickel et al. 1997), where m < n samples are sampled with return from the database of size n. As Bickel et al. (1997) and others discuss, this is an alternative bootstrap approach, which can lead to similar insights to the standard bootstrap, and is superior in certain situations when the standard (n out of n) bootstrap is not effective for various reasons. Our setting is different from theirs, in that not only the bootstrap-based inference, but also the estimation itself, cannot be performed from the full data set. Thus we are taking advantage of the m out of n bootstrap for both estimation and inference.
In our approach, we empirically try different values of m, giving rise to distributions of estimators of the mutation rates. We evaluate them on the basis of their empirical spread (variance) and their bias in estimating the true rates. We discuss strategies for estimating these quantities in the next section.
Statistical inference:
The goal of inference is to interpret and understand the performance of our estimation procedure and validate the underlying assumptions. Our first inference goal is to get an idea of the relationship between our estimates and the “real” values. The second is to test the hypothesis of site independence underlying our method (and much of the analysis of genetic information).
Bias and variance estimation based on a simulation–bootstrap hybrid:
A key question regarding our methodology is, How reliable are our mutation rate estimates? Asymptotic theory can be used to derive approximate confidence intervals for the ML estimates we derive (see McCullagh and Nelder 1989 for details). However, our modeling problem seems to be far from “asymptopia” and these intervals are not reliable. Also, CLL-link binomial regression has inherent bias (McCullagh and Nelder 1989, Chap. 15). We try, therefore, to investigate the error in our estimates through a combination of resampling-based and simulation approaches.
The parametric bootstrap (Efron and Tibshirani 1994) allows us to investigate properties of our estimators through a plug-in approach as follows: generate multiple data sets from the model we estimated, reestimate the model from these data sets, and investigate the consistent error (bias) and instability (variance) of these estimators. The main problem with application of the parametric bootstrap in our case is the implicit assumption it makes, that our estimated model is “close” to the true model and generates data with similar properties. This assumption is clearly violated in our case in one respect: we are able to estimate rates only for sites in HVS-I that are polymorphic in our data (292 of 553). However, the other 261 sites clearly do not have probability 0 of mutating. Rather, it is the luck of the draw that determines which portion of the slowly mutating sites in HVS-I are polymorphic in our data. If we now draw a parametric bootstrap sample, using our estimated rates, we expect that many of the sites that are polymorphic in our data would never mutate in this bootstrap sample, and the number of polymorphic sites in every bootstrap sample would be much smaller than the number in our original data set. It should be noted that the nonparametric bootstrap does not alleviate this problem and is even more problematic since bootstrap sampling of the original samples would cause only slight perturbations in the binary variables bik, which are the real inputs to our modeling approach.
On the other hand, we have at our disposal information about the “prior” distribution of the mutation rates in HVS-I. Tamura and Nei (1993) originally showed that a Gamma prior with shape parameter ∼α = 0.1 is appropriate for the distribution of mutation rates in the entire control region of the mtDNA (which includes HVS-I, as well as HVS-II and their intermediate region). Later authors, including Excoffier and Yang (1999) and others, have suggested different values of α may be more appropriate for HVS-I alone. We reestimate this parameter from our Hg-level data, using a methodology in the spirit of Tamura and Nei (1993), as follows.
As discussed above, we assume that the sites that are nonpolymorphic in all our Hg's have never mutated. Furthermore, sites that are polymorphic in one Hg only can reasonably be assumed to have mutated only once, since the fact that they are nonpolymorphic in all other Hg's is indicative of their low mutation rate. While this assumption may not be completely accurate, it is “close enough” to obtain a rough estimate of α. So, assuming we know how many sites have mutated 0, 1, and >1 times in our complete data, we can now estimate α by a “method of moments” requiring that the empirical distribution matches the posterior probabilities for these three groups under the negative binomial distribution. As we show below, this method leads us to an estimate of α = 0.25 for the shape parameter based on our data.
For simulating our process and estimating its variance, we can now simulate a set of “true” rates by drawing a sample of size 553 from our hypothesized distribution Gamma(α, β), where α = 0.25 is our estimate of the shape parameter and β is the scale parameter, which we can tune, for example, by imposing the constraint 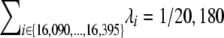 from Forster et al. (1996) for calibration. We can then use these 553 rates to generate multiple data sets, for which we know the correct rates, and then examine our algorithm's performance on these.
from Forster et al. (1996) for calibration. We can then use these 553 rates to generate multiple data sets, for which we know the correct rates, and then examine our algorithm's performance on these.
To generate simulated data (that is bik's) that are like our actual data, we also need the tk's, i.e., the Hg tree sizes. For this purpose, we can take advantage of the parametric bootstrap and use our estimated tk's to generate the simulation data sets (we could then also quantify the bias our method suffers in estimating these quantities, although this is not the main focus of this article).
We can then apply our estimation methodology to multiple samples drawn via this simulation–bootstrap hybrid methodology and obtain estimates of the bias inherent in this methodology for data “like” the genetic data we have.
To summarize our bias estimation methodology, given an estimation methodology E, and a data set D, it proceeds as follows:
Apply E to D to obtain estimates
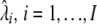 and
and 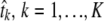 . If E contains m of n bootstrap sampling embedded in it, apply it to multiple bootstrap samples according to this methodology.
. If E contains m of n bootstrap sampling embedded in it, apply it to multiple bootstrap samples according to this methodology.Draw a sample of true rates λi, i = 1, … , I from Γ(α, β).
- Repeat the following r times:
- Create a new data set D* by drawing bik ∀i, k using our simulation–bootstrap hybrid and Equation 3.
- Apply our methodology E to D* to obtain estimates
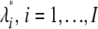 .
.
Calculate empirically the bias of these estimates compared to the (known) λi.
If E contains m of n bootstrap sampling, use bootstrap variance estimates. If not, use the simulation–bootstrap hybrid repeated samples to estimate the variance.
Evaluate the overall relationship between λi and bias and variance, to generate a bias correction that is a function of the magnitude of λi.
Hypothesis testing about site independence:
A fundamental question about our methodology and many other methods in phylogenetics is, To what extent are the molecular clock and site independence assumptions we make realistic? In our ML framework, we can actually test the site-independence assumption statistically, against the alternative that mutation mechanisms in one site may depend on the nucleotide value in another site (or multiple sites, potentially).
Unfortunately, we cannot similarly test the lineage-independence hypothesis, since change in the rate of the mutational clock is indistinguishably confounded with the tree sizes tk.
Assume we want to test whether site r affects site s. Denote as before by brk, bsk the indicator variables for sites r, s being nonpolymorphic in Hg k, respectively. Given a null hypothesis of site independence between r, s, we can express the “alternative” that site s is more likely to be nonpolymorphic if site r is nonpolymorphic, by adding a parameter expressing this dependence to our formulation, as follows:
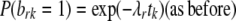 |
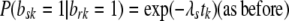 |
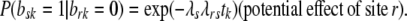 |
Under the null of no dependence, we have λrs = 1 and we go back to the formulation in Equation 2, while under the alternative we can rewrite the likelihood as
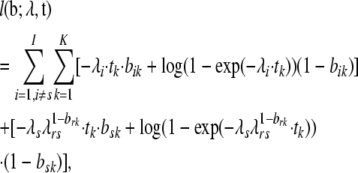 |
(4) |
where the last part in Equation 4 allows an extra parameter for the cross-effect between the two sites. We can then test the hypothesis H0: λrs = 1 via a generalized likelihood-ratio test with 1 d.f., comparing the ML solutions of Equation 2 and Equation 4.
When we apply this testing methodology for all pairs of sites, we are performing a large number of tests, and we need to take into account the issue of multiple comparisons when evaluating the outcome of our tests. For that purpose, we employ the false discovery rate multiple-comparisons correction at 5%, which guarantees that the expected rate of falsely rejected null hypotheses is at most 5% of all rejected hypotheses, possibly less, under some types of dependence (Benjamini and Hochberg 1995). This correction is slightly less conservative than the standard Bonferroni correction (i.e., allows us to reject more nulls), but similar in spirit.
The main advantage of our testing methodology is that it aligns naturally with our modeling approach and specifically that it does not require detailed phylogenetic reconstruction. It should be noted, however, that it cannot expose every type of nonindependence, and it may have limited power to expose others. For example, if a specific combination of nucleotide values in two sites has a strong affinity, and hence once one site mutates into this state, and the other follows closely, our method can identify this affinity only if this phenomenon has happened in many of the Hg's. A detailed phylogenetic analysis could have more power to identify and characterize these relationships.
Genographic mtDNA data:
Each mitochondrial DNA sample submitted to the Genographic Project goes through the standard classification process (Behar et al. 2007):
Sequencing of a number of coding-region markers: the number has increased during the project and currently is at 22.
Sequencing of the full extended HVS-I, defined as sites 16,024–16,569 of the samples aligned to revised Cambridge Reference Sequence (rCRS).
On the basis of step 1, determine a Hg designation by SNPs into one of 23 Hg's: L0/1, L2, L3(xM, N), M, C, D, N, N1, A, I, W, X, R, R9, R0, HV, H, V, J, T, U, K, B.
On the basis of steps 1 and 2, determine a haplogroup designation into one of 87 Hg's.
Table 2 of Behar et al. (2007) shows a summary of Hg distribution for the 16,609 samples used in our analysis (the reference database). Following assumption 1 in the Statistical estimation approach section, we assume that the 23-Hg nomenclature labels are all correct. Since they are based on coding-region SNPs and the careful classification protocol discussed in Behar et al. (2007), this assumption is likely to be true. It is less likely to be accurate for the 87-Hg nomenclature. However, as the 87-Hg version allows us to get much better resolution in our analysis, we also use it with the implicit assumption that its classification is accurate and compare and discuss the results from using both nomenclatures.
TABLE 2.
Results of generalized likelihood-ratio tests for site independence
| Effect | Raw P-value | Corrected |
|---|---|---|
| 16,182 ⇒ 16,183 | 7.7 × 10−12 | <0.0001 |
| 16,183 ⇒ 16,182 | 2.2 × 10−9 | <0.0001 |
| 16,114 ⇒ 16,526 | 0.0000012 | 0.03 |
| 16,212 ⇒ 16,153 | 0.000027 | 0.66 |
| 16,266 ⇒ 16,148 | 0.000033 | 0.8 |
| 16,304 ⇒ 16,163 | 0.000039 | 0.95 |
| 16,184 ⇒ 16,335 | 0.000045 | 1 |
| 16,104 ⇒ 16,111 | 0.000053 | 1 |
| 16,327 ⇒ 16,163 | 0.000068 | 1 |
| 16,526 ⇒ 16,114 | 0.00009 | 1 |
| ⋮ | ⋮ | ⋮ |
Supplemental Table 4 of Behar et al. (2007) contains all the information required to calculate the bik values for the full data set. We can see that some of the sites are completely saturated for the 23-Hg nomenclature: 16,129, 16,189, and 16,519 are polymorphic in all 23 Hg's and several other sites are polymorphic in at least 20 Hg's. Thus, to model the rates reliably from these data we have to resort to our subsampling methodology.
With the 87-Hg nomenclature, we clearly have a lot more information about the mutation rates in our data, but a less reliable Hg classification. Site 16,519 is polymorphic in the most Hg's: 65 of the 87. Thus, on the basis of these data we could estimate the rates directly without resorting to subsampling. The quality of estimates will be hampered by the uncertainty about the correctness of the Hg labels.
One issue about the data that is highly relevant to our analysis below is the problems in sequencing around the poly-cytosine (poly-C) region created by the transition T16,189C (relative to rCRS). This comes up in the dependence we identify below between sites 16,182 and 16,183 in our sequences, which we suspect may be due to sequencing problems. Mutations in these two sites always occur in concordance with the adjacent polymorphism T16,189C that creates a poly-C stretch that causes significant reading difficulties of this region, using standard sequencing procedures (Figure 3). These difficulties relate to a technical sequencing problem in which DNA strands that differ in the number of cytosine repeats are assembled and thus overlapping positions subsequent to T16,189C are impossible to be appreciated since they are affected by the shift created by the variable number of cytosines in the different DNA strands. Therefore, the positions around the poly-C stretch are usually removed from analysis (Behar et al. 2007). A different question relates to our ability to correctly understand the number of adenosines that immediately precede the poly-C region (four in the rCRS). Figure 3 shows that different numbers of adenosines are associated with the poly-C stretch. Since most of the mutations we observe in 16,182 and 16,183 are transversions between adenosine and cytosine, it is possible that the poly-C stretch creates a technical problem here as well despite the unquestionable reads we get for these positions. We successfully used fragment-length analysis techniques, similar to those used to count the number of repeats in short tandem repeats, to understand the real number of cytosine repeats in various samples and found no clear evidence for mistakes in the number of preceding adenosines (data not shown). Nevertheless, caution mandates the questioning of the authenticity of our results for positions 16,182 and 16,183 and the possibility that the poly-C stretch plays a role in creating artificial dependence.
Figure 3.—

The poly-C stretch. Position 16,189 is highlighted and five sequences are shown. A sequence identical to the rCRS in the presented region is shown at the top. Below it, four sequences containing the T16,189C polymorphism are arranged to show one to four adenosines preceding the poly-C stretch. A typical chromatogram of the sequence after the poly-C stretch is also demonstrated.
Mutation rate estimation protocols:
Considering the discussion above about the various Hg nomenclatures we have at our disposal and the subsampling approaches, we implemented four different protocols to estimate mutation rates from our data: (1) subsampling-based estimates, using 100 repeated samples of 1000 sequences of our 16,609 total sequences and the 23-Hg nomenclature; (2) subsampling-based estimates, using 100 repeated samples of 3000 sequences of our 16,609 total sequences and the 23-Hg nomenclature; (3) subsampling-based estimates, using 100 repeated samples of 4000 sequences of our 16,609 total sequences and the 23-Hg nomenclature; and (4) estimates with no subsampling, using the 87-Hg nomenclature.
We then used the glm function in R to calculate the ML estimates of 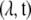 in Equation 2. See McCullagh and Nelder (1989) for discussion of the theory of GLMs and Venables and Ripley (1994) for discussion of the glm function in S+, which is the predecessor of R.
in Equation 2. See McCullagh and Nelder (1989) for discussion of the theory of GLMs and Venables and Ripley (1994) for discussion of the glm function in S+, which is the predecessor of R.
Running the binomial regression, and applying the constraint 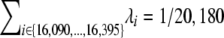 from Forster et al. (1996) for calibration, we obtain ML estimates in each setting (in the subsampling protocols 1–3, we actually obtain a whole distribution of estimates in each setting). We then apply our bias correction (which turns out to be small, see below) and use the empirical range of estimates from the bootstrap samples (for protocols 1–3) or the estimated variance from the simulation–bootstrap hybrid (for protocol 4) to calculate confidence intervals.
from Forster et al. (1996) for calibration, we obtain ML estimates in each setting (in the subsampling protocols 1–3, we actually obtain a whole distribution of estimates in each setting). We then apply our bias correction (which turns out to be small, see below) and use the empirical range of estimates from the bootstrap samples (for protocols 1–3) or the estimated variance from the simulation–bootstrap hybrid (for protocol 4) to calculate confidence intervals.
Investigating mutation rates at TAS loci:
Having high-quality site-specific rate estimates facilitates investigation of possible function in HVS-I. For example, HVS-I includes a series of cis-acting TAS located at the 5′ end of the control region (Doda et al. 1981). These short DNA stretches of ∼15 nucleotides are believed to play an important role in the regulation between replication termination and elongation of the mtDNA (Falkenberg et al. 2007). While this modeling process is not fully resolved, in vivo footprints of protein-binding sites overlapped with the TAS loci in mtDNA positions 16,158–16,173 on the H strand and 16,305–16,318 and 16,331–16,353 on the L strand (Roberti et al. 1998). Previous studies identified the TAS elements by comparing the mtDNA control region from various mammals for conserved sequences, using a small number of samples and seeking well-preserved regions (Sbisa et al. 1997). Our estimation methodology combined with the large database available for us allows us to critically examine the level of conservation of these regions in modern humans. The validity of our statistical model clearly depends on neutrality assumptions, leading us to treat the preservation and functionality search problem as a hypothesis-testing problem. Under the null of no functional constraints, the mutation rates of the TAS regions should be “no different on average” than the rest of HVS-I. If this null is wrong, it invalidates the statistical model, and the estimates we get are no longer valid mutation rate estimates. However, they are clearly expected to be smaller than the true mutation rates in these TAS regions (due to purifying selection). Hence we can still formally use them to test the function hypothesis.
To test this hypothesis, we consider the four TAS loci identified by Roberti et al. (1998), in sites 16,097–16,107 (TAS E), 16,158–16,173 (TAS D), 16,305–16,318 (TAS C), and 16,331–16,353 (TAS A and TAS B, which overlap). As mentioned above, protein binding was detected in the latter three only. We compare the mutation rates in these loci to the rest of HVS-I, both as individual loci and for all four combined.
It should be clarified that our test (like all statistical tests) does not have power against all possible alternatives. For example, if there are some sites in TAS loci with a tendency toward frequent mutations, and these mutations then get gradually weeded out by selection, then our approach may fail to identify this situation as “nonnull.” In that case, an alternative approach, which uses statistics related to the prevalence of the rarer states in polymorphic sites, may be more effective. As this is not a natural extension of our methodology, we leave this approach as a topic for future research.
RESULTS
Table 1 (first four columns) gives an estimate and confidence interval of mutation rates for the 48 quickest mutating sites in HVS-I, from several different variants of our approach (the complete list is given in supplemental Table 1). We see that the fastest mutating site, 16,519, is estimated to mutate once about every 200,000–500,000 years (depending on which of our estimates is used). The 10th fastest site mutates ∼4 times slower, and the slowest site in this list mutates ∼10 times slower. Thus, for example, two individuals whose time to the most recent common mtDNA ancestor (TMRCA) is 20,000 years have a probability of ∼exp(−40,000/350,000) = 0.87 to have the same nucleotide in site 16,519 due to identity by descent. The total probability that they share the same nucleotide is of course slightly higher, since they may also have it due to homoplasy. Figure 4 shows a graphical representation of the rate estimates as they physically appear on HVS-I (using the estimates from the 3000-samples version, as in the third column of Table 1). We can see the relatively uniform spread of the fastest mutating sites, perhaps with a cluster around the poly-C region in 16,184–16,189, and the relative dearth of fast sites after 16,370, and especially in the range 16,400–16,518. This dearth is also statistically significant: a Wilcoxon rank sum test (Hollander and Wolfe 1973) for the region 16,400–16,500 compared to the rest of HVS-I gives a P-value of 1.4 × 10−9 for the null that these two regions have the same distribution of mutation rate estimates. Even allowing for the fact that we chose this region by looking at the data, this result is still significant.
TABLE 1.
Mutation rate estimates (in mutations per million years) and 90% confidence intervals for the fastest sites in HVS-I from some versions of our method and Bandelt et al. (2006)
| Est. [90% C.I.]
|
|||||
|---|---|---|---|---|---|
| Locus | 1000 samples | 3000 samples | 4000 samples | 87 Hg | Bandelt et al. (2006) |
| 16,051 | 0.54 [0.33–0.85] | 0.5 [0.30–0.82] | 0.54 [0.35–0.84] | 0.55 [0.39–0.79] | 0.67 [0.31–1.3] |
| 16,086 | 0.35 [0.12–0.7] | 0.49 [0.25–0.8] | 0.55 [0.31–0.87] | 0.81 [0.59–1.1] | 0.29 [0.08–0.74] |
| 16,092 | 0.56 [0.32–0.96] | 0.57 [0.34–0.88] | 0.54 [0.35–0.88] | 0.54 [0.38–0.77] | 0.57 [0.25–1.1] |
| 16,093 | 1.6 [0.91–2.5] | 1.7 [1.1–2.3] | 1.8 [1.0–3.2] | 2.8 [2.1–3.9] | 3.2 [2.3–4.3] |
| 16,111 | 0.64 [0.37–1.0] | 0.58 [0.37–0.86] | 0.64 [0.37–1.1] | 0.7 [0.5–0.98] | 0.71 [0.35–1.3] |
| 16,126 | 0.52 [0.28–1] | 0.68 [0.45–1] | 0.66 [0.44–0.9] | 0.53 [0.37–0.75] | 0.43 [0.16–0.94] |
| 16,129 | 1.9 [1.1–2.8] | 1.8 [1.2–3] | 1.7 [1.2–2.9] | 1.3 [0.93–1.7] | 1.8 [1.1–2.6] |
| 16,145 | 0.56 [0.31–1.2] | 0.61 [0.39–0.94] | 0.64 [0.44–0.95] | 0.71 [0.51–1] | 0.67 [0.31–1.3] |
| 16,148 | 0.34 [0.19–0.56] | 0.32 [0.21–0.47] | 0.3 [0.20–0.45] | 0.36 [0.24–0.53] | 0.38 [0.13–0.87] |
| 16,172 | 1.8 [1.2–2.8] | 1.6 [1.1–2.6] | 1.5 [0.93–2.3] | 1.2 [0.89–1.7] | 0.86 [0.45–1.5] |
| 16,182 | 0.64 [0.36–1.1] | 0.68 [0.39–0.98] | 0.64 [0.44–0.89] | 0.67 [0.48–0.94] | 0.095 [0.005–0.45] |
| 16,183 | 1.8 [1.1–3] | 1.9 [1.2–2.9] | 1.8 [1.3–2.4] | 1.2 [0.86–1.6] | 0 [0–0.29] |
| 16,184 | 0.21 [0.06–0.49] | 0.32 [0.17–0.58] | 0.35 [0.21–0.56] | 0.55 [0.39–0.78] | 0.095 [0.005–0.45] |
| 16,189 | 2.5 [1.6–3.7] | 2.4 [1.7–3.4] | 2.2 [1.3–3.8] | 2.5 [1.8–3.4] | 2.2 [1.5–3.1] |
| 16,192 | 1.1 [0.6–1.7] | 0.94 [0.6–1.4] | 0.88 [0.63–1.3] | 1.0 [0.75–1.4] | 1.4 [0.89–2.2] |
| 16,209 | 0.41 [0.21–0.68] | 0.43 [0.26–0.68] | 0.46 [0.28–0.73] | 0.48 [0.33–0.69] | 0.43 [0.16–0.94] |
| 16,213 | 0.26 [0.11–0.57] | 0.32 [0.18–0.55] | 0.34 [0.2–0.55] | 0.28 [0.18–0.43] | 0.52 [0.22–1.1] |
| 16,218 | 0.28 [0.12–0.54] | 0.35 [0.19–0.53] | 0.36 [0.23–0.52] | 0.47 [0.32–0.67] | 0 [0–0.29] |
| 16,223 | 0.46 [0.18–0.91] | 0.57 [0.34–0.93] | 0.64 [0.38–0.98] | 0.64 [0.46–0.9] | 0.86 [0.45–1.5] |
| 16,234 | 0.52 [0.21–0.95] | 0.68 [0.42–1.2] | 0.68 [0.41–1.1] | 0.72 [0.52–1] | 0.43 [0.16–0.94] |
| 16,239 | 0.36 [0.20–0.6] | 0.35 [0.21–0.55] | 0.32 [0.21–0.48] | 0.43 [0.3–0.63] | 0.19 [0.03–0.6] |
| 16,249 | 0.5 [0.25–0.81] | 0.54 [0.31–0.88] | 0.54 [0.36–0.8] | 0.49 [0.34–0.7] | 0.38 [0.13–0.87] |
| 16,256 | 0.54 [0.32–1] | 0.64 [0.41–1.0] | 0.62 [0.4–1.0] | 0.88 [0.64–1.2] | 0.86 [0.45–1.5] |
| 16,260 | 0.21 [0.06–0.48] | 0.28 [0.15–0.43] | 0.26 [0.16–0.44] | 0.47 [0.32–0.67] | 0.19 [0.03–0.6] |
| 16,261 | 0.65 [0.33–1.1] | 0.64 [0.42–1.0] | 0.6 [0.41–0.86] | 0.58 [0.41–0.83] | 1.0 [0.59–1.7] |
| 16,265 | 0.45 [0.22–0.83] | 0.44 [0.28–0.64] | 0.44 [0.31–0.64] | 0.57 [0.4–0.8] | 0.48 [0.19–1] |
| 16,266 | 0.34 [0.13–0.67] | 0.5 [0.25–0.85] | 0.5 [0.3–0.86] | 0.74 [0.53–1.0] | 0.38 [0.13–0.87] |
| 16,270 | 0.48 [0.31–0.7] | 0.32 [0.22–0.5] | 0.29 [0.19–0.43] | 0.23 [0.15–0.37] | 0.24 [0.06–0.67] |
| 16,274 | 0.7 [0.39–1.2] | 0.81 [0.47–1.3] | 0.81 [0.56–1.1] | 1.4 [1.0–1.9] | 0.76 [0.38–1.4] |
| 16,278 | 1.1 [0.7–1.7] | 0.93 [0.55–1.5] | 0.86 [0.6–1.2] | 1.0 [0.75–1.4] | 1.1 [0.66–1.9] |
| 16,290 | 0.17 [0.05–0.42] | 0.3 [0.13–0.52] | 0.31 [0.17–0.52] | 0.42 [0.29–0.61] | 0.38 [0.13–0.87] |
| 16,291 | 0.65 [0.38–1.1] | 0.66 [0.42–0.98] | 0.68 [0.45–0.95] | 0.8 [0.58–1.1] | 1.0 [0.59–1.7] |
| 16,292 | 0.42 [0.22–0.8] | 0.43 [0.25–0.69] | 0.40 [0.25–0.62] | 0.47 [0.33–0.67] | 0.67 [0.31–1.3] |
| 16,293 | 0.31 [0.18–0.59] | 0.31 [0.19–0.55] | 0.29 [0.16–0.46] | 0.44 [0.3–0.63] | 0.76 [0.38–1.4] |
| 16,294 | 0.74 [0.44–1.1] | 0.72 [0.42–1.0] | 0.75 [0.44–1.1] | 0.7 [0.5–0.97] | 0.29 [0.08–0.74] |
| 16,295 | 0.32 [0.13–0.57] | 0.36 [0.23–0.58] | 0.33 [0.21–0.52] | 0.35 [0.23–0.52] | 0.48 [0.19–1] |
| 16,298 | 0.41 [0.23–0.7] | 0.36 [0.23–0.57] | 0.32 [0.22–0.47] | 0.25 [0.16–0.39] | 0.57 [0.25–1.1] |
| 16,304 | 0.49 [0.31–0.79] | 0.4 [0.26–0.68] | 0.4 [0.27–0.59] | 0.41 [0.28–0.6] | 0.57 [0.25–1.1] |
| 16,311 | 2.3 [1.5–3.5] | 2.4 [1.6–3.9] | 2.6 [1.6–5.8] | 2.6 [1.9–3.6] | 2.8 [2–3.8] |
| 16,319 | 0.8 [0.4–1.6] | 0.81 [0.51–1.3] | 0.82 [0.54–1.3] | 0.62 [0.44–0.88] | 0.48 [0.19–1] |
| 16,320 | 0.53 [0.29–0.86] | 0.43 [0.3–0.64] | 0.40 [0.27–0.6] | 0.42 [0.29–0.62] | 0.8 [0.41–1.4] |
| 16,325 | 0.66 [0.28–1.1] | 0.65 [0.43–0.94] | 0.6 [0.38–0.82] | 0.63 [0.45–0.89] | 0.33 [0.10–0.8] |
| 16,355 | 0.41 [0.19–0.73] | 0.45 [0.27–0.75] | 0.46 [0.25–0.77] | 0.6 [0.42–0.84] | 0.38 [0.13–0.87] |
| 16,362 | 2.4 [1.4–4.1] | 2.4 [1.7–3.1] | 2.2 [1.6–3.0] | 2.3 [1.7–3.1] | 1.8 [1.2–2.7] |
| 16,390 | 0.49 [0.25–0.87] | 0.54 [0.33–0.9] | 0.52 [0.33–0.76] | 0.68 [0.48–0.95] | |
| 16,399 | 0.38 [0.20–0.69] | 0.39 [0.24–0.57] | 0.41 [0.25–0.64] | 0.57 [0.4–0.8] | |
| 16,519 | 3.6 [2.4–6.1] | 2.9 [1.9–4.9] | 3.0 [1.7–4.7] | 4.4 [3.1–6.2] | |
| 16,527 | 0.31 [0.11–0.62] | 0.36 [0.21–0.55] | 0.32 [0.24–0.47] | 0.45 [0.31–0.65] | |
Figure 4.—

Graphical representation of mutation rates along HVS-I.
Bias-variance analysis:
To quantify how biased our derived estimates are, we employ the bootstrap–simulation approach we described above. The first step is to decide on a reasonable prior distribution for the mutation rates. To accomplish that, we find the shape parameter α that would be most consistent with the counts of sites that have mutated 0, 1, and >1 times, as described above. The resulting estimate is 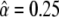 .
.
We then draw a sample of mutation rates from this prior and use the estimated t̂k's from our method (supplemental Table 2) to implement the bias estimation methodology. Figure 5 shows the estimated bias as a function of the true mutation rate for each one of our four estimation settings. The points are means of the estimates from 100 runs of our simulation–bootstrap algorithm, and the lines are LOESS smoothed estimates of the bias (Cleveland et al. 1992). These plots are shown on the log scale; i.e., they represent the ratio of the mutation rate to the bias in its estimates from the different methods. We can observe that the bias has some interesting behavior and no clear consistent pattern (although an obvious tendency to be negative and more pronounced for lower mutation rates). However, encouragingly we can observe that in the region of higher mutation rates that is of interest of us, the bias is almost invariably <0.2 in absolute value on the log scale and therefore no bigger than ∼20% in our rate estimates.
Figure 5.—




Smoothed bias estimation curves for our various estimation protocols, using our simulation–bootstrap hybrid. The smoothing was done using LOESS (Cleveland et al. 1992).
Hypothesis testing:
For hypothesis testing of site independence, we utilized the 87-Hg nomenclature, since the additional information in the more detailed phylogeny is critical for our chances of identifying true dependence. We applied the generalized likelihood-ratio (GLR) test described above to all pairs of sites that are polymorphic in at least 5 of the 87 Hgs—a total of 156 sites, giving us a total of 156 × 155 = 24,180 tests.
Table 2 contains the 10 pairs of sites that gave the lowest P-values for the GLR test and their false discovery rate (FDR)-corrected P-values (Benjamini and Hochberg 1995) (although we used the more powerful FDR scheme, the conclusions would have been the same from using the simple Bonferroni correction). We observe that after the FDR correction, we are left with only three cases where we can reject the site-independence hypothesis at P = 0.05. We now analyze these cases in more detail.
The two-way relationship 16,182 ⇔ 16,183 is by far the strongest nonindependence effect our methodology identifies in our sequences. As we discussed above, it is unclear to what extent sequencing ambiguity persists in this position as a result of its proximity to the poly-C region. However, since most of the mutations we observe in these two sites are between A ⇔ C, i.e., transversions, it seems possible that the poly-C sequence plays a role in creating artificial dependence.
The remaining significant effect is the pair 16,114 ⇒ 16,526. Examining our raw sequences, this significant Hg-level relationship does not seem to follow from easily detectable sequence-level relationships; i.e., we do not observe a consistent tendency for mutations in sites 16,526 and 16,114 to coappear. We therefore lean toward attributing this discovery to chance and not to a real dependence.
So while our hypothesis-testing framework did identify three significant nonindependence relationships in our data, further analysis of these suggests that uncertainty about sequencing issues persists for two of them, while the third is probably due to pure chance.
Our results are encouraging in that they support the validity of site-independence assumptions in analyzing mtDNA HVS-I data. Any dependence that exists is not strong enough to discover with our testing methodology, using our very large database and most detailed (87-Hg) phylogenetic protocol.
Mutation rates at TAS loci:
We compare the estimated mutation rates in the four TAS loci to the rest of HVS-I. We use the standard definition of HVS-I as comprising nucleotides 16,024–16,365 only, given our previous finding that the 16,400–16,500 region has significantly slower mutation rate estimates. We employ two nonparametric tests to quantify the results of these comparisons: the Wilcoxon rank sum test and the Kolmogorov–Smirnov test (Hollander and Wolfe 1973). Table 3 shows the results of our analysis, using the estimates from the 87-Hg protocol. Values in italics indicate significantly lower estimates in the given TAS at level P = 0.05. As we can see, the TAS loci seem to have a tendency toward slightly lower estimated rates than average, but this is most evident in TAS E, which is the only one not showing evidence of protein binding (Roberti et al. 1998). It is not exactly clear what the appropriate multiple-comparison correction to the P-values of the individual tests would be here. A conservative approach, of correcting for the execution of 10 tests, would leave none of our results significant. More relaxed multiple-comparison correction approaches may conceivably conclude that TAS E and/or the entire set of TAS loci combined have a tendency for slightly lower mutation rates than the rest of HVS-I.
TABLE 3.
Statistical analysis of mutation rate estimates in TAS loci, compared to the rest of HVS-I
| TAS site | Positions | Wilcoxon P-value | K–S P-value |
|---|---|---|---|
| TAS E | 16,097–16,107 | 0.020 | 0.013 |
| TAS D | 16,158–16,173 | 0.172 | 0.082 |
| TAS C | 16,305–16,318 | 0.464 | 0.693 |
| TAS A + B | 16,331–16,353 | 0.037 | 0.135 |
| All combined | 0.082 | 0.049 |
After multiple-comparisons correction, arguably none of these results are significant (see text). Values in italics indicate significantly lower estimates in the given TAS at level P = 0.05. K–S, Kolmogorov–Smirnov.
We conclude that, if these loci do contain some patterns whose conservation is critical for replication termination, these patterns are likely to be complex and include dependencies that are not significantly reflected at the individual-site level.
DISCUSSION
We discuss several issues related to the quality of our methodology and our estimates and their usefulness.
Importance of rate estimation:
The mutation dynamics of the human genome in general and mtDNA in particular have experienced a surge of interest in recent years (Torroni et al. 2006). Many articles deal with the real or apparent “slowdown” effect in the molecular clock for older time periods (e.g., Ho et al. 2005). Since we share Bandelt et al.'s (2006) opinion that there is no convincing evidence for a molecular clock slowdown (other than saturation causing these apparent effects), we view this issue as unrelated to our analysis in this article.
Reliable mutation rate estimates are clearly important for several widely accepted reasons. Understanding the function of various regions in the genome and the mutual influence between different regions, which may be caused by either a functional relation or a physical or chemical one, is one of the key challenges of the field of genomics and, indeed, one of the most important scientific questions of our time (Hardison 2003; Hapmap Consortium 2005). Creating a better understanding of the mutation mechanisms and potential dependencies in those is an important step in this process, as it may help to separate nongenic areas that have function (and are therefore preserved) from ones that do not and to discover the relationships between regions within our genome. Our investigation of the TAS loci and the observation that they do not demonstrate the high degree of preservation previously attributed to them is an example of a function-related observation whose validity is tied to the quality of rate estimates available. Our testing (and mostly acceptance) of the site-independence assumption also has potential function-related implications, as it suggests lack of significant interaction between different loci in HVS-I.
Mutation rates can be used to improve phylogeny estimation algorithms and sequence quality checking (Bandelt et al. 2002). It should be clarified, however, that these rates are not very useful for time estimation on known phylogenies. As Rosset (2007) has shown, under a simple substitution model like the one we assume here, the individual rates are of no consequence for time estimation, only their sum. This is a direct consequence of the fact that the sum of independent Poisson random variables is still Poisson distributed. Under more complex models, the individual rates may have a minor effect on time estimates.
We have also recently used our estimates reported here to improve the accuracy of the mtDNA Hg classification protocol in the Genographic Project (Behar et al. 2007).
An interesting aspect of our mutation rate estimation methodology is the estimates we derive of tk, the total length of the coalescent tree of the samples we have in each Hg (it should be reiterated that this is not the TMRCA of the Hg, but the sum of the lengths of all branches in the coalescent tree). These can be used for inference on the age and demographic history of the Hg's. Table 4 gives some estimates of tk, derived from our calculations based on the 87-Hg protocol (the full list is available in supplemental Table 2). Detailed discussion of these results is beyond the scope of this article, but we can clearly see the difference between Hg M* (255 samples, estimate of tk is ∼6 million years) and Hg V (471 samples, estimate of tk is only 1.7 million years), implying that our samples from M* are much more diverse than those from V, a difference that demonstrates the older age of the polyphyletic Hg M* and its more ancient expansion.
TABLE 4.
Coalescent tree size estimates
| Hg | No. samples | Total tree length (yr) |
|---|---|---|
| A | 361 | 4,628,667 |
| B | 301 | 5,624,497 |
| C | 229 | 3,089,925 |
| D | 147 | 2,692,974 |
| H | 6,232 | 36,186,219 |
| M* | 255 | 5,878,315 |
| V | 471 | 1,726,071 |
Haplogroup classification vs. detailed phylogeny:
Most of the approaches for estimating individual mutation rates in HVS-I we mentioned above are based on a reconstruction of the full phylogenetic tree through a ML approach (Excoffier and Yang 1999), quartet puzzling (Meyer and von Haeseler 2003), or maximum parsimony (Bandelt et al. 2006).
In our case, if we were able to obtain a full phylogeny (like in Figure 1B), we would be able to observe the actual mik values (at least up to uncertainty about repeated mutations on tree branches), use Equation 1 for modeling, and most likely get better-quality results than our modeling based on Equation 2. However, the fundamental idea behind our approach is that reliable Hg classification on a tree whose general structure is known (such as the human mtDNA tree) is a much simpler task than identifying the complete phylogeny of a large set of samples. Building detailed phylogenies for large samples presents significant computational and, more importantly, statistical difficulties. The resulting phylogenies may be highly underdetermined and uncertain (Felsenstein 2003). Use of ML methodology like that of Excoffier and Yang (1999) would also require parametric assumptions about the mutation rates.
For example, the data set we use here is composed of 16,609 HVS-I samples of mtDNA. The Hg classification is primarily based on a set of coding-region SNPs and is therefore very reliable. On the other hand, relying on HVS-I to build detailed, reliable phylogenies within Hg's, with hundreds or even thousands of samples per Hg, is an overwhelming task.
A more relevant question might be whether we would gain from having more phylogenetic information, in the form of more detailed SNP-based phylogeny. The qualitative answer is that more detailed phylogeny clearly leads to better estimates and to avoiding the saturation problem that precludes us from using the full data set at once. In our analysis we can see this by considering Table 1 and Figure 6 below. The 87-Hg nomenclature clearly leads to smaller C.I.'s and therefore apparently to better estimates than the 23-Hg nomenclature. However, these estimates are not as reliable due to the (unquantifiable) uncertainty in the 87-Hg classification based on HVS-I rules in addition to coding-region SNPs.
Figure 6.—

Comparison of the estimates (black circles) and confidence intervals from two of our variants and Bandelt et al. (2006). Note that the y-axis is on a logarithmic scale.
Comparison to estimates from the literature:
As mentioned in the Introduction, previous efforts to estimate site-specific rates and dependencies in HVS-I include several that are statistically appealing but use small numbers of samples (Yang 1995; Yang and Wang 1995; Nielsen 1997). More recent efforts were mostly based on phylogenetic reconstruction and counting (Excoffier and Yang 1999; Meyer and von Haeseler 2003; Bandelt et al. 2006).
Of all these, the method of Bandelt et al. (2006) uses by far the most data (873 samples, compared to our 16,609), with extensive manual work on phylogeny reconstruction minimizing the dependence on modeling assumptions and approximations. We therefore compare our estimates to those from Bandelt et al. (2006). Since they used the limited definition of HVS-I as 16,051–16,365, we concentrate on the region that is common to our study and theirs. As can be seen in Table 1, the estimates are similar in spirit. Since the estimates given by Bandelt et al. (2006) are based on mutation counting on a “known” phylogeny, they have a Poisson distribution (Equation 1) under our assumptions on the substitution model. We can thus use standard Poisson inference methodology to build confidence intervals for them (Johnson and Kotz 1969), which we do in Table 1. We also normalize their estimates to be on the same scale as ours, by constraining their sum to be the same as the sum of our estimates for the same range (16,051–16,365). We observe that the confidence intervals from their estimates are slightly smaller than ours for the fastest sites, but get much larger than ours as the rates decrease. For example, if we consider the first four rows in Table 1, we see that in rows 1–3, where the rates are relatively small, the confidence intervals from all variants of our methodology are smaller than those based on Bandelt et al. (2006). In row 4, which corresponds to 16,093, one of the fastest sites in HVS-I (and coincidentally one of the sites where the rate estimate of Bandelt et al. 2006 most disagrees with ours), the confidence interval based on Bandelt et al. (2006) is smaller than those our methods generate. We can infer that our approach, which uses less phylogenetic information but a much larger number of samples overall, has advantages for estimating fast—but not the fastest—sites compared to Bandelt et al. (2006). Qualitatively, our estimates and theirs seem to agree well, and the confidence intervals almost invariably overlap. A graphical representation of the confidence interval relationships in five randomly selected sites can be seen in Figure 6.
Extensions of the methodology:
In this article we have discussed and demonstrated the application of our methodology to single-nucleotide polymorphisms in human mtDNA HVS-I. This is a natural application because these sites are highly polymorphic, large amounts of data are available, and Hg classification is relatively easy to obtain. The natural question is, What other domains would comply with these same conditions?
A simple extension is application to mtDNA HVS-I across species, as long as site-specific mutation rates are assumed similar between species, and we have coding-SNP verified Hg allocation for each species.
A more interesting extension may be to short tandem polymorphisms on the Y chromosome (Y-STRs), which comply with all three conditions. The mutation rates (and, more generally, mechanisms) of these patterns have been under intense study for several years, but progress is difficult to make, unless some highly nonrealistic assumptions are made (for more details, see, for example, Zhivotovsky 2001; Calabrese and Sainudiin 2005). Our methodology would be directly applicable to Y-STR if we could assume that the mutation rate of each Y-STR does not depend on its state (repeat count). This is a slightly more general assumption than the stepwise mutation model, identical, for example, to the STR mutation model assumed by Zhivotovsky (2001). In that case, our approach can be immediately applied to calculate this probability, using the same assumption, that bik = 1 if and only if the STR count is fixed for all samples in an Hg.
The real challenge is to accommodate dependence on count number in the STR mutation model. This can be done by assuming a different rate λil for each site i and count l and defining binomial variables bilk that allow count dependence. The only remaining question is which value l would be used for (site, Hg) combinations where this STR count is polymorphic. Several approaches come to mind, but detailed discussion and experimentation are a topic for future research.
Acknowledgments
We thank the Genographic Project participants who donated their genetic information. This study was supported by the National Geographic Society, IBM, and the Waitt Family Foundation. S.R. is partially supported by European Union grant MIRG-CT-2007-208019; C.T.S. is supported by The Wellcome Trust.
References
- Bandelt, H., L. Quintana-Murci, A. Salas and V. Macaulay, 2002. The fingerprint of phantom mutations in mitochondrial DNA data. Am. J. Hum. Genet. 71(5): 1150–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt, H. J., Q. P. Kong, M. Richards and V. Macaulay, 2006. Estimation of mutation rates and coalescence times: some caveats, pp. 47–90 in Human Mitochondrial DNA and the Evolution of Homo sapiens, edited by H. J. Bandelt, V. Macaulay and M. Richards. Springer, Berlin.
- Behar, D. M., S. Rosset, J. Blue-Smith, O. Balanovsky, S. Tzur et al., 2007. The genographic project public participation mitochondrial DNA database. PLoS Genet. 3(6): e104. [DOI] [PMC free article] [PubMed]
- Benjamini, Y., and Y. Hochberg, 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 57 289–300. [Google Scholar]
- Bickel, P., F. Gotze and W. van Zwet, 1997. Resampling fewer than n observations: gains, losses and remedies for losses. Stat. Sin. 7 1–31. [Google Scholar]
- Calabrese, P., and R. Sainudiin, 2005. Models of microsatellite evolution, Chap. 10 in Statistical Methods in Molecular Evolution, edited by R. Nielsen. Springer, Berlin/Heidelberg, Germany/New York.
- Cleveland, W., E. Grosse and W. Shyu, 1992. Local regression models, Chap. 8 in Statistical Models in S, edited by J. Chambers and T. Hastie. Wadsworth & Brooks/Cole, Belmont, CA.
- Doda, J. N., C. T. Wright and D. A. Clayton, 1981. Elongation of displacement-loop strands in human and mouse mitochondrial DNA is arrested near specific template sequences. Proc. Natl. Acad. Sci. USA 78 6116–6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron, B., and R. Tibshirani, 1994. An Introduction to the Bootstrap. Chapman & Hall/CRC, London/New York.
- Excoffier, L., and Z. Yang, 1999. Substitution rate variation among sites in the mitochondrial hypervariable region i of humans and chimpanzees. Mol. Biol. Evol. 16 1357–1368. [DOI] [PubMed] [Google Scholar]
- Falkenberg, M., M. G. Larsson and C. M. Gustafsson, 2007. Dna replication and transcription in mammalian mitochondria. Annu. Rev. Biochem. 76 679–699. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J., 2003. Inferring Phylogenies. Sinauer Associates, Sunderland, MA.
- Forster, P., R. Harding, A. Torroni and H. Bandelt, 1996. Origin and evolution of native American mtDNA variation: a reappraisal. Am. J. Hum. Genet. 59 935–945. [PMC free article] [PubMed] [Google Scholar]
- Hapmap Consortium, 2005. A haplotype map of the human genome. Nature 437 1299–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardison, R., 2003. Comparative genomics. PLoS Biol. 1(2): e58. [DOI] [PMC free article] [PubMed]
- Ho, S., M. Phillips, A. Cooper and A. Drummond, 2005. Time dependency of molecular rate estimates and systematic overestimation of recent divergence times. Mol. Biol. Evol. 22 1561–1568. [DOI] [PubMed] [Google Scholar]
- Hollander, M., and D. Wolfe, 1973. Nonparametric Statistical Inference. John Wiley & Sons, New York.
- Johnson, N., and S. Kotz, 1969. Discrete Distributions. Houghton Mifflin Company, Boston.
- Jukes, T., and C. Cantor, 1969. Evolution of protein molecules, pp. 21–132 in Mammalian Protein Metabolism, Vol. 3, edited by H. Munro. Academic Press, New York.
- Kimura, M., 1980. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16 111–120. [DOI] [PubMed] [Google Scholar]
- Kimura, M., 1981. Estimation of evolutionary distances between homologous nucleotide sequences. Proc. Natl. Acad. Sci. USA 78 454–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateiu, L., and B. Rannala, 2006. Inferring complex DNA substitution processes on phylogenies using uniformization and data augmentation. Syst. Biol. 55 259–269. [DOI] [PubMed] [Google Scholar]
- Mayrose, I., D. Graur, N. Ben-Tal and T. Pupko, 2004. Comparison of site-specific rate-inference methods for protein sequences: empirical Bayesian methods are superior. Mol. Biol. Evol. 21 1781–1791. [DOI] [PubMed] [Google Scholar]
- McCullagh, P., and J. Nelder, 1989. Generalized Linear Models. Chapman & Hall, London.
- Meyer, S., and A. von Haeseler, 2003. Identifying site-specific substitution rates. Mol. Biol. Evol. 20 182–189. [DOI] [PubMed] [Google Scholar]
- Nielsen, R., 1997. Site-by-site estimation of the rate of substitution and the correlation of rates in mitochondrial DNA. Syst. Biol. 46 346–353. [DOI] [PubMed] [Google Scholar]
- Roberti, M., C. Musicco, P. L. Polosa, F. Milella, M. N. Gadaleta et al., 1998. Multiple protein-binding sites in the TAS-region of human and rat mitochondrial DNA. Biochem. Biophys. Res. Commun. 243 36–40. [DOI] [PubMed] [Google Scholar]
- Rosset, S., 2007. Efficient inference on known phylogenetic trees using Poisson regression. Bioinformatics 23 e142–e147. [DOI] [PubMed] [Google Scholar]
- Sbisa, E., F. Tanzariello, A. Reyes, G. Pesole and C. Saccone, 1997. Mammalian mitochondrial d-loop region structural analysis: identification of new conserved sequences and their functional and evolutionary implications. Gene 205 125–140. [DOI] [PubMed] [Google Scholar]
- Siepel, A., and D. Haussler, 2005. Phylogenetic hidden Markov models, pp. 325–351 in Statistical Methods in Molecular Evolution, edited by R. Nielsen. Springer, Berlin/Heidelberg, Germany/New York.
- Tamura, K., and M. Nei, 1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10 512–526. [DOI] [PubMed] [Google Scholar]
- Torroni, A., A. Achilli, V. Macaulay, M. Richards and H. J. Bandelt, 2006. Harvesting the fruit of the human mtDNA tree. Trends Genet. 22 339. [DOI] [PubMed] [Google Scholar]
- Venables, W., and B. Ripley, 1994. Modern Applied Statistics With S-Plus. Springer, New York.
- Yang, Z., 1993. Maximum likelihood estimation of phylogeny from DNA sequences when substitution rates differ over sites. Mol. Biol. Evol. 10 1396–1401. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1994. Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39 306–314. [DOI] [PubMed] [Google Scholar]
- Yang, Z., 1995. A space-time process model for the evolution of DNA sequences. Genetics 139 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., and T. Wang, 1995. Mixed model analysis of DNA sequence evolution. Biometrics 51 552–561. [PubMed] [Google Scholar]
- Zhivotovsky, L. A., 2001. Estimating divergence time with the use of microsatellite genetic distances: impacts of population growth and gene flow. Mol. Biol. Evol. 18 700–709. [DOI] [PubMed] [Google Scholar]