

Abstract
It has been widely appreciated that natural selection opposes the progress of inbreeding in small populations, thus limiting the actual inbreeding depression for fitness traits. However, no method to account for the consequences of this process has been given so far. I give a simple and intuitive method to predict inbreeding depression, taking into account the increase in selection efficiency against recessive alleles during inbreeding. It is based on the use of a “purged inbreeding coefficient” gt that accounts for the reduction of the probability of the deleterious homozygotes caused by the excess d of detrimental effect for deleterious alleles in the homozygous condition over its additive expectation. It is shown that the effect of purging can be important even for relatively small populations. For between-loci variable deleterious effects, accurate predictions can be obtained using the effective homozygous deleterious excess de, which can be estimated experimentally and is robust against variation of the ancestral effective population size. The method can be extended to any trait and it is used to predict the evolution of the mean viability or fecundity in a conservation program with equal or random family contributions.
INBREEDING depression is the reduction of the value of a trait that occurs in offspring of related parents, and it is caused by an increase in their probability of homozygosity by descent (f). It is one of the genetic properties of populations that is more relevant to breeding and conservation as well as to the cultural evolution of human social rules. Furthermore, it has been invoked as a driving force for the evolution of many biological phenomena, such as breeding systems or diploidy (Charlesworth and Charlesworth 1987; Kondrashov and Crow 1991). For a relatively small population, the average kinship among breeders will increase over time, and there is an obvious interest in predicting the expected reduction of the mean of different traits through generations.
Inbreeding depression occurs when the alleles reducing the expression of the trait tend to be recessive, so that their expression is greater under increased homozygosity. This is a main genetic property of fitness and fitness-related traits, as spontaneous mutations (the ultimate source of genetic diversity) having an appreciable deleterious effect tend to be recessive and, furthermore, because the more recessive the deleterious effect is, the less efficient becomes natural selection in removing it (García-Dorado et al. 2004). Yet, even inbreeding depression for fitness itself is generally predicted using the neutrally expected f value. Of course, for very small effective population sizes, drift is the leading force governing the evolution of gene frequency (excepting lethal alleles) and the neutral approach gives reliable predictions, but this is obviously not the general case, as the neutral prediction for f does not inform on the actual probability of homozygosity by descent for alleles under natural selection. In particular, the increased expression of recessive alleles during inbreeding, which is responsible for inbreeding depression, is also responsible for an increased efficiency of natural selection against them that is known as purge, which tends to reduce the actual inbreeding depression rate. Both inbreeding depression and purge are a natural consequence of this increased expression of recessive alleles, and we here set aside the unknown fraction of inbreeding depression that might be ascribed to overdominant loci.
Significant effects of purging have been repeatedly reported for plants and animals (see the recent survey by Gulisija and Crow 2007) but attempts to empirically derive a predictive approach of its magnitude have proved elusive (Boakes et al. 2007). Furthermore, it has been traditionally considered that the analytical predictions of the consequences of selection during the inbreeding process require a knowledge of the joint distribution of allele frequencies and homozygous and heterozygous deleterious effects, as well as the use of cumbersome mathematical procedures, such as transition matrix methods. Recently, a simpler method was proposed on the basis of results derived for the mutation–selection–drift balance (García-Dorado 2007), which was based on predicting the increased efficiency of natural selection against recessive genes in small populations from the corresponding transient increase of the additive variance ascribed to dominance. Nevertheless, the prediction of the transient increase in additive variance is relatively cumbersome and, furthermore, it involves assumptions leading to some underestimate of the purge. In the cases explored in García-Dorado (2007), underestimation was at most 10%, but analysis of its Appendix B reveals that this underestimate will become larger for very recessive deleterious alleles, which can make substantial contributions to the actual depression.
Here I present a simple and intuitive approach that reliably predicts inbreeding depression for fitness (or for other traits), taking into account the effect of purge. It is based on the idea that purge is caused by the reduction in homozygosity by descent specifically due to the recessive nature of the deleterious effects.
METHODS AND RESULTS
The basic model:
Consider an infinite hermaphrodite panmictic population at the mutation–selection balance that undergoes a reduction of its effective size to a stable value N during a period for which fitness inbreeding depression should be predicted. Assume that for each of the partially recessive alleles m segregating at relatively low frequency q in the ancestral population, the disadvantages for relative fitness for the homozygous and the heterozygous genotype are s and hs, respectively, (fitnesses 1, 1 − hs, and 1 − s for genotypes ++, +m, and mm). Thus, s is the homozygous selection coefficient and h is the coefficient of dominance. On the basis of the heterozygous disadvantage (hs), the additive expectation for the deleterious effect of the deleterious homozygote would be 2hs, so that the deleterious excess of the homozygote over its additive expectation is s − 2hs = 2d, where d = s(1/2 − h) is Falconer's heterozygous genotypic d value for relative fitness (Falconer and Mackay 1996) and can be viewed as the per copy deleterious excess in the homozygous condition. The contribution of this gene to the ancestral rate of fitness inbreeding depression δ, defined as the percentage of fitness decay per 1% increase of the inbreeding coefficient (f) under relaxed selection, is of course 2dq(1 − q). Overall inbreeding depression rate will be predicted ignoring linkage disequilibrium and adding up predictions over loci.
Ancestral recessive alleles, which were held at low frequencies by selection in the original population, became increasingly expressed during the period of reduced size due to their increased probability of homozygosity. This causes inbreeding depression as well as purge, which can be defined as the increase in selection against deleterious alleles that is due to inbreeding. To predict the consequences of purge, we define a purged inbreeding coefficient gt that accounts for the accumulated purge of the excess of deleterious homozygotes. Due to purge, each generation, the excess of the frequency of deleterious copies that are in homozygous condition after natural selection is (1 − d/wt) times that before selection, where wt is the average fitness at the generation considered. As wt is usually close to 1, we assume (1 − d/wt) ≈ (1 − d), although this can lead to a slight underestimate of the purge in some instances. As usual, when generation t + 1 is obtained through panmixia, any given deleterious copy can become homozygous by descent with probability 1/2N by joining a copy of the same individual allele at generation t, or by joining a copy of a different individual allele, sampled with probability (1 − 1/2N) from adults surviving natural selection at generation t, that happens to be identical by descent to it. Therefore, the purged inbreeding coefficient of the deleterious allele undergoes each generation a reduction by a (1 − d) factor due to purge and an increase at a rate 1/2N due to the finite population number, so that
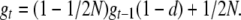 |
This is a simple equation in differences with solution
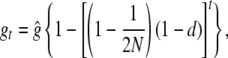 |
(1) |
where
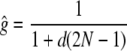 |
(2) |
is the equilibrium value that gt approaches as t increases, which of course goes to 1 as d goes to 0. Note that Equation 1 can also be written as 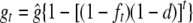 , where ft = 1 − (1 − 1/2N)t is the standard inbreeding coefficient.
, where ft = 1 − (1 − 1/2N)t is the standard inbreeding coefficient.
Therefore, if ΔWt is the fitness decay from generation t to t + 1 and decays were additive over generations, the overall inbreeding depression up to generation t would be approximately
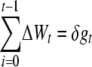 |
(3) |
instead of the classical prediction 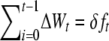 .
.
More realistic predictions:
In a more realistic situation, the ancestral population will be finite with effective size N0, so that it could be assumed to be at the mutation–selection–drift (MSD) balance, rather than at the mutation–selection balance. We use a shortcut approximation to infer the corresponding ancestral inbreeding depression rate (García-Dorado 2007). This simple approach considers that, at the equilibrium, the rate of increase of heterozygosity over the whole genome ΔΣ(pq) (where p = 1 − q and the summation is over the loci that can mutate to deleterious alleles) should be zero, so that the increase from mutation should be canceled out by the rate of loss due to drift and by the rate of elimination due to selection:
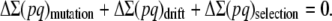 |
For relatively small frequency of the deleterious alleles
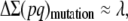 |
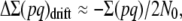 |
and
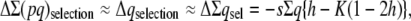 |
where K = Σq2/Σq is the proportion of deleterious copies that undergo selection in the homozygous condition, and it can be computed as 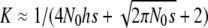 (García-Dorado 2007). Therefore, at the MSD balance
(García-Dorado 2007). Therefore, at the MSD balance 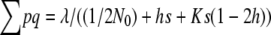 , where λ is the rate of deleterious mutation per gamete and generation, and the ancestral inbreeding depression rate over the whole genome can be readily computed as
, where λ is the rate of deleterious mutation per gamete and generation, and the ancestral inbreeding depression rate over the whole genome can be readily computed as
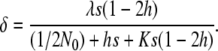 |
(4) |
At this ancestral equilibrium, selection would compensate for the corresponding mutational input and inbreeding depression, except for the ancestral rate of fitness decline from fixation of deleterious alleles. However, we consider that the reduction in effective size is large enough that the ancestral per-generation inbreeding depression and the ancestral fitness decline from deleterious fixation can be neglected.
Furthermore, spontaneous mutation continues to occur at the same rate during the period of reduced size, so that the population genetic architecture will change from its ancestral equilibrium to a new mutation–selection–drift balance as f approaches 1. At this new balance, selection against newly arisen deleterious mutations cancels out the fitness decline both from the new mutational input and from inbreeding depression, except for the decay due to the corresponding rate of deleterious fixation, which we denote by Dm. Therefore, for any given ft value, the rate of decline from deleterious fixation will be Dm at a population fraction ft in which all the variation is due to new mutation that occurred during this inbreeding process. Thus, adding the rates of fitness decline up to generation t during the period of reduced population size gives
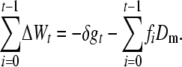 |
Since 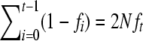 and
and 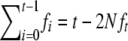 , the above expression can be rewritten as
, the above expression can be rewritten as 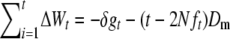 .
.
Regarding fitness traits, it is more realistic to consider a multiplicative model, where the fitness decay per generation is relative to the standing mean fitness average at that generation. Then, mean fitness at generation t relative to that in the ancestral population can be predicted as 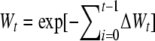 , i.e., as
, i.e., as
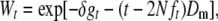 |
(5) |
where the rate of fitness decline from deleterious mutation in the new MSD balance is Dm = 2NλU, in which U is the ultimate fixation probability of a deleterious mutation and can be computed from diffusion theory. It is worthwhile to note that the analytically tractable additive prediction computed using the homozygous deleterious effect (U = [1 − exp(2Nsq0)]/[1 − exp(2Ns)], with q0 = 1/2N) (Kimura 1969) is quite reliable even for recessive deleterious mutations (results not shown).
For moderate values of the new effective population size, the term (t − 2Nft)Dm can also be neglected in the short to medium term, and we can use
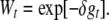 |
(6) |
Between-loci variable deleterious effects:
Finally, we must consider that h and s values are variable across loci, so that exact prediction implies the integration of expression (5) or (6) over the joint distribution of h and s for spontaneous deleterious mutation. When this distribution is unknown, Equation 5 (or Equation 6 if a short period is considered) should allow the inference of an effective d value (de) by assaying the association of purge with the effective population size. This de value could then be used to obtain approximated predictions from Equation 5 for different effective population sizes and generation numbers. The accuracy of this approximation is checked later.
Extension to populations maintained under equal family contribution:
The above approach can be readily extended to a population where genetic management is used to equalize the number of offspring contributed by the different parents to the next generation. This is a common strategy in ex situ conservation programs, as its immediate consequence is to approximately double the effective population size, so that inbreeding is slowed (Frankham et al. 2002.). We denote by ftE = 1 − (1 − 1/4N)t the inbreeding coefficient at generation t under this strategy. The method, however, relaxes natural selection upon fecundity traits and reduces natural selection for viability to that occurring within families.
The expected mean fecundity after t generations given by García-Dorado (2007) can be rewritten as
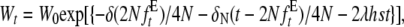 |
(7) |
where δN is the expected rate of inbreeding depression at the neutral mutation–drift balance that is finally attained after relaxed selection for fecundity caused by equal family contribution (δN = 4Nλs(1− 2h)).
For viability, only half of the available additive variance is used under equal family contribution (EC), so that the effective homozygous deleterious effect is 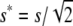 and, since effective population size is doubled, the ultimate deleterious fixation rate becomes smaller than under no management (NM). Furthermore, this affects the purged inbreeding coefficient, which becomes
and, since effective population size is doubled, the ultimate deleterious fixation rate becomes smaller than under no management (NM). Furthermore, this affects the purged inbreeding coefficient, which becomes 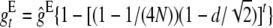 ,
, 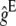 being the equilibrium value for gt under equal family contribution (
being the equilibrium value for gt under equal family contribution (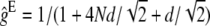 ). It also affects nonpurging selection as, according to Fisher's theorem, the per-generation increase in fitness due to selection in a hypothetical infinite ancestral population under no management amounts to its fitness additive variance. At the equilibrium, this increase should equal the per generation mutational decay (2λhs). However, if equal family contribution is established in this hypothetical infinite population, natural selection is limited to the half part of the fitness additive variance that is expressed within families (λhs), thus compensating for just half of the mutational decay, so that mean fitness would decline, at an initial rate λhs, until a new equilibrium is attained with twofold additive variance and a larger segregating load. Therefore, during the period of reduced size with equal contribution, we can approximately assume that at generation t, the (1 − ftE) panmictic fraction of the ancestral population suffers a λhs decline. Adding up over generations, the overall fitness decline from this source up to generation t gives
). It also affects nonpurging selection as, according to Fisher's theorem, the per-generation increase in fitness due to selection in a hypothetical infinite ancestral population under no management amounts to its fitness additive variance. At the equilibrium, this increase should equal the per generation mutational decay (2λhs). However, if equal family contribution is established in this hypothetical infinite population, natural selection is limited to the half part of the fitness additive variance that is expressed within families (λhs), thus compensating for just half of the mutational decay, so that mean fitness would decline, at an initial rate λhs, until a new equilibrium is attained with twofold additive variance and a larger segregating load. Therefore, during the period of reduced size with equal contribution, we can approximately assume that at generation t, the (1 − ftE) panmictic fraction of the ancestral population suffers a λhs decline. Adding up over generations, the overall fitness decline from this source up to generation t gives 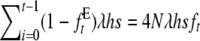 (a more formal demonstration can be found in García-Dorado 2007).
(a more formal demonstration can be found in García-Dorado 2007).
Therefore, taking into account partially relaxed selection, inbreeding depression, purge, and new mutation, the expected fitness at generation t is
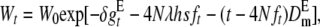 |
(8) |
where
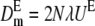 |
is the rate of viability decline due to deleterious fixation in the new balance, which is obtained using 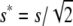 and N* = 2N to compute the corresponding deleterious fixation probability UE.
and N* = 2N to compute the corresponding deleterious fixation probability UE.
Extension to other traits:
The above approach can also be used to predict inbreeding depression for other traits different from fitness. Thus, for a given trait X in a population for which the effective size has been reduced to N, the overall inbreeding depression by generation t can also be computed using Equation 3, where, in this case, δ is the ancestral inbreeding depression rate for X (computed using the d value for the trait), while gt is computed using the d values for fitness for the loci responsible for the inbreeding depression of X.
Checking the predictions:
To illustrate the importance of the reduction in fitness inbreeding depression due to purge and to check the precision of our predictions, I computed the expected average fitness given by Equation 5 using a wide range of mutation rates and of distributions of mutational effects. The expected mean fitness under equal family contribution is also computed assuming that selection occurs through fecundity or viability (Equations 7 or 8, with W0 = 1). For simplicity, Dm and 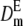 are computed using the homozygous deleterious effect s to determine the approximated final fixation probability from the corresponding diffusion results for the additive case (Kimura 1969), which has been shown to give reasonable predictions for recessive deleterious mutations (results not shown). Therefore, we use U = [1 − exp(2Nesq0)]/[1 − exp(2Nes)], with q0 = 1/2N and Ne = N to compute Dm or with
are computed using the homozygous deleterious effect s to determine the approximated final fixation probability from the corresponding diffusion results for the additive case (Kimura 1969), which has been shown to give reasonable predictions for recessive deleterious mutations (results not shown). Therefore, we use U = [1 − exp(2Nesq0)]/[1 − exp(2Nes)], with q0 = 1/2N and Ne = N to compute Dm or with 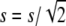 and Ne = 2N to compute
and Ne = 2N to compute 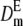 . The classical neutral predictions computed as Wt = exp[−δft ] are also given.
. The classical neutral predictions computed as Wt = exp[−δft ] are also given.
Studied cases have been chosen to allow comparison to the simulation results published by Fernández and Caballero (2001). Therefore, simulation results come from their Table 1, where fitness averages for the NM strategy (their R strategy) are those for natural selection that occurred through fecundity. The reason is that, in the case where selection occurred through viability, the simulation program computed averages after natural selection each generation, while our equations predict average fitness after panmixia but before selection. Both in the simulation procedure and in the predictions, s is assumed to be gamma distributed and, for each s value, h is uniformly distributed between zero and exp[−ks], where k is adjusted to obtain the desired average h value. In each case, predictions from Equations 5, 7, and 8 are averaged over 104 mutations sampled from the corresponding distributions for s and h.
TABLE 1.
Average fitness after 50 generations with population size N for different mutational models and management strategies
| Mean fitness
|
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| δ MSD (N0)b | NMe
|
EC-Vf
|
EC-Fg
|
||||||||||||
| Case | N0 | δ MSa | N | λ | E(s)c | Shaped | E(h)c | P, classicalh | P, Eq. 5i | Sj | P, Eq. 8i | Sj | P, Eq. 7i | Sj | |
| a | 500 | 3.75 | 3.86 | 25 | 2 | 0.01 | 0.07 | 0.35 | 0.086 | 0.22 | 0.24 | 0.38 | 0.38 | 0.20 | 0.19 |
| b | 1,000 | 3.82 | 3.90 | 25 | 1 | 0.025 | 0.25 | 0.35 | 0.084 | 0.27 | 0.34 | 0.39 | 0.40 | 0.16 | 0.15 |
| c | 2,000 | 1.65 | 1.63 | 25 | 0.5 | 0.05 | 1 | 0.35 | 0.355 | 0.51 | 0.51 | 0.52 | 0.52 | 0.27 | 0.23 |
| d | 10,000 | 0.74 | 0.71 | 25 | 0.1 | 0.1 | 0.5 | 0.35 | 0.637 | 0.86 | 0.88 | 0.84 | 0.86 | 0.62 | 0.59 |
| e | 25,000 | 0.19 | 0.19 | 25 | 0.03 | 0.264 | 2.3 | 0.35 | 0.886 | 0.97 | 0.98 | 0.89 | 0.95 | 0.72 | 0.70 |
| h | 25,000 | 0.51 | 0.48 | 25 | 0.03 | 0.264 | 2.3 | 0.20 | 0.737 | 0.94 | 0.97 | 0.92 | 0.95 | 0.72 | 0.72 |
| i | 2,000 | 1.66 | 1.64 | 100 | 0.5 | 0.05 | 1 | 0.35 | 0.695 | 0.82 | 0.84 | 0.66 | 0.69 | 0.43 | 0.47 |
| j | 25,000 | 0.50 | 0.47 | 100 | 0.03 | 0.264 | 2.3 | 0.20 | 0.901 | 0.98 | 0.98 | 0.94 | 0.96 | 0.84 | 0.83 |
Ancestral inbreeding depression for simulated data computed under mutation–selection balance using Equation 6.2.6 in Crow and Kimura (1970, p. 260).
Ancestral inbreeding depression for the MSD balance for N0 computed from Equation 4.
E stands for expectation.
Shape parameter for the gamma distribution of homozygous deleterious effects s.
No management.
Equal family contribution with natural selection through viability.
Equal family contribution with natural selection through fecundity.
Classical neutral prediction obtained as exp[−δft ], using the MSD δ-value.
Predictions from the specified equation (underlined).
Simulation results from Fernández and Caballero (italics).
In their simulation procedure, Fernández and Caballero started from frequencies corresponding to the deterministic equilibrium between recurrent mutation and selection for a set of 5800 linked loci spread over 20 M, and they simulated natural selection and nonrecurrent mutation during 50 generations with 25 or 100 breeding individuals. To test for the robustness of predictions, we start from García-Dorado's approach to the mutation–selection–drift (MSD) balance for the same mutational parameter, using Equation 4 to compute δ. To obtain predictions comparable to simulation results published by Fernández and Caballero, we adjust the ancestral population size to obtain the ancestral inbreeding depression rate corresponding to the mutation–selection balance they used.
Table 1 presents the predictions together with the published simulation results for a wide range of mutational models. Under no management, our predictions from Equation 5 closely fit the corresponding simulation results, and comparison to predictions from the classical neutral approach show that the effect of purging is generally very important. Furthermore, these predictions are more accurate than those based on the approach previously given by García-Dorado (2007). In that article, comparison was restricted to case c (for which Fernandez and Caballero gave the behavior of mean fitness components during 50 generations), and a downward bias up to 10% was reported for mean viability. However, averaging over all the cases considered in Table 1, that approach underestimates mean fitness by 21% at generation 50 (results not shown), while the average underestimate with the present approach is <5%. Results from Equations 7 and 8 for the evolution of the mean under EC also predict quite accurately the corresponding simulation values.
DISCUSSION
In a recent article (García-Dorado 2007), simple equations were proposed to compute the main fitness genetic properties at the MSD balance, and a method was developed to predict the behavior of the fitness mean and additive variance through a period of reduced population size, during which purge was computed from the increase in this additive variance ascribed to dominance. Although the inferences regarding the MSD balance were accurate, that for the increase in additive variance after population size reduction was downwardly biased for substantially recessive deleterious allele, resulting in a similar bias for the effect of purge. Here I present a simple and intuitive prediction for inbreeding depression with purge that does not rely upon the prediction of the additive variance. It can be computed from an estimable parameter and can be extended to any trait. The prediction of the additive variance will be addressed elsewhere.
It has been usually accepted that inbreeding depression from deleterious alleles of large effect should be more efficiently purged than that due to mildly deleterious alleles (Hedrick 1994; García-Dorado 2003). Our approach shows that the critical parameter determining the purge is not the homozygous deleterious effect but its excess above the corresponding additive expectation. According to this approach, the actual inbreeding depression can be computed using the purged inbreeding coefficient (gt) for deleterious alleles, which accounts for the reduction of their probability of homozygosis by descent caused by the excess d of their homozygous deleterious effect.
Therefore, alleles whose deleterious effect is important but only slightly recessive may be less efficiently purged than other alleles with smaller but more recessive deleterious effect although, obviously, the former should have lower frequencies in the ancestral population. Furthermore, for a given ancestral rate of inbreeding depression δ, the actual inbreeding depression will be smaller when δ is due to fewer deleterious alleles with larger d values segregating at lower initial frequency.
The causes of the different results obtained under different strategies in Table 1 are obscured by the multiple differences between the mutational models. However, the general behavior of the mean fitness can be explained considering that (i) the per locus inbreeding depression rate at the ancestral balance increases for small h values, (ii) the efficiency of purge increases with d, and (iii) the rate of decline due to mutation when selection is relaxed is λs. Therefore, our equations predict that, in the short to medium term, EC results in higher fitness compared to NM when most deleterious alleles contribute to δ, having small h values but d values low enough to not induce important purge. In this scenario, the decline from new mutation under EC is small compared to the actual depression under NM. In fact, when selection acts through viability, EC is at an advantage until its larger segregating load outweighs its slowed inbreeding. The corresponding advantage for fecundity is usually smaller (or nonexistent), lasting for fewer generations and becoming irreversible as deleterious fixation progresses. This fitness advantage is more important if the ancestral population was very large and if the new size is quite small (results not shown).
The predictions obtained here imply that purge can become very relevant even for quite small populations and should be taken into consideration in conservation programs. However, purge did not account for an increase in fitness above the ancestral noninbred value, supporting the current advice discouraging deliberate inbreeding (Hedrick and Kalinowski 2000), at least when this is achieved using a small panmictic population. An obvious development of this approach should be its extension to predict inbreeding depression for pedigree data, where the probability of being homozygous by descent for recessive deleterious alleles may also be reduced by a factor d at each pedigree knot.
This approach relies on the assumption that the ancestral population was at equilibrium between mutation and selection or between mutation, selection, and drift. If the ancestral population contains an excess of copies of deleterious alleles compared to the equilibrium case, an increase in fitness might occur even in the absence of any reduction in effective population size, due to natural selection using up the corresponding excess of fitness additive variance. This excess in natural selection should also be added to predict the fitness changes after a reduction in population size, but it cannot be considered part of the purge, a concept that has been generally used to describe the increase in selection efficiency against recessive alleles due to the increased probability of homozygous genotypes during bottlenecking or inbreeding.
The predictions presented here are accurate and can be readily computed in terms of the ancestral inbreeding depression rate δ, the effective population size N, and the excess in homozygous disadvantage d. To be precise, the joint distribution of δ and d contributed by each segregating locus should be known. A way to achieve this is by averaging the predictions based on the equilibrium inbreeding depression rate inferred for the ancestral population over the distribution of mutational deleterious effects. Unfortunately, only extrapolated mutational parameters would be generally available. Fortunately, accurate predictions can be obtained from the above equations in terms of overall δ and of the “effective homozygous deleterious excess de,” which we denote by de and that can be defined as the d value accounting for the purging observed under specified conditions. Both δ and de can be experimentally estimated by assaying depression in at least two different situations. Ideally, one situation could be a severe bottleneck not allowing for relevant purging, from which δ could be estimated. The other situation should provide an estimate of the actual depression for an effective size large enough to allow for purge, but small enough to cause substantial inbreeding within a reasonable timescale. Figure 1 gives predictions obtained from Equation 6 for models c and h, using the actual distribution of s and h (thick line) or using de values that were fitted to account for the mean fitness that was predicted after five generations with effective size 10 using the actual s, h distribution (de = 0.04 for c and de = 0.19 for h, thin line in Figure 1). In both cases, this de value gives accurate predictions for mean fitness over a wide range of effective sizes (4, 10, and 25) and generation numbers (up to 50), to the point that the thick and thin lines often overlap. Of course, practical applicability of this procedure depends on the precision of the estimates of δ and de, which may be improved if more than two situations are involved in the estimate. An obvious task is to develop estimating procedures for de that make optimal use of all the information available concerning inbreeding depression.
Figure 1.—

Mean fitness predictions without purge and using d or de to predict purge. Predicted mean fitness obtained from Equation 6 by using the distribution of s and h (thick line) or by using the effective homozygous deleterious excess de computed to fit the above mean fitness prediction after five generations with N = 10 (thin line) is shown. Classical neutral predictions are also given for comparison (dotted line).
It should also be taken into consideration that the ancestral inbreeding depression rate δ is sensitive to the size of the ancestral populations, so that extrapolation of estimates may not be appropriate unless ancestral effective numbers are similar. However, it is interesting to note that de seems to be quite robust against N0. For example, for cases c and h, we have used the de values computed for the ancestral population size used for Table 1 and Figure 1 (de = 0.04 computed for case c with N0 = 2000 and de = 0.19 for case h with N0 = 25,000) to compute inbreeding depression with purge using Equation 6 for several ancestral effective sizes in each case. For case c, δ ranged from 0.81 to 4.50 when N0 ranged from 2 × 102 to 2 × 106, and for case h, δ ranged from 0.19 to 0.76 when N0 ranged from 2.5 × 102 to 2.5 × 106. Over these ranges for N0, the predictions obtained using the above de (estimated for the N0 in Table 1) together with the δ appropriate to the N0 considered are, in all cases (N ranging from 4 to 25 during the inbreeding process), as accurate as in Figure 1. In the future, it will be worthwhile to inquire into the relationship of de with the joint distribution of s and h for new mutations and to explore what can be inferred about the genetic architecture of fitness from empirical estimates of de and δ.
The approach given here provides simple predictions for the effect of purge on the actual inbreeding depression for fitness and quantitative traits during a period of reduced population size. These predictions can be computed as a function of parameters that can be empirically estimated and are relevant on evolutionary and applied grounds. This approach could allow one to introduce purge into the theory analyzing the consequences of inbreeding depression on the evolution of main biological features, as population breeding systems. Furthermore, it allows one to predict inbreeding depression for traits different from fitness and should improve the efficiency of genetic breeding programs. Finally, but of particular importance, it shows that purge can become very relevant for the evolution of fitness even in quite small populations. Therefore, although deliberate inbreeding is not advised, the present approach should allow one to include purging into the prospect of in situ scenarios for endangered populations or in the evaluation of breeding strategies in ex situ conservation programs, allowing one to devise conservation strategies in a much more realistic genetic context.
Acknowledgments
I am grateful to Armando Caballero, Antonio Cuevas, and Carlos López-Fanjul for helpful suggestions. This work was supported by grant CGL2008-02343/BOS from the Ministerio de Ciencia e Innovación.
References
- Boakes, E., J. Wang and W. Amos, 2007. An investigation of inbreeding depression and purging in captive pedigreed populations. Heredity 98 172–182. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D., and B. Charlesworth, 1987. Inbreeding depression and its evolutionary consequences. Annu. Rev. Ecol. Syst. 14 237–268. [Google Scholar]
- Crow, J. F, and M. Kimura, 1970. An Introduction to Population Genetics Theory. Harper & Row, NY.
- Falconer, D. S., and T. Mackay, 1996. Introduction to Quantitative Genetics, Ed. 4. Longman, Essex, England.
- Fernández, J., and A. Caballero, 2001. Accumulation of deleterious mutations and equalization of parental contributions in the conservation of genetic resources. Heredity 86 480–488. [DOI] [PubMed] [Google Scholar]
- Frankham, R., J. D. Ballou and D. A. Briscoe, 2002. Introduction to Conservation Genetics. Cambridge University Press, Cambridge, UK.
- García-Dorado, A., 2003. Tolerant versus sensitive genomes: the impact of deleterious mutation on fitness and conservation. Conserv. Genet. 4 311–324. [Google Scholar]
- García-Dorado, A., 2007. Shortcut predictions for fitness properties at the MSD balance and for its build-up after size reduction under different management strategies. Genetics. 176 983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Dorado, A., C. López-Fanjul and A. Caballero, 2004. Rates and effects of deleterious mutations and their evolutionary consequences, pp. 20–32 in Evolution: From Molecules to Ecosystems, edited by A. Moya and E. Font. Oxford University Press, Oxford.
- Gulisija, D., and J. F. Crow, 2007. Inferring the purging from pedigree data. Evolution 61 1043–1051. [DOI] [PubMed] [Google Scholar]
- Hedrick, P. W., 1994. Purging inbreeding depression and the probability of extinction: full-sib mating. Heredity 73 363–372. [DOI] [PubMed] [Google Scholar]
- Hedrick, P. W., and S. T. Kalinowski, 2000. Inbreeding depression in conservation biology. Annu. Rev. Ecol. Syst. 31 139–162. [Google Scholar]
- Kimura, M., 1969. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashov, A. S., and J. F. Crow, 1991. Haploidy or diploidy: Which is better? Nature 351 314–315. [DOI] [PubMed] [Google Scholar]