

Abstract
Dicer-substrate small interfering RNAs (DsiRNAs) are synthetic RNA duplexes that are processed by Dicer into 21-mer species and show improved potency as triggers of RNA interference, particularly when used at low dose. Chemical modification patterns that are compatible with high potency 21-mer small interfering RNAs have been reported by several groups. However, modification patterns have not been studied for Dicer-substrate duplexes. We therefore synthesized a series of chemically modified 27-mer DsiRNAs and correlated modification patterns with functional potency. Some modification patterns profoundly reduced function although other patterns maintained high potency. Effects of sequence context were observed, where the relative potency of modification patterns varied between sites. A modification pattern involving alternating 2′-O-methyl RNA bases was developed that generally retains high potency when tested in different sites in different genes, evades activation of the innate immune system, and improves stability in serum.
Introduction
RNA interference (RNAi) is a natural pathway in mammalian cells where double-stranded RNAs (dsRNAs) suppress expression of genes with complementary sequence (Hannon and Rossi, 2004; Meister and Tuschl, 2004). Long dsRNAs are processed by the RNase-III class nuclease Dicer into 21–23 bp products having 2-base 3′-overhangs and 5′-phosphates (Bernstein et al., 2001). These species, called small interfering RNAs (siRNAs), are the actual molecular triggers of RNAi and direct target specificity of the RNA-induced silencing complex (RISC) (Elbashir et al., 2001). Synthetic 21-mer RNA duplexes can be introduced into cells and will function as mimics of the natural siRNAs that result from Dicer processing. Different RNA structures can also be used to suppress gene expression via RNAi. For example, short hairpin RNAs that have a 19–21 base duplex domain and a 4–9 base loop can be effective triggers of RNAi. Another approach is to use longer synthetic linear RNA duplexes that are substrates for Dicer. These longer duplexes (typically 25–30 bp length) are processed by Dicer into 21-mer siRNAs, enter RISC, and trigger RNAi. Synthetic Dicer-substrate RNAs can have increased potency when compared with 21-mer duplexes, possibly owing to effects relating to linkage of Dicer processing and RISC loading (Kim et al., 2005).
SiRNAs are routinely used today as an experimental tool to suppress gene expression in vitro and many thousands of research reports have been published using RNAi in cell culture. SiRNAs are also being used in vivo for investigational purposes and are under development as therapeutics (BEHLKE, 2006). To realize the full potential of siRNAs in vivo, it may be necessary to chemically modify the RNA duplexes to improve nuclease stability, alter pharmacokinetics, and prevent recognition by the innate immune system. Even though dsRNAs are more stable than single-stranded RNAs (ssRNAs), siRNAs are still rapidly degraded in serum and can benefit from chemical modification. A wide variety of novel chemical modifications have been developed and tested for use in nucleic acids applications such as antisense (AS) and ribozyme technologies (KURRECK, 2003). Many of these modifications may have similar utility for use in siRNAs.
Although extensive studies have been published relating to chemical modification of 21-mer siRNAs, similar studies exploring the modification patterns compatible with function of Dicer-substrate RNAs have not been reported. We previously described optimization of unmodified RNA substrates for human Dicer (Rose et al., 2005). A series of chemically modified Dicer-substrate small interfering RNAs (DsiRNAs) starting from this basic design were synthesized using combinations of RNA, 2′-O-methyl (2′OMe) RNA, 2′-fluoro (2′-F) RNA, and DNA bases and were compared for functional potency in knockdown of endogenous targets or reporter constructs in vitro. Some of the modification patterns retained similar high potency compared with the unmodified parent DsiRNA, whereas other patterns showed reduced potency or were wholly inactive. The functional behavior of chemically modified siRNAs can vary with sequence context, so some of the DsiRNA modification patterns were tested against different endogenous genes as well as reporter constructs to assess to what extent correlations between potency and specific modification patterns might be sequence specific. A modification pattern that includes use of 2′OMe modified bases was identified that generally had minimal impact on potency at multiple sites tested. The DsiRNAs made with this modification pattern did not trigger immune responses in human immune cells in vitro and showed improved serum stability.
Materials and Methods
Chemically synthesized siRNAs
All RNA oligonucleotides described in this study were synthesized using t-Butyl-dimethylsilyl (TBDMS) chemistry and purified using HPLC (Integrated DNA Technologies, Coralville, IA). All oligonucleotides were QC tested by electrospray-ionization mass spectrometry (ESI-MS) and were within ±0.02% predicted mass. Duplexes were also QC tested by analytical HPLC and were >90% pure. Final duplexes were prepared as sodium salts.
In vitro dicing assays
RNA duplexes (100 pmol) were incubated in 20 μL of 20 mM Tris pH 8.0, 200 mM NaCl, 2.5 mM MgCl2 with or without 1 unit of recombinant human Dicer (Stratagene, La Jolla, CA) at 37°C for 18–24 hours. Samples were desalted using a Performa SR 96-well plate (Edge Biosystems, Gaithersburg, MD). Electrospray-ionization liquid chromatography mass spectroscopy (ESI-LCMS) of duplex RNAs pre- and post-treatment with Dicer was done using an Oligo HTCS system (Novatia, Princeton, NJ; Hail et al., 2004), which consisted of a ThermoFinnigan TSQ7000, Xcalibur data system, ProMass data processing software and Paradigm MS4 HPLC (Michrom BioResources, Auburn, CA). All dicing experiments were performed at least twice.
HeLa cell culture, transfections, qRT-PCR, and western blots
HeLa cells were split into 24-well plates at 40% confluency and were transfected the next day with TriFECTin (Integrated DNA Technologies, Coralville, IA) using 1 μL per 100 μL OptiMEM I (Invitrogen, Carlsbad, CA) with RNA duplexes at the indicated concentrations. All transfections were performed three times or more. RNA was prepared 24 hours after transfection using the SV96 Total RNA Isolation Kit (Promega, Madison, WI). RNA quality was verified using a Bioanalyzer 2100 (Agilent, Palo Alto, CA) and cDNA was synthesized using 150 ng total RNA with SuperScript-II Reverse Transcriptase (Invitrogen, Carlsbad, CA) per the manufacturer's instructions using both random hexamer and oligo-dT priming.
Quantitative real-time PCR assays were done using 10 ng cDNA per 10 μL reaction, Immolase DNA polymerase (Bioline, Randolph, MA), 200 nM primers, and 200 nM probe. Hypoxanthine phosphoribosyltransferase (HPRT1) (NM_000194) specific primers were HPRT-For 5′ GACTTTGCTTTCCTTGGTCAGGCA, HPRT-Rev 5′ GGCTTATATCCAACACTTCGTGGG and probe HPRT-P 5′ FAM-ATGGTCAAGGTCGCAAGCTTGCTGGT-IowaBlackFQ (IBFQ). STAT1 (NM_007315) specific primers were STAT1-For 5′ AACCGCATGGAAGTCAGGTT, STAT1-Rev 5′ ATGGGCTTCATCAGCAAGGA, and probe STAT1-P 5′ FAM-AGCTCTCACTGAACCGCAGCAGGAA-IBFQ. Cycling conditions employed were 95°C for 10 minutes followed by 40 cycles of 2-step PCR with 95°C for 15 seconds and 60°C for 1 minute. The PCR and fluorescence measurements were done using an ABI Prism 7900 Sequence Detector (Applied Biosystems Inc., Foster City, CA). All data points were performed in triplicate. Expression data were normalized to levels of an internal control gene, human acidic ribosomal phosphoprotein P0 (RPLP0; NM_001002), which was measured in separate wells in parallel using primers RPLP0-For 5′ GGCGACCTGGAAGTCCAACT, RPLP0-Rev 5′ CCATCAGCACCACAGCCTTC, and probe RPLP0-P 5′ FAM-ATCTGCTGCATCTGCTTGGAGCCCA-IBFQ (Bieche et al., 2000). Copy number standards were run in parallel using linearized cloned amplicons for both the HPRT and RPLP0 assays. Unknowns were extrapolated against standards to establish absolute quantitative measurements.
Total protein was extracted from HeLa cells using Radioimmunoprecipitation buffer (RIPA) buffer (50 mM Tris–HCl pH 7.4, 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, protease Inhibitor Cocktail [Sigma-Aldrich, #P8340]), and centrifuged. Aliquots of 15μg of protein for each sample were run on a 10% polyacrylamide gel with 0.1% sodium dodecyl sulfate (SDS–PAGE) and transferred to a PVDF membrane (Bio-Rad, Hercules, CA). After blocking with 5% powdered milk, the membrane was incubated with the primary antibody (1 hour), washed with TBST (10 mM Tris pH 8, 150 mM NaCl, 0.1% Tween-20), incubated with the secondary horseradish-peroxidase-conjugated antibody (1 hour) and washed again with TBST. The detection reaction was prepared with SuperSignal West Dura Extended Duration Substrate (Pierce, Rockford, IL), and the protein bands were visualized using a Kodak Image Station 440CF (Kodak, Rochester, NY). For quantification of band intensities, Kodak 1D 3.6 software was used. Band densities were averaged from three separate experiments (three separate transfection wells run on separate lanes) and normalized to internal β-actin controls. The antibodies used were anti-HPRT (ab10479, Abcam, 1:1000, 1 hour incubation) (Abcam, Cambridge, MA), anti-β-actin (ab8226, Abcam, 1:2000, 1 hour incubation), goat antirabbit HRP conjugated (12–348, Upstate, 1:5000, 1 hour incubation) and goat antimouse (12–349, Upstate, 1:3000, 1 hour incubation) (Upstate, Millipore, Billerica, MA).
Cotransfection of HCT116 cells with psiCheck luciferase reporter plasmids and DsiRNA
The pLuc-EGFP vector, in which a portion of enhanced green fluorescent protein (EGFP) was cloned into the 3′-UTR of the Renilla expression cassette in the psiCHECK-2 vector (Promega, Madison, WI) has been described previously (Rose et al., 2005). The HCT116 colon carcinoma cell line was grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 20 U/mL penicillin/streptomycin, and 2 mM L-Gln (DMEM-complete). Cells were grown in a humidified, 5% CO2 incubator at 37°C. HCT116 cells were seeded in 48-well plates and transfected 24 hours later (∼70% confluency) using 0.5 μL of Lipofectamine 2000 (Invitrogen, Carlsbad, CA) per well. Each well received 100 ng of psi-GFP and DsiRNAs at the indicated concentrations (100 pM, 1 nM, and 10 nM). All samples were transfected in duplicate or triplicate, and the experiment was performed three times. Luciferase assays were performed 24 hours after transfection using the Dual Luciferase Assay system according to the manufacturer (Promega, Madison, WI). Renilla luciferase activities in relative light units (RLUs) are reported as normalized values (i.e., Renilla luciferase activity units/Firefly luciferase activity units).
For studies involving mouse Tissue Factor target (F3), HCT116 cells were cotransfected in suspension in triplicate wells in 96-well plates using 25 ng reporter DNA, 0.15 μL Lipofectamine 2000 and 10–250 pM siRNA, for a 100 μL final transfection volume per well. Triplicate batch complexations were performed as follows: 3 μL DsiRNA at 100× final concentration was diluted with 15 μL 5 ng/μL Opti-MEM-diluted reporter DNA (prepared in one large batch for all samples) and mixed with an equal volume of a batch dilution of Lipofectamine 2000 in Opti-MEM (for 10 samples: 145 μL Opti-MEM + 4.5 μL LF2000). Complexes were allowed to form at room temperature for 30 minutes. During complexation, near-confluent HCT116 cells were detached by trypsinization, resuspended in full media, centrifuged to remove residual trypsin, and resuspended at a cell density sufficient to achieve approximately 80%–90% confluency equivalence upon plating. Complexes were mixed with 270 μL cell suspension, and 3 × 100 μL of the complexes/cell mixtures were plated in triplicate wells of a 96-well plate. The day after transfection, the medium was removed, cells lysed with 40 μL Passive Lysis Buffer (Promega), and dual luciferase assays performed on a Veritas Luminometer (Turner Biosystems, Sunnyvale, CA), using 10 μL lysate and 50 μL of each substrate reagent (Promega). Target-specific Renilla reporter expression was standardized to internal nontarget Firefly luciferase expression, and normalized to values for samples treated with a scrambled control siRNA.
For construction of the mouse Tissue Factor reporter, a fragment of the F3 gene (NM_010171) spanning bases 154–1591 was cloned into SpeI sites in the 3′-UTR of the Renilla luciferase gene in the psiCHECK-2 vector (Promega) using primers mTF-for 5′-gatgattctagactcgagGAGACCTCGCCTCCAGCC-3′ and mTF-rev 5′-gatgatgtcgactagtATCACAAAGATGCCCCAAGC-3′ (uppercase letters are complementary to the F3 target gene). All siRNAs studied were located between bases 211 and 1371 and were included within the subcloned fragment.
Serum stability assay
Aliquots of 2 nmol of each RNA duplex were incubated at 37°C in 200 μL buffer (PBS, pH 7.4, with 2 mM MgCl2) containing 50% (v/v) of fetal bovine serum (Invitrogen, Carlsbad, CA), resulting in a concentration of 10 μM siRNA duplex. The serum was not heat inactivated. The reactions were stopped with addition of 100 μL of phenol/chloroform/isoamyl alcohol (25/24/1 v/v/v) and the mixture was stored at −80°C. When all incubations were done, samples were extracted and ethanol precipitated. For gel analysis, 20 pmol (approximately 0.0067 OD260) of each sample was loaded onto 20% polyacrylamide nondenaturing gels in TBE buffer. Following electrophoresis, the gels were stained with 1× GelStar (Lonza, Rockland, ME) and visualized on a UV-transilluminator.
Detection of human interferon-α (IFN-α) in PBMC culture supernatants
Human peripheral blood was obtained from normal healthy donors (Ullevaal University Hospital Blood Center, Oslo, Norway), and peripheral blood mononuclear cells (PBMCs) were isolated by standard Isopaque-Ficoll (Lymphoprep; Nycomed) gradient centrifugation. Cells were counted and resuspended at 3.0 × 106 per ml. DsiRNAs were complexed with Lipofectamine 2000 for triplicate suspension transfections at 500 μL 100 nM siRNA in 24-well plates. The suspension of 300 pmole siRNA (5.1 μg) was diluted with 200 μL serum-free RPMI medium (SFM; Gibco, Invitrogen, Carlsbad, CA) and mixed with 200 μL of SFM-diluted Lipofectamine 2000 (10.5 μL). Following 30 minutes incubation at room temperature, the complexes were mixed by resuspension with 1.1 mL of 3.0 × 106/mL PBMC and 3 × 0.50 mL of the cell/complexes mixture seeded into triplicate wells of 24-well plates. Medium was collected from samples 20 hours posttransfection, transferred to 1 mL tubes on ice, centrifuged for 5 minutes at 400 × g at 4°C, and diluted with one part dilution buffer provided in the ELISA kit (PBL Biomedical Laboratories, Piscataway, NJ). All samples were assayed in duplicate. The amount of IFN-α was quantified from a standard curve according to the manufacturer's high sensitivity protocol.
Cytokine assays in T98G cells
T98G cells were obtained from ATCC and maintained under standard culture conditions in high glucose DMEM containing 10% fetal calf serum and 1% pen/strep. Cells were plated at 3 × 104 per well in a 24-well plate the day before transfection in 500 μL complete media without antibiotics. Transfections were conducted using 0.6 μL per well of siLentFect (Bio-Rad, Hercules, CA) and appropriate amount of siRNA duplex to give either 10 nM or 100 nM final concentration. Media was not changed to remove siRNA complexes. Following incubation at 37°C, cell supernatants were collected and placed in tubes and stored at −20°C until cytokine assays were run as a group.
Secreted cytokine levels were measured 24 hours post-transfection using Luminex xMAP technology with Beadlyte reagents (IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, TNF-α, IFN-γ, and GM-CSF) from Upstate (Millipore, Billerica, MA) or IFN-α Biosource (Invitrogen, Carlsbad, CA) on a Luminex 100 system (Luminex, Austin, TX). Standard curves for the Beadlyte assays were generated using Beadlyte Human Multi-Cytokine Standard 3 (5000–6.9 pg/mL) from Upstate. IFN-α standard curves were generated using the Hu Cytokine II Standard (7100–9.7 pg/mL) from Biosource. Mean Fluorescence Intensity (MFI) was measured using gate settings of 7500–13,500, sample size 75 μL and 75 detection events per bead set. Sample concentrations (pg/mL) were determined by extrapolation from the standards using a 5-PL curve.
Results
Testing and optimization of modification patterns
We previously described optimization of synthetic RNA substrates for human Dicer. The final preferred structure was an asymmetric duplex with a 25-mer sense (S) strand and a 27-mer AS strand having a single 2-base 3′-overhang positioned on the AS strand and two DNA residues at the 3′-end of the S strand (Rose et al., 2005). This design results in a single, predictable product from Dicer processing and improves relative loading of the AS strand into RISC. A series of chemically modified DsiRNAs were synthesized at a known effective site in the human STAT1 gene (NM_007315) employing combinations of RNA, 2′OMe RNA, 2′-F RNA, and DNA bases (Table 1).Site selection was performed using accepted design criteria (PEEK, 2007; Peek and Behlke, 2007). Previously, end modification of blunt 27-mer RNA duplexes with a bulky, hydrophobic dye (fluorescein) was found to interfere with Dicer processing and significantly reduced functional potency (Kim et al., 2005). We therefore avoided chemical modifications adjacent to the predicted site of Dicer processing (Fig. 1) so as to not interfere with cleavage.
Table 1.
STAT1 27-mer Modified DsiRNAs

FIG. 1.
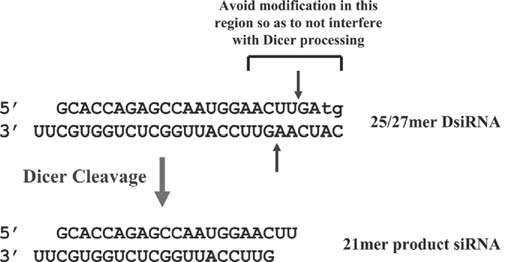
Scheme for designing chemically modified Dicer-substrate siRNAs (DsiRNAs). The region of the RNA duplex surrounding the expected cleavage site (Rose et al., 2005) was left as unmodified RNA. Sequence 5′- to the cleavage site were modified in the sense strand and sequence 3′- to the cleavage site were modified in the antisense strand. In all cases, two DNA residues were placed at the 3′-end of the sense strand.
The series of STAT1 chemically modified DsiRNAs were transfected into HeLa cells at 10 nM, 1 nM, and 0.1 nM concentrations and relative knockdown of STAT1 expression was assessed by qRT-PCR at 24 hours (Fig. 2). This site showed high potency and the unmodified construct suppressed gene expression by more than 85% at 0.1 nM concentration (Sequence 1 in Table 1 and Fig. 2). Several of the modification patterns showed potency nearly identical to the unmodified version at high (10 nM) and medium (1 nM) doses (such as Sequences 2, 4, 6, 8, 10, and 11), whereas other modification patterns showed reduced activity (Sequences 9, 14, 16, and 17). At low dose (0.1 nM), alternating 2′OMe bases generally showed the best potency; however none of the modified duplexes was as potent as the original unmodified duplex. These results were not surprising; many chemical modification patterns have been described for 21-mer siRNAs that can similarly reduce effectiveness of the RNA duplex as a trigger of RNAi. These effects can vary with sequence context, making it important to examine the behavior of modification patterns at different sites in different gene targets.
FIG. 2.
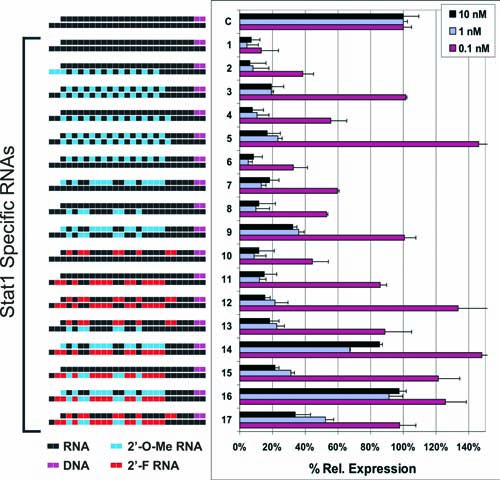
Relative potency of various chemical modification patterns for human STAT1 DsiRNAs. RNA duplexes were transfected into HeLa cells at 10, 1, and 0.1 nM concentration and relative STAT1 mRNA levels were assessed at 24 hours using qRT-PCR. The chemical modification patterns employed are illustrated schematically on the left where black represents RNA, Blue represents 2′OMe RNA, red represents 2′-F RNA, and purple represents DNA bases.
Since the alternating 2′OMe RNA modification pattern appeared promising in the STAT1 series, modified DsiRNA using this pattern were synthesized specific to a second target, the reporter gene EGFP (Fig. 3a). These duplexes were cotransfected into HCT116 cells with a luciferase expression plasmid containing EGFP sequences cloned into the 3′-UTR of the firefly luciferase gene, which was used as a reporter to assess relative efficacy of knockdown (Fig. 3b). As before, duplexes having an alternating 2′OMe pattern in either the S or AS strands showed high potency (in this case identical to the unmodified version). However, the duplex having modifications present on both S and AS strands was largely inactive, highlighting the potential for a modification pattern to perform well in one sequence context (Fig. 2, Duplexes 3 and 5) but not in a different context (Fig. 3b, Duplex 4). For general research applications, a modification pattern that is generally effective irrespective of sequence context is desirable. For DsiRNAs, selective modification of a single strand (either S or AS) may therefore be preferable to designs having both strands modified. Of note, in the EGFP series modification pattern #EGFP-5 included placement of 2′OMe bases at the 5′-end of the AS strand so that modifications flanked the expected Dicer cut site. This pattern showed potency similar to the unmodified and other modified duplexes that did not span the cut site.
FIG. 3.
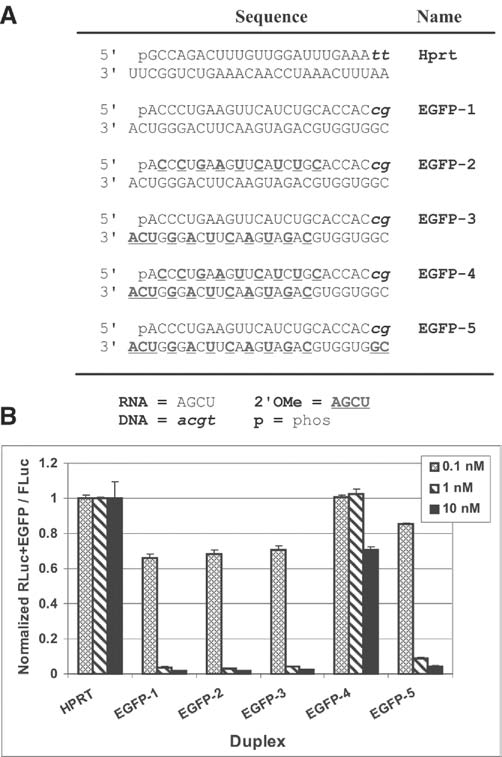
Relative potency of 2′OMe modification patterns for EGFP DsiRNAs. (A) Sequence of the EGFP-specific and control (HPRT) DsiRNAs are shown. Black uppercase letters are RNA, bold uppercase underlined letters are 2′OMe RNA, and lowercase italic letters are DNA bases. (B) DsiRNA duplexes were cotransfected into HCT116 cells at the concentrations indicated with a psiCHECK-2 vector containing EGFP sequence in the 3′-UTR of firefly luciferase. Luciferase assays were performed 24 hours posttransfection and relative FLuc signal was normalized against an internal Renilla luciferase control.
Since a modification pattern that could be generally employed at any site was desired, the utility of the alternating 2′OMe pattern was tested in the sequence context of additional sites on two new target genes. A site in the human HPRT1 gene (NM_000194) was studied comparing an unmodified DsiRNA with duplexes having the antisense-strand modified (ASm) or both strands modified (Sm/ASm; Fig. 4a). Duplexes were transfected into HeLa cells at 10 nM, 1 nM, and 0.1 nM concentrations and the relative level of HPRT mRNA levels was assessed by qRT-PCR at 24 hours (Fig. 4b). The ASm duplex had similar potency to the unmodified duplex and, in this case, the duplex having both strands modified was nearly as potent. Active sites in the mouse F3 gene (Mus musculus coagulation factor III, NM_010171) were identified by screening a series of candidate RNA duplexes (data not shown). The three most potent sites were studied in greater detail, comparing the functional potency of unmodified and ASm duplexes (Fig. 5a). HCT116 cells were cotransfected with a luciferase expression plasmid containing F3 sequence cloned into the 3′-UTR of the firefly luciferase gene, which was used as a reporter to assess relative efficacy of knockdown. Functional studies for two duplexes are shown (Fig. 5b). The DsiRNAs studied were extremely potent, having IC50 concentrations of 10–20 pM. For these hyperfunctional sites, the ASm duplexes were equally potent to the unmodified duplexes.
FIG. 4.
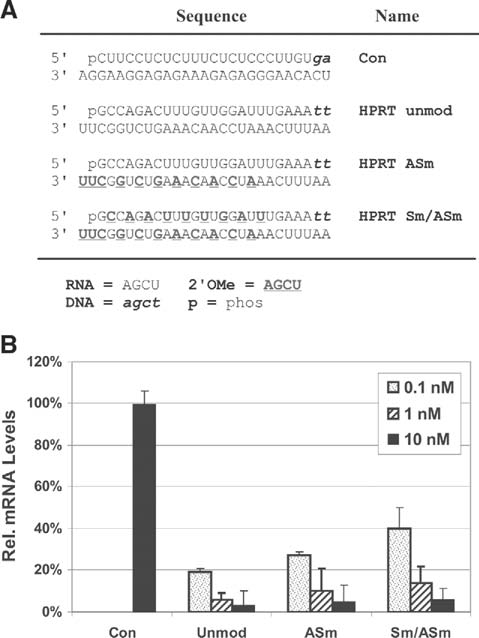
Relative potency of 2′OMe modification patterns for human HPRT DsiRNAs. (A) Sequence of the HPRT-specific and control DsiRNAs are shown. Black uppercase letters are RNA, bold uppercase underlined letters are 2′OMe RNA, and lowercase italic letters are DNA bases. (B) DsiRNA duplexes were cotransfected into HeLa cells at the concentrations indicated. RNA was prepared at 24 hours posttransfection and qRT-PCR was performed to assess expression of HPRT mRNA.
FIG. 5.
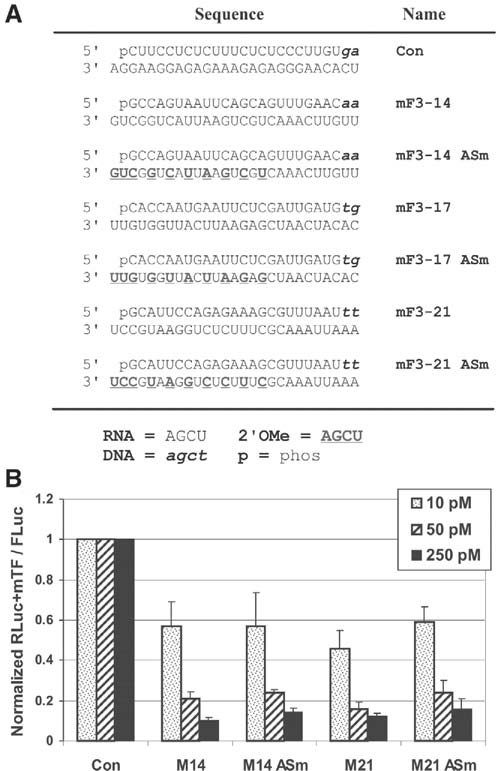
Relative potency of 2′OMe modification patterns for mouse F3 DsiRNAs. (A) Sequence of the F3 specific and control DsiRNAs are shown. Black uppercase letters are RNA, bold uppercase underlined letters are 2′OMe RNA, and lowercase italic letters are DNA bases. (B) DsiRNA duplexes were cotransfected into HCT116 cells at the concentrations indicated with a psiCHECK-2 vector containing F3 sequence in the 3′-UTR of firefly luciferase. Luciferase assays were performed 24 hours posttransfection and relative FLuc signal was normalized against an internal Renilla luciferase control.
Dicer processing, stability, and kinetics
Ideally, the chemical modification patterns employed should not alter processing of the RNA duplexes as Dicer substrates (Fig. 1). To verify cleavage patterns, mass spectrometry analyses of the products of in vitro dicing reactions were performed (Rose et al., 2005) to identify the actual species produced by cleavage of each of the HPRT-specific DsiRNAs (Fig. 4a) when incubated with recombinant human Dicer. Results are summarized in Table 2. DsiRNAs are designed to offer the PAZ domain of Dicer a single 3′-overhang and help orient the substrate so that cleavage occurs at a predictable site 21–22 bases from the 3′-end of the overhang (Rose et al., 2005). A mix of 21-mer and 22-mer products is often seen. For the unmodified HPRT DsiRNA duplex, mass analysis of cleavage products was consistent with the 27-mer substrate being cleaved by Dicer at the expected position, resulting in a single 21-mer RNA species with symmetric 2-base 3′-overhangs and 5′-phosphate groups at each end (i.e., a mature siRNA). A similar processing pattern was observed for the 2′OMe modified ASm and Sm/ASm duplexes except that a mix of both 21-mer and 22-mer species was produced. Therefore, limited chemical modification as used here does not significantly interfere with Dicer processing of short substrate RNAs.
Table 2.
ESI-MS Analysis of in vitro Dicing Products

The 2′OMe modification has been used to improve the stability of 21-mer siRNAs in the presence of serum nucleases. The modification pattern employed here was designed to leave an unprotected domain so that the RNA duplexes could be cleaved by the endoribonuclease Dicer. This modification pattern might or might not confer stability to other nucleases. Unmodified and 2′OMe modified duplexes (Fig. 6a) were incubated in fetal calf serum (which was not heat inactivated), and the degradation products were resolved using polyacrylamide gel electrophoresis (PAGE; Fig. 6b). No intact 21-mer siRNA is seen after 6 hours incubation in fetal calf serum. Consistent with earlier reports, the unmodified 27-mer duplexes were somewhat more nuclease resistant than 21-mer duplexes at the same site (Kubo et al., 2007), and small amounts of the unmodified 27-mer DsiRNA were still present at 6 hours. In contrast, an appreciable amount of the 2′OMe ASm modified 27-mer DsiRNA remained intact after 24 hours incubation in serum. Thus modification with 2′OMe bases in even the limited ASm pattern improved nuclease stability of the DsiRNA. Greater stabilization was seen when the alternating pattern of 2′OMe bases was extended through the full length of the duplex (data not shown); however, this extensive modification also blocked Dicer processing and was not studied further.
FIG. 6.
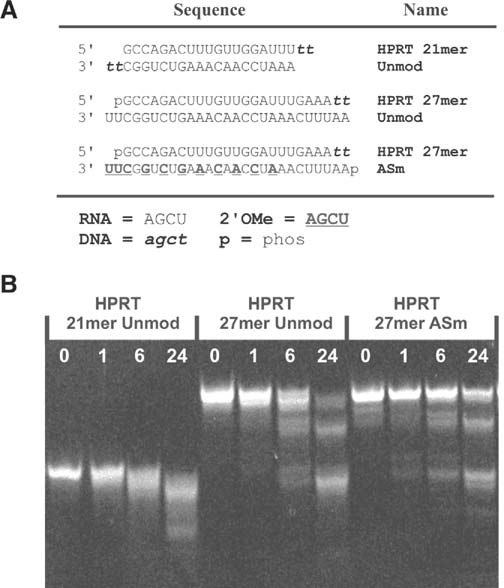
Stability of 2′OMe modified DsiRNAs in serum. (A) Sequences of HPRT-specific 21-mer siRNA and 27-mer DsiRNAs are shown. Uppercase letters are RNA, bold uppercase underlined letters are 2′OMe RNA, and lowercase italic letters are DNA bases. (B) RNA duplexes were incubated in fetal calf serum for the length of time indicated (hours) and extracted with phenol/chloroform. Degradation products were separated using PAGE and visualized with fluorescent staining.
We next examined the initial rate of target knockdown and duration of silencing using the HPRT DsiRNAs. The unmodified HPRT DsiRNA was transfected into HeLa cells at 10 nM concentration and RNA was prepared at 3, 6, 12, 24, and 48 hours posttransfection. HPRT mRNA levels were assessed using qRT-PCR (Fig. 7a). Degradation of HPRT mRNA was rapid, showing a 56% reduction at 3 hours, 90% reduction at 6 hours, and 97% reduction at 12 hours. Cell extracts were prepared from parallel cultures at 24, 48, and 72 hours, and HPRT protein levels were assessed by western blot analysis. HPRT protein is relatively stable (Steen et al., 1991) so changes in protein levels lagged well behind changes in RNA levels, finally reaching 90% reduction at 3 days posttransfection.
FIG. 7.
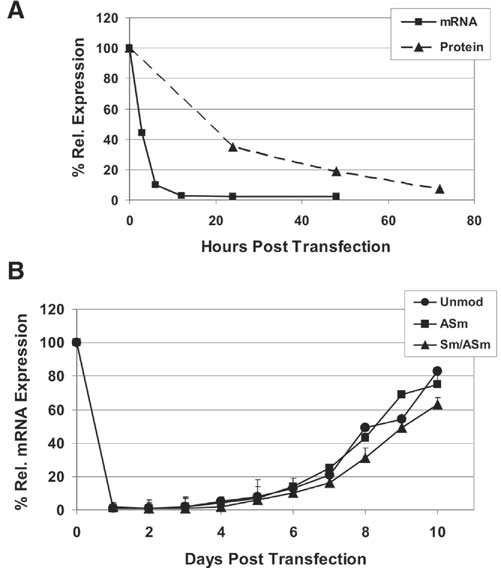
Time course of HPRT knockdown. (A) The unmodified HPRT DsiRNA was transfected into HeLa cells at 10 nM concentration and mRNA levels were assessed by qRT-PCR at 3, 6, 12, 24, and 48 hours. Cell extracts were prepared from parallel cultures at 24, 48, and 72 hours and separated by SDS–PAGE. Western blots were performed and relative levels of HPRT protein expression were assessed by densitometry normalized to an internal β-actin standard. (B) The unmodified, ASm modified, and Sm/ASm modified HPRT DsiRNAs (Fig. 4a) were transfected into HeLa cells at 10 nM concentration and mRNA levels were assessed by qRT-PCR daily for 10 days.
A 27-mer DsiRNA targeting EGFP was previously reported to reduce fluorescence from a cotransfected EGFP reporter plasmid for 9–10 days in HEK293 cells (Kim et al., 2005). We examined the duration of silencing of an endogenous gene, HPRT1, and compared activity of the unmodified versus 2′OMe modified duplexes. The unmodified, ASm, and Sm/ASm HPRT DsiRNAs (Fig. 4a) were transfected into HeLa cells at 10 nM concentration and multiple parallel cultures were maintained for 10 days by serial passage without additional treatment. RNA was prepared daily and relative HPRT mRNA levels were assessed by qRT-PCR (Fig. 7b). Effective suppression of HPRT mRNA was achieved with 98%–99% knockdown seen in days 1–3. HPRT mRNA levels remained < 75% control levels through the first week and had not yet returned to baseline by day 10. No significant difference in duration of silencing was observed between the unmodified and 2′OMe modified duplexes tested here.
Innate immune stimulation
Synthetic nucleic acids, such as dsRNAs, can stimulate the innate immune system and trigger a Type I Interferon response (Marques and Williams, 2005; Schlee et al., 2006). In vivo, all cell types (and receptors) are present, so the risk of triggering the immune system is significant, especially if the dsRNA is administered using a lipid-based delivery tool. Lipid-based delivery approaches maximizes exposure of the cargo to the endosomal compartment where Toll-like Receptors (TLRs) 3, 7, and 8 reside (Heil et al., 2004; Hornung et al., 2005; SIOUD, 2005), which appear to be the primary molecules responsible for immune recognition of siRNAs. In vitro, the risk of triggering an immune response is highly dependent on the specific cell line employed, and many tissue culture lines lack the immune receptors necessary to respond to siRNAs. However, certain cell types express receptors that recognize and respond to the presence of longer dsRNAs, such as the DsiRNAs employed here, that do not respond to 21-mer siRNAs (Reynolds et al., 2006). For example, the T98G neuroblastoma cell line has been shown to respond to 27-mer but not to 21-mer dsRNAs, and it is thought that this responsiveness relates to recognition of blunt ends on longer RNAs by the cytoplasmic receptor RIG-I (Marques et al., 2006).
Chemical modification of an RNA duplex can block the immune response to a sequence that is normally immunostimulatory, even in vivo (Morrissey et al., 2005b). Although a variety of different modifications can help evade immune detection (Kariko et al., 2005), the 2′OMe modification is effective, inexpensive, and may also confer nuclease resistance to the sequence. In particular, 2′OMe U and G bases seem to be potent in preventing immune recognition, and it is now thought that this occurs via direct competitive inhibition of TLR binding unmodified RNAs by 2′OMe-containing RNAs (Judge et al., 2006; Robbins et al., 2007). Although the utility of the 2′OMe modification to evade immune detection has been well described for 21-mer siRNAs, it has not been well studied in the context of longer 27-mer DsiRNAs. It is also not known whether 2′OMe RNA can block responses via different signaling pathways that do not respond to 21-mer siRNAs, such as RIG-I. We therefore examined the capacity of unmodified and 2′OMe-modified DsiRNA to trigger an immune response in a mixed population of human PBMCs and in T98G cells.
The PBMCs are a natural mixed lymphocyte population and are more representative of the natural spectrum of immune receptors that are encountered in vivo than individual tissue culture cell lines. Three different DsiRNAs specific for murine tissue factor (F3) were studied for their ability to trigger IFN-α secretion, including unmodified and 2′OMe ASm duplexes (Fig. 5a). The RNAs were transfected using cationic lipids and culture supernatants were tested for IFN-α by ELISA 20 hours posttransfection (Fig. 8). All three unmodified RNA duplexes stimulated IFN-α secretion. Use of 2′OMe modification blocked this response for all three sequences. Immune response to siRNAs in these cells is thought to be primarily dependent upon interactions with TLR3 and TLR7/8. Modification with 2′OMe RNA has been shown to block IFN-α responses in this cell population using 21-mer siRNAs, and we have now demonstrated using three different sequences that the same holds true for 27-mer DsiRNAs. Interaction with the cytoplasmic RNA receptor RIG-I was next studied in T98G cells.
FIG. 8.
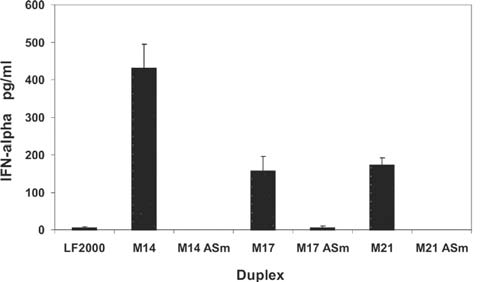
Unmodified but not 2′OMe-modified DsiRNAs stimulate secretion of interferon-α (IFN-α) from human PBMCs. Human PBMCs were transfected with 100 nM F3-specific DsiRNA duplexes (Fig. 5a) using a cationic lipid and supernatants were measured for IFN-α levels by ELISA 20 hours posttransfection.
The unmodified and the ASm 2′OMe-modified versions of the HPRT DsiRNA (Fig. 4a) were transfected at 10 nM and 100 nM concentrations into T98G cells using cationic lipids and cell culture supernatants were tested for the release of IFN-α using a fluorescent bead sandwich assay (Luminex xMAP technology). No IFN-α secretion was detectable at 4 or 24 hours posttransfection. The experiment was repeated using a panel of 10 cytokine assays (IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, TNF-α, IFN-γ, and GM-CSF). No cytokine response was detected at 4 hours. Strong secretion of IL-6 was seen at 24 hours in cultures transfected with the unmodified HPRT DsiRNA but not with the 2′OMe ASm DsiRNA. No other cytokine in this assay panel was detected above background. IL-6 levels for experiments done in T98G cells using 100 nM of the HPRT DsiRNA are shown in Fig. 9. Thus modification of 27-mer DsiRNAs with 2′OMe RNA on only a single strand is sufficient to block stimulation of an innate immune response in T98G cells, which is thought to occur through interaction with the cytoplasmic receptor RIG-I. In summary, four different unmodified sequences were studied, all of which trigger an innate immune response when delivered into certain cell types using cationic lipid mediated transfection. Limited chemical modification of a single strand of the duplex with 2′OMe RNA was sufficient to prevent interferon or other cytokine secretion for all four sequences.
FIG. 9.
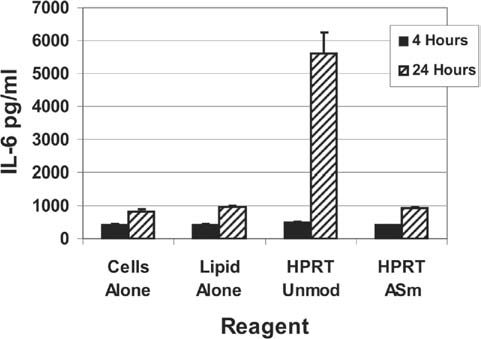
T98G cells secrete IL-6 in response to unmodified but not 2′OMe modified DsiRNAs. T98G cells were transfected with 100 nM HPRT DsiRNAs (Fig. 4a) using the cationic lipid siLentFect and culture supernatants were tested at 4 and 24 hours posttransfection for levels of IL-6 using the Luminex xMAP system with Beadlyte cytokine assay reagents. Quantitative cytokine measurements were calculated by extrapolation of relative fluorescence measurements against a standard curve run in parallel.
Discussion
A variety of groups have studied chemical modification patterns in 21-mer siRNAs with the overall goal of improving nuclease resistance without compromising potency of the RNA duplex as a trigger of RNAi (Nawrot and Sipa, 2006; Zhang et al., 2006). Modification of a few residues is usually well tolerated; however, widespread modification of the siRNA can lead to reduced activity compared to unmodified RNA. Further, these effects can be sequence-context dependent. In spite of these challenges, many examples of highly potent modified siRNAs have been reported.
The primary nuclease activity present in serum is a 3′-exonuclease and modification of the 3′-end of DNA oligonucleotides will slow or prevent degradation (Eder et al., 1991). Phosphorothioate (PS) or boranophosphate modifications can be used to stabilize the phosphate backbone. Boranophosphate-modified RNAs have improved nuclease resistance and can function as siRNAs. However, boranophosphate-modified RNAs cannot be easily manufactured by chemical synthesis methods and instead are made using enzymatic approaches, which limits widespread use (Hall et al., 2004; Hall et al., 2006). PS modifications can be introduced using standard synthesis methods and can be selectively placed at any desired position. The PS modification can show sequence nonspecific toxicity, so most investigators favor limited incorporation in siRNAs at the 3′-ends (Amarzguioui et al., 2003; Braasch et al., 2003; Chiu and Rana, 2003; Harborth et al., 2003). Although extensive PS modification can be compatible with siRNA function, the use of other modification strategies, such as 2′-modified sugars, may be superior (Choung et al., 2006).
Different chemical groups can replace the 2′-hydroxyl of the ribose which increase both duplex stability (Tm) and confer nuclease resistance (Freier and Altmann, 1997; Egli et al., 2005). 2′-O-methyl RNA is a naturally occurring modification found in mammalian ribosomal RNAs and transfer RNAs. This modification is easily incorporated into synthetic RNAs and can improve nuclease resistance; its use has been thoroughly investigated in 21-mer siRNAs (Amarzguioui et al., 2003; Chiu and Rana, 2003; Czauderna et al., 2003; Harborth et al., 2003; Choung et al., 2006). The precise modification pattern employed is important and RNA duplexes entirely made of 2′OMe bases are inactive as siRNAs. A pattern of alternating 2′OMe bases on both the S and AS strands generally retains high potency and is substantially protected against nuclease degradation (Czauderna et al., 2003; Choung et al., 2006). Unnatural 2′-modifications can also be used. The 2′-F RNA modification has been successfully employed and is most commonly inserted at pyrimidine positions. The use of mixed 2′OMe and 2′-F modifications allows synthesis of heavily modified duplexes that can be stable in serum for days and yet retain high potency as triggers of RNAi (Allerson et al., 2005; Morrissey et al., 2005a, 2005b; Prakash et al., 2005; Kraynack and Baker, 2006). Locked nucleic acids (LNAs) represent a relatively new class of 2′-modification that significantly increase Tm. The positions within a siRNA that can be modified with LNA bases without compromising potency are more limited than for 2′OMe or 2′-F bases (Braasch et al., 2003; Grunweller et al., 2003; Elmen et al., 2005; Mook et al., 2007), but LNAs can be used if minimal (as opposed to extensive) modification of the siRNA is employed.
In this study, we examined chemical modification patterns of 27-mer DsiRNA duplexes, testing their relative potency, ability to be cleaved by Dicer, and potential for immune activation. A chemical modification pattern using alternating 2′OMe residues that does not span the expected Dicer cut site in the 27-mer sequence was found to generally have high potency at six sites tested in four different gene targets when compared with the parent unmodified duplexes. This modification pattern was most uniformly active when a single strand was modified, and ASm was used for most of the studies reported herein. Given the extent to which functional performance of chemically modified siRNAs and DsiRNAs varies with sequence context, we would not be surprised if new sites are found where the successful modification patterns described here show reduced activity. Similar modification patterns were reported to perform well in the context of 21-mer siRNAs by other groups (Czauderna et al., 2003; Choung et al., 2006). The modification pattern as employed here in the context of 27-mer DsiRNAs, however, only provides for modest improvements in nuclease stability. This is not surprising, as only 9–11 modified bases were incorporated in the RNA duplex, and an unmodified nuclease-sensitive domain was intentionally placed at the expected site of Dicer cleavage, so as to not impede functional processing of the RNA duplexes within the natural RNAi pathway. This strategy was generally successful and Dicer processing of the modified duplexes produced 21- to 22-mer species, as assessed by mass spectrometry analysis of in vitro dicing reactions. We do note that, at the one site studied, use of the ASm 2′OMe modification pattern did lead to accumulation of both 21-mer and 22-mer products after in vitro dicing, whereas the unmodified duplex was cleaved to a single 21-mer species. Spacing the modifications further away from the dicing site (using 9 or 10 modified bases, instead of the 11 employed in this example) may avoid this effect.
We expect that more extensively modified sequences could be developed for individual sites that would have increased nuclease stability and yet retain high potency. Heavily modified compounds of this kind may be desirable for pharmaceutical applications. The effort needed to perform extensive site-specific optimization for each target site of interest is feasible for drug development projects but not for general research needs. The goal of the present study was to define modification patterns that would be useful for research purposes that would remain active at most sites regardless of sequence context.
The innate immune system comprises a variety of receptors that can recognize and respond to foreign nucleic acids with an interferon or cytokine response (Marques and Williams, 2005; Schlee et al., 2006). For siRNAs, members of the TLR family are probably the most important innate immune sensors. Three TLRs (TLR3, 7, and 8) specifically interact with single-stranded or double-stranded RNAs. These receptors primarily reside within endosomal compartments and delivery methods that gain access to these locations maximize the risk of triggering an immune response. Other methods of delivery present varying degrees of risk, and sequences which trigger a response in vivo using lipid delivery can be safely administered using a mechanical delivery approach which avoids entry into endosomal compartments (Heidel et al., 2004). It has been shown that longer dsRNAs, such as the 27-mer DsiRNAs studied here, can trigger the innate immune system in some cell types when corresponding 21-mer siRNAs are undetected (Reynolds et al., 2006). This response may be all or in part due to interaction with the cytoplasmic RNA helicase, RIG-I (Marques et al., 2006). Chemical modification can prevent immune recognition of 21-mer siRNAs when transfected into immune-responsive cells in vitro or when administered in vivo (Kariko et al., 2005; Judge et al., 2006; Robbins et al., 2007; Morrissey, 2005, p. 258).
We describe here that chemical modification of 27-mer DsiRNAs can similarly block recognition by the innate immune system in two different in vitro systems using cationic lipid mediated delivery. The duplexes studied had 10–11 2′OMe bases present in the AS strand of the duplex, of which at least two comprised G or U residues (which are more effective in preventing TLR activation; Judge et al., 2006). Importantly, this limited modification pattern was sufficient to prevent immune activation of T98G cells, where RNAs of this class have been shown to trigger cytokine responses when 21-mer siRNAs do not (Marques et al., 2006). Further, in a normal mixed human PBMC population, unmodified 27-mer DsiRNA duplexes were strongly immunostimulatory when delivered with cationic lipids, whereas no IFN-α secretion was detected for the 2′OMe modified duplexes.
Immune response studies are reported here for four different DsiRNA sequences (one human HPRT1 site and three mouse F3 sites), comparing unmodified with 2′OMe modified RNA duplexes. A set of DsiRNA duplexes specific for a single site in the human STAT1 gene was tested for cytokine responses in the laboratory of Bryan Williams (this study was performed using a subset of the duplexes shown in Table 1; Zamanian-Daryoush et al., 2008) and included examination of both 2′OMe and 2′-F modified duplexes. In this sequence context, both 2′OMe and 2′-F modifications blocked IFN-α and TNF-α secretion in human PBMC cultures. However, in RIG-I dependent T98G cells, the 2′-F modified duplexes triggered an immune response, whereas 2′OMe modified duplexes did not. It therefore appears that the 2′OMe modification may provide more generalized protection from immune responses than other 2′-modifications. Modification patterns of this kind should have utility for both research and therapeutic applications of DsiRNAs.
Acknowledgments
We gratefully acknowledge members of the research group at IDT for helpful discussions and assistance with the manuscript. This work was supported by grants from the National Institutes of Health HL 07470 and the W.H. Keck foundation (J.J.R.) and the Norwegian Research Council (M.T.W.).
References
- ALLERSON C.R. SIOUFI N. JARRES R. PRAKASH T.P. NAIK N. BERDEJA A. WANDERS L. GRIFFEY R.H. SWAYZE E.E. BHAT B. Fully 2′-modified oligonucleotide duplexes with improved in vitro potency and stability compared to unmodified small interfering RNA. J. Med. Chem. 2005;48:901–904. doi: 10.1021/jm049167j. [DOI] [PubMed] [Google Scholar]
- AMARZGUIOUI M. HOLEN T. BABAIE E. PRYDZ H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003;31:589–595. doi: 10.1093/nar/gkg147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEHLKE M.A. Progress towards in vivo use of siRNAs. Mol. Ther. 2006;13:644–670. doi: 10.1016/j.ymthe.2006.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERNSTEIN E. CAUDY A.A. HAMMOND S.M. HANNON G.J. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- BIECHE I. NOGUES C. PARADIS V. OLIVI M. BEDOSSA P. LIDEREAU R. VIDAUD M. Quantitation of hTERT gene expression in sporadic breast tumors with a real-time reverse transcription-polymerase chain reaction assay. Clin. Cancer Res. 2000;6:452–459. [PubMed] [Google Scholar]
- BRAASCH D.A. JENSEN S. LIU Y. KAUR K. ARAR K. WHITE M.A. COREY D.R. RNA interference in mammalian cells by chemically-modified RNA. Biochemistry. 2003;42:7967–7975. doi: 10.1021/bi0343774. [DOI] [PubMed] [Google Scholar]
- CHIU Y.L. RANA T.M. siRNA function in RNAi: a chemical modification analysis. RNA. 2003;9:1034–1048. doi: 10.1261/rna.5103703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOUNG S. KIM Y.J. KIM S. PARK H.O. CHOI Y.C. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun. 2006;342:919–927. doi: 10.1016/j.bbrc.2006.02.049. [DOI] [PubMed] [Google Scholar]
- CZAUDERNA F. FECHTNER M. DAMES S. AYGUN H. KLIPPEL A. PRONK G.J. GIESE K. KAUFMANN J. Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003;31:2705–2716. doi: 10.1093/nar/gkg393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDER P.S. DEVINE R.J. DAGLE J.M. WALDER J.A. Substrate specificity and kinetics of degradation of antisense oligonucleotides by a 3′ exonuclease in plasma. Antisense Res. Dev. 1991;1:141–151. doi: 10.1089/ard.1991.1.141. [DOI] [PubMed] [Google Scholar]
- EGLI M. MINASOV G. TERESHKO V. PALLAN P.S. TEPLOVA M. INAMATI G.B. LESNIK E.A. OWENS S.R. ROSS B.S. PRAKASH T.P. MANOHARAN M. Probing the influence of stereoelectronic effects on the biophysical properties of oligonucleotides: comprehensive analysis of the RNA affinity, nuclease resistance, and crystal structure of ten 2′-O-ribonucleic acid modifications. Biochemistry. 2005;44:9045–9057. doi: 10.1021/bi050574m. [DOI] [PubMed] [Google Scholar]
- ELBASHIR S.M. HARBORTH J. LENDECKEL W. YALCIN A. WEBER K. TUSCHL T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- ELMEN J. THONBERG H. LJUNGBERG K. FRIEDEN M. WESTERGAARD M. XU Y. WAHREN B. LIANG Z. ORUM H. KOCH T. WAHLESTEDT C. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 2005;33:439–447. doi: 10.1093/nar/gki193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREIER S.M. ALTMANN K.H. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRUNWELLER A. WYSZKO E. BIEBER B. JAHNEL R. ERDMANN V.A. KURRECK J. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Res. 2003;31:3185–3193. doi: 10.1093/nar/gkg409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAIL M.E. ELLIOTT B. ANDERSON K. High-throughput analysis of oligonucleotides using automated electrospray ionization mass spectrometry. Am. Biotechnol. Lab. 2004;12:12–14. [Google Scholar]
- HALL A.H. WAN J. SHAUGHNESSY E.E. RAMSAY SHAW B. ALEXANDER K.A. RNA interference using boranophosphate siRNAs: structure-activity relationships. Nucleic Acids Res. 2004;32:5991–6000. doi: 10.1093/nar/gkh936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HALL A.H. WAN J. SPESOCK A. SERGUEEVA Z. SHAW B.R. ALEXANDER K.A. High potency silencing by single-stranded boranophosphate siRNA. Nucleic Acids Res. 2006;34:2773–2781. doi: 10.1093/nar/gkl339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HANNON G.J. ROSSI J.J. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- HARBORTH J. ELBASHIR S.M. VANDENBURGH K. MANNINGA H. SCARINGE S.A. WEBER K. TUSCHL T. Sequence, chemical, and structural variation of small interfering RNAs and short hairpin RNAs and the effect on mammalian gene silencing. Antisense Nucleic Acid Drug Dev. 2003;13:83–105. doi: 10.1089/108729003321629638. [DOI] [PubMed] [Google Scholar]
- HEIDEL J.D. HU S. LIU X.F. TRICHE T.J. DAVIS M.E. Lack of interferon response in animals to naked siRNAs. Nat. Biotechnol. 2004;22:1579–1582. doi: 10.1038/nbt1038. [DOI] [PubMed] [Google Scholar]
- HEIL F. HEMMI H. HOCHREIN H. AMPENBERGER F. KIRSCHNING C. AKIRA S. LIPFORD G. WAGNER H. BAUER S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- HORNUNG V. GUENTHNER-BILLER M. BOURQUIN C. ABLASSER A. SCHLEE M. UEMATSU S. NORONHA A. MANOHARAN M. AKIRA S. DE FOUGEROLLES A. ENDRES S. HARTMANN G. Sequence-specific potent induction of IFN-alpha by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005;11:263–270. doi: 10.1038/nm1191. [DOI] [PubMed] [Google Scholar]
- JUDGE A.D. BOLA G. LEE A.C. MACLACHLAN I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Mol. Ther. 2006;13:494–505. doi: 10.1016/j.ymthe.2005.11.002. [DOI] [PubMed] [Google Scholar]
- KARIKO K. BUCKSTEIN M. NI H. WEISSMAN D. Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity. 2005;23:165–175. doi: 10.1016/j.immuni.2005.06.008. [DOI] [PubMed] [Google Scholar]
- KIM D.H. BEHLKE M.A. ROSE S.D. CHANG M.S. CHOI S. ROSSI J.J. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 2005;23:222–226. doi: 10.1038/nbt1051. [DOI] [PubMed] [Google Scholar]
- KRAYNACK B.A. BAKER B.F. Small interfering RNAs containing full 2′-O-methylribonucleotide-modified sense strands display Argonaute2/eIF2C2-dependent activity. RNA. 2006;12:163–176. doi: 10.1261/rna.2150806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUBO T. ZHELEV Z. OHBA H. BAKALOVA R. Modified 27-nt dsRNAs with dramatically enhanced stability in serum and long-term RNAi activity. Oligonucleotides. 2007;17:445–464. doi: 10.1089/oli.2007.0096. [DOI] [PubMed] [Google Scholar]
- KURRECK J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003;270:1628–1644. doi: 10.1046/j.1432-1033.2003.03555.x. [DOI] [PubMed] [Google Scholar]
- MARQUES J.T. DEVOSSE T. WANG D. ZAMANIAN-DARYOUSH M. SERBINOWSKI P. HARTMANN R. FUJITA T. BEHLKE M.A. WILLIAMS B.R. A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat. Biotechnol. 2006;24:559–565. doi: 10.1038/nbt1205. [DOI] [PubMed] [Google Scholar]
- MARQUES J.T. WILLIAMS B.R. Activation of the mammalian immune system by siRNAs. Nat. Biotechnol. 2005;23:1399–1405. doi: 10.1038/nbt1161. [DOI] [PubMed] [Google Scholar]
- MEISTER G. TUSCHL T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- MOOK O.R. BAAS F. DE WISSEL M.B. FLUITER K. Evaluation of locked nucleic acid-modified small interfering RNA in vitro and in vivo. Mol. Cancer Ther. 2007;6:833–843. doi: 10.1158/1535-7163.MCT-06-0195. [DOI] [PubMed] [Google Scholar]
- MORRISSEY D.V. BLANCHARD K. SHAW L. JENSEN K. LOCKRIDGE J.A. DICKINSON B. MCSWIGGEN J.A. VARGEESE C. BOWMAN K. SHAFFER C.S. POLISKY B.A. ZINNEN S. Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology. 2005a;41:1349–1356. doi: 10.1002/hep.20702. [DOI] [PubMed] [Google Scholar]
- MORRISSEY D.V. LOCKRIDGE J.A. SHAW L. BLANCHARD K. JENSEN K. BREEN W. HARTSOUGH K. MACHEMER L. RADKA S. JADHAV V. VAISH N. ZINNEN S. VARGEESE C. BOWMAN K. SHAFFER C.S. JEFFS L.B. JUDGE A. MACLACHLAN I. POLISKY B. Potent and persistent in vivo anti-HBV activity of chemically modified siRNAs. Nat. Biotechnol. 2005b;23:1002–1007. doi: 10.1038/nbt1122. [DOI] [PubMed] [Google Scholar]
- NAWROT B. SIPA K. Chemical and structural diversity of siRNA molecules. Curr. Top. Med. Chem. 2006;6:913–925. doi: 10.2174/156802606777303658. [DOI] [PubMed] [Google Scholar]
- PEEK A.S. Improving model predictions for RNA interference activities that use support vector machine regression by combining and filtering features. BMC Bioinformatics. 2007;8:182. doi: 10.1186/1471-2105-8-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEEK A.S. BEHLKE M.A. Design of active small interfering RNAs. Curr. Opin. Mol. Ther. 2007;9:110–118. [PubMed] [Google Scholar]
- PRAKASH T.P. ALLERSON C.R. DANDE P. VICKERS T.A. SIOUFI N. JARRES R. BAKER B.F. SWAYZE E.E. GRIFFEY R.H. BHAT B. Positional effect of chemical modifications on short interference RNA activity in mammalian cells. J. Med. Chem. 2005;48:4247–4253. doi: 10.1021/jm050044o. [DOI] [PubMed] [Google Scholar]
- REYNOLDS A. ANDERSON E.M. VERMEULEN A. FEDOROV Y. ROBINSON K. LEAKE D. KARPILOW J. MARSHALL W.S. KHVOROVA A. Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA. 2006;12:988–993. doi: 10.1261/rna.2340906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBBINS M. JUDGE A. LIANG L. MCCLINTOCK K. YAWORSKI E. MACLACHLAN I. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 2007;15:1663–1669. doi: 10.1038/sj.mt.6300240. [DOI] [PubMed] [Google Scholar]
- ROSE S.D. KIM D.H. AMARZGUIOUI M. HEIDEL J.D. COLLINGWOOD M.A. DAVIS M.E. ROSSI J.J. BEHLKE M.A. Functional polarity is introduced by Dicer processing of short substrate RNAs. Nucleic Acids Res. 2005;33:4140–4156. doi: 10.1093/nar/gki732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLEE M. HORNUNG V. HARTMANN G. siRNA and isRNA: two edges of one sword. Mol. Ther. 2006;14:463–470. doi: 10.1016/j.ymthe.2006.06.001. [DOI] [PubMed] [Google Scholar]
- SIOUD M. Induction of inflammatory cytokines and interferon responses by double-stranded and single-stranded siRNAs is sequence-dependent and requires endosomal localization. J. Mol. Biol. 2005;348:1079–1090. doi: 10.1016/j.jmb.2005.03.013. [DOI] [PubMed] [Google Scholar]
- STEEN A.M. SAHLEN S. LAMBERT B. Expression of the hypoxanthine phosphoribosyl transferase gene in resting and growth-stimulated human lymphocytes. Biochim. Biophys. Acta. 1991;1088:77–85. doi: 10.1016/0167-4781(91)90155-f. [DOI] [PubMed] [Google Scholar]
- ZAMANIAN-DARYOUSH M. MARQUES J.T. GANTIER M.P. BEHLKE M.A. JOHN M. RAYMAN P. FINKE J. WILLIAMS B.R. Determinants of cytokine induction by small interfering RNA in human peripheral blood mononuclear cells. J. Interferon Cytokine Res. 2008;28:221–233. doi: 10.1089/jir.2007.0090. [DOI] [PubMed] [Google Scholar]
- ZHANG H.Y. DU Q. WAHLESTEDT C. LIANG Z. RNA Interference with chemically modified siRNA. Curr. Top. Med. Chem. 2006;6:893–900. doi: 10.2174/156802606777303676. [DOI] [PubMed] [Google Scholar]