

Abstract
Factor VIII is a multidomain protein composed of A1, A2, B, A3, C1, and C2 domains. Deficiency or dysfunction of factor VIII causes hemophilia A, a bleeding disorder. Administration of exogenous recombinant factor VIII as a replacement leads to development of inhibitory antibodies against factor VIII in 15−30% of hemophilia A patients. Hence, less immunogenic preparations of factor VIII are highly desirable. Inhibitory antibodies against factor VIII are mainly directed against immunodominant epitopes in C2, A3, and A2 domains. Further, several universal epitopes for CD4+ T-cells have been identified within the C2 domain. The C2 domain is also known to interact specifically with phosphatidylserine-rich lipid vesicles. Here, we have investigated the hypothesis that complexation of O-phospho-l-serine, the head group of phosphatidylserine, with the C2 domain can reduce the overall immunogenicity of factor VIII. The biophysical (circular dichroism and fluorescence) and biochemical studies (ELISA and size exclusion chromatography) showed that O-phospho-l-serine binds to the phospholipid-binding region in the C2 domain, and this interaction causes subtle changes in the tertiary structure of the protein. O-Phospho-l-serine also prevented aggregation of the protein under thermal stress. The immunogenicity of the factor VIII-O-phospho-l-serine complex was evaluated in hemophilia A mice. The total and inhibitory antibody titers were lower for factor VIII-O-phospho-l-serine complex compared with factor VIII alone. Moreover, factor VIII administered as a complex with O-phospho-l-serine retained in vivo activity in hemophilia A mice. Our results suggest that factor VIII-O-phospho-l-serine complex may be beneficial to increase the physical stability and reduce immunogenicity of recombinant factor VIII preparations.
Factor VIII (FVIII) is a multidomain protein that functions as a cofactor in the coagulation cascade. FVIII is composed of six domains, NH2-A1-A2-B-A3-C1-C2-COOH. It is synthesized as a 2351-residue single chain precursor protein, which is subsequently cleaved at residue 1680 to form the heavy chain (A1-A2-B) and the light chain (A3-C1-C2) (1-3). The light chain has a molecular mass of 80 kDa. The heavy chain undergoes further processing and is cleaved at several sites between the A2 and B domains, generating polypeptides with molecular masses ranging from 90 to 180 kDa (2). The heavy chain and the light chain are held together by a divalent metal ion such as calcium (4).
The deficiency or dysfunction of FVIII activity causes hemophilia A, a life-threatening bleeding disorder. Replacement therapy with preparations of recombinant FVIII (rFVIII)1 is the treatment of choice (5). The administration of exogenous FVIII, however, leads to the development of inhibitory antibodies in 15−30% of hemophilia A patients, complicating the replacement therapy (6, 7). Numerous treatment strategies, such as factor VIIa (a bypass agent), porcine FVIII (8), immune tolerance therapy (ITT) with high doses of rFVIII (9), etc., are currently employed clinically to manage patients with inhibitors. However, rFVIII preparations that prevent the formation of inhibitors present alternate clinical approaches.
The epitope regions and the mechanisms by which FVIII antibodies inhibit clotting activity are well understood. Most immunodominant epitopes of rFVIII are located in the C2, A3, and A2 domains of FVIII (7, 10, 11). The immunoprecipitation and inhibitor neutralization assays of the inhibitor plasma from hemophilia A patients clearly indicated that the anti-light chain antibody titer was the highest (12). More recently, Reding et al. (13) and Pratt et al. (14) have identified several universal epitopes for CD4+ T-cells in the 2291−2330 region of the C2 domain using proliferation assays with CD4+ cells from normal humans, hemophilia A patients (13), and mice (14). Three-dimensional models proposed based on crystallo-graphic studies and mutational analysis show that the C2 domain also contains 2−4 hydrophobic loops and other charged residues that promote lipid binding (Fig. 1) (15-17). Further, the C2 domain has structural features characteristic of universal immunodominant CD4+ epitopes (shown as sticks in Fig. 1) (18).
Fig. 1. The three-dimensional structure of C2 domain.
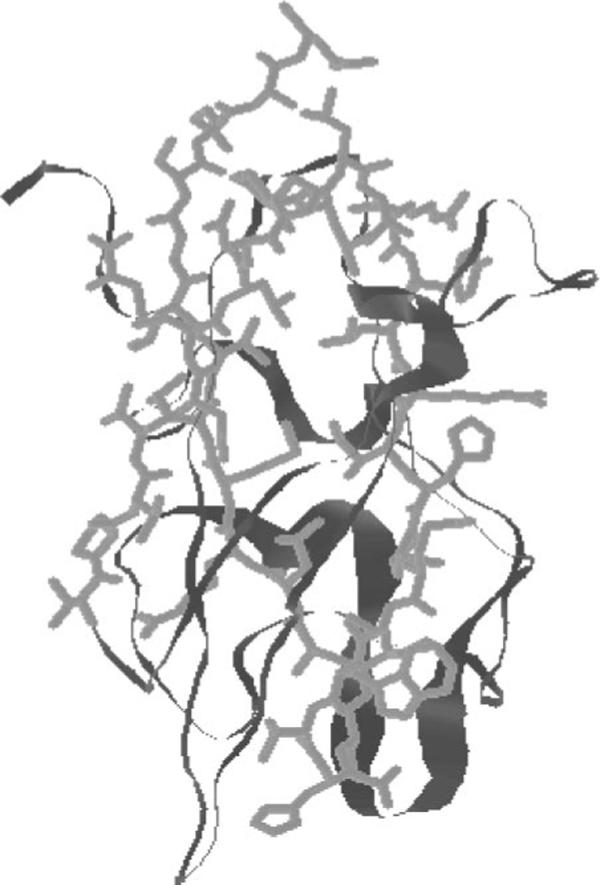
The immunodominant epitope 2291−2330, which encompasses the lipid binding site (the putative binding site for OPLS), is shown as sticks. The figure was regenerated from Protein Data Bank coordinates submitted by Pratt et al. (15).
In the blood coagulation cascade, FVIII binds to the membrane surface of activated platelets via specific interaction between phosphatidylserine and the C2 domain (19, 20). The specificity of the interaction is mediated by O-phospho-l-serine (OPLS) the head group of phosphatidylserine (21). Since OPLS is known to interact with C2 domain that contains immunodominant epitopes, it would be of significance to know the effect of OPLS on the immunogenicity of rFVIII. Here we have investigated the effects of OPLS on the conformation, folding, and stability of rFVIII using physical and biochemical methods. We have also evaluated the impact of OPLS on the activity and immunogenicity of rFVIII in vivo in FVIII knock-out (hemophilia A) mice. The results suggest that binding of OPLS to immunodominant epitopes of rFVIII results in improved stability and reduction in immunogenicity of rFVIII in hemophilia A mice.
EXPERIMENTAL PROCEDURES
Materials
We used recombinant human factor VIII (Baxter, Glendale, CA) for in vivo experiments. Monoclonal antibodies ESH4 and ESH8 were obtained from American Diagnostica Inc. (Greenwich, CT). Normal coagulation control plasma and FVIII deficient plasma for the activity assays were purchased from Trinity Biotech (County Wicklow, Ireland). Platelin L reagent used in activated partial thromboplastin time and Bethesda assays was purchased (BioMerieux, Durham, NC). The activated partial thromboplastin time and Bethesda assays were performed using a COAG-A-MATE coagulation analyzer (Organon Teknika Corp., Durham, NC). Diethanolamine, OPLS, phosphocholine calcium salt (PC), and glycerol-1-phosphate (PA) were obtained from Sigma. p-Nitrophenyl phosphate was purchased from Pierce. All buffer salts were purchased from Fisher and used without further purification.
Preparation of rFVIII·OPLS Complexes
The l-form of O-phosphoserine (OPLS) was selected for our studies, since this form has been shown to interact with rFVIII to a greater extent than the d-form (21). The rFVIII·OPLS complex was prepared by diluting rFVIII stock solution in buffer composed of 5, 10, or 20 mm OPLS, 25 mm Tris, 5 mm CaCl2, and 300 mm NaCl, pH 7.0. The solution was incubated at room temperature for 30 min. rFVIII complexes with PA or PC were prepared as described for rFVIII·OPLS complex by diluting with Tris buffer containing 10 mm PA or PC. For immunogenicity studies, all solutions were prepared using pyrogen-free water and were sterile filtered prior to use.
Complexation of rFVIII with OPLS can also be accomplished during purification of rFVIII. Briefly, rFVIII was expressed transiently by transfecting COS-7 cells with the plasmid containing the cDNA of human FVIII. The transfected cells were cultured in serum free media. The medium was subjected to a two-step ion exchange chromatography (22). The factor VIII-containing medium was loaded on a HiPrep 16/10 SP FF column (Amersham Biosciences) and eluted using a linear gradient from 100−650 mm NaCl in HEPES-CaCl2 buffer containing 10 mm OPLS. Fractions containing rFVIII were loaded on a Resource Q column (Amersham Biosciences) and eluted with a 200 mm to 1 m NaCl linear gradient in the HEPES-CaCl2 buffer. The purity of the isolated protein was assessed by silver-stained SDS-PAGE and size exclusion chromatography (SEC) using a Biosep SEC-S-4000 (Phenomenex, Torrance, CA) (details to be published elsewhere). It was also found that inclusion of OPLS in the elution buffer enhanced the recovery of rFVIII.
Effect of OPLS on the Tertiary Structure of rFVIII
rFVIII was used at a concentration of 5 μg/ml (0.017 μm) in Tris buffer (Tris (25 mm), sodium chloride (300 mm), and calcium chloride (5 mm), pH 7). OPLS was dissolved in Tris buffer, and pH was adjusted to 7.0. Stock solution of OPLS at 1 mm concentration was prepared in Tris buffer containing 5 μg/ml rFVIII. Required volumes (as calculated by the alligation method) of the above solution were mixed with a 5 μg/ml solution of rFVIII in Tris buffer to obtain various concentrations of OPLS. Following each addition, the sample was allowed to equilibrate for 5 min before the fluorescence intensity was measured. Changes in tertiary structure of the protein in the presence of various concentrations of OPLS were monitored using a PTI-Quantamaster fluorescence spectrophotometer (Photon Technology International, Lawrenceville, NJ). Excitation wavelength was set at 285 nm, and emission was monitored at 335 nm (23). An I-shaped cuvette was used to minimize any inner filter effect (24). The fluorescence intensity (F) at 335 nm in the presence of a given concentration of OPLS was normalized to the fluorescence intensity of rFVIII (Fo), to obtain the F/Fo ratio. The F/Fo is related to rFVIII and lipid concentrations by the following set of equations,
| (Eq. 1) |
| (Eq. 2) |
where P represents protein, L is OPLS, PL is protein-OPLS complex, [L] is free OPLS concentration, PT is total protein concentration, n is stoichiometry, and Fmax is maximal change in F/Fo.
The data were plotted as F/Fo versus OPLS concentration (μm) and fitted using WinNonlin (Pharsight, Mountainview, CA) with the following expression derived from the above equations to obtain estimates for KD.
| (Eq. 3) |
The above equation assumes a stoichiometry (n) of 1 and a single binding site.
Sandwich ELISA: Interaction of OPLS with rFVIII
Binding of OPLS to the phospholipid-binding region of rFVIII was confirmed by evaluating the ability of OPLS to compete with ESH4, an antibody against the phospholipid-binding region of the C2 domain (25) in a sandwich ELISA. Nunc-Maxisorb 96-well plates were coated with ESH4 antibody by incubating 50 μl/well solution of the antibody at a concentration of 5 μg/ml in carbonate buffer (0.2 m, pH 9.4) overnight at 4 °C. The plate was then washed with phosphate buffer containing 0.05% Tween 20 (PBT consisting of 10 mm Na2HPO4, 1.8 mm KH2PO4, 0.14 mm NaCl, 2.7 mm KCl, and 0.02% NaN3). The remaining nonspecific protein binding sites on the plastic's adsorptive surface were blocked by incubating 200 μl of blocking buffer consisting of 1% bovine serum albumin in phosphate buffer (consisting of 10 mm Na2HPO4, 1.8 mm KH2PO4, 0.14 mm NaCl, and 2.7 mm KCl) for2hat room temperature. The plates were washed with PBT, and 50 μl of 100 ng/ml rFVIII in the presence or absence of OPLS in blocking buffer was added and incubated at 37 °C for 1 h. The plates were washed with PBT and incubated with 50 μl of biotinylated ESH8 at a 1 μg/ml concentration and 50 μl of a 1:1000 dilution of avidin-alkaline phosphatase conjugate, both in blocking buffer at room temperature for 1 h. The plates were washed with PBT and 100 μl of 1 mg/ml p-nitrophenyl phosphate solution in diethanolamine buffer (consisting of 1 m diethanolamine and 0.5 mm MgCl2). The plates were incubated at room temperature for 30 min, and the reaction was quenched by adding 100 μl of 3 n NaOH. Optical density at 405 nm was monitored using a plate reader.
Effect of OPLS on Thermal Denaturation of rFVIII
Circular Dichroism Spectroscopy
CD spectroscopy was used to monitor the unfolding of rFVIII upon thermal stress. CD spectra were acquired on a JASCO-715 spectropolarimeter calibrated with d-10-camphor sulfonic acid. Thermal denaturation was performed in the presence and in the absence of 5 mm OPLS at a controlled heating rate of 60 °C/h from 20 to 80 °C. Protein concentrations typically used were 25 μg/100 μl in Tris buffer. Ellipticity was monitored at 215 nm, and the spectrum was obtained from 250 to 208 nm in a 0.1-cm quartz cuvette. The shorter path length cuvette was used to minimize the contribution of OPLS on rFVIII signal. Further the CD spectrum of rFVIII was corrected by subtracting the base-line spectrum of 5 mm OPLS buffer. Higher concentrations of OPLS were not investigated as it contributed significantly to the buffer base line. It is appropriate to mention here that thermal denaturation studies were carried out in Tris buffer because of its low metal ion binding capacity (26). However, Tris buffer has a high temperature coefficient, which causes pH changes at elevated temperatures. Hence the observed changes at elevated temperatures could be due to combination of temperature and pH changes.
Steady State Fluorescence Anisotropy Studies
Changes in the steady-state emission anisotropy (r) of rFVIII upon thermal denaturation in the presence and in the absence of OPLS were measured by a PTI-Quantamaster fluorescence spectrophotometer, equipped with a Peltier unit and motorized Glan-Thompson polarizing prisms. The concentration of the protein used was typically 5 μg/ml, and a variable path length cuvette was used to minimize the inner filter effect. The G-factor was determined at 20 °C and was applied to data collected at higher temperatures. Thermal denaturation was conducted at a heating rate of 60 °C/h. Studies were carried out over the temperature range of 20−80 °C with a holding time of 1−2 min every 5 °C. Samples were excited at 280 nm, and the emission was monitored at 335 nm. Excitation and emission slit widths were set at 4 nm.
Anisotropy was calculated using the following equation (24),
| (Eq. 4) |
where represents emission intensity with the emission polarizer oriented parallel to the excitation polarizer, is emission intensity with the emission polarizer oriented perpendicular to the excitation polarizer, and G is instrument-specific G-factor .
Data were represented as percentage change in anisotropy (r)asa function of temperature. The percentage change in anisotropy (r) was computed as follows,
| (Eq. 5) |
where ro represents the anisotropy of the protein at 20 °C, and rt is the anisotropy at a given temperature.
Size Exclusion Chromatography
SEC was performed to confirm the onset of aggregation observed in the CD experiments. SEC was performed using a Biosep-SEC4000S 300 × 4.6-mm (Phenomenex, Torrance, CA) column with an exclusion limit of 2,000,000 daltons. The column was calibrated using a standard protein mixture of known molecular weight from Bio-Rad (Hercules, CA). The chromatograph consisted of a Waters 510 isocratic pump (Waters, Milford, MA), equipped with a Shimadzu (Shimadzu, Braintree, MA) autoinjector, a column oven, fluorescence detector, and an integrator. Tris buffer was used as the mobile phase, and eluent was monitored at an excitation of 285 nm and emission of 335 nm. Typical sample injection volumes were 50 μl. SEC profiles of rFVIII (20 μg/ml) in the presence and in the absence of OPLS (5 mm) at different temperatures of thermal denaturation were obtained by heating the protein at controlled heating rates of 60 °C/h. Samples were withdrawn at 25, 35, 45, 50, 55, 60, 65, 70, and 75 °C and stored at 4 °C prior to injection onto the column.
Animals
A colony of hemophilia A mice (C57BL/6J with a target deletion in exon 16 of the FVIII gene) was established with breeding pairs from the original colony (27). Equal numbers of adult male and female mice, aged 8−12 weeks, were used for the studies. The sex of the animal has no impact on the immune response (28).
Determination of rFVIII·OPLS Complex Activity in Vivo
The in vivo activity of rFVIII·OPLS complex was confirmed by the tail clip method in the hemophilia A mice (29). Groups of animals (n = 3) were administered subcutaneously either with Tris buffer or with rFVIII alone or rFVIII·OPLS complex (10 mm). The dose of rFVIII used was ∼2 μg of protein (9.85 IU) per animal. The tip (1 cm) of the tail of each animal was cut off with a sharp scalpel about 2 h after administration. The animals were then monitored for the next 18 h and the survival was noted at the end of the period.
Immune Response
Immunization and Sampling
Immunization of hemophilia A mice consisted of two intravenous injections that were 2 weeks apart or four subcutaneous injections 1 week apart of rFVIII or rFVIII·OPLS complex (2 μg in 100 μl of Tris buffer). The subcutaneous route of administration was investigated mainly to amplify the immune response. Further, Reipert et al. (30) have reported that the IgG subtype levels were identical after subcutaneous and intravenous administration in hemophilia A mice, suggesting an identical mechanism of immune response for subcutaneous and intravenous routes. Blood samples were obtained in acid citrate dextrose buffer at various time points up to 6 weeks by cardiac puncture. Immunogenicity of rFVIII with PA or PC was also evaluated after subcutaneous administration. All studies were performed in accordance with the guidelines of the Institutional Animal Care and Use Committees at the University at Buffalo.
Measurement of Total and Inhibitory Anti-rFVIII Antibody Titers
Antibody titers were determined by standard antibody capture ELISA (31). Briefly, Nunc-Maxisorb 96-well plates were coated overnight at 4 °C with 50 μl/well of 5 μg/ml rFVIII and subsequently blocked with 1% bovine serum albumin (blocking buffer). 50 μl/well of various dilutions of the sample and standard concentrations (12.5−150 μg/ml) of ESH8 antibody in blocking buffer were incubated at 37 °C for 1 h. The plates were washed and incubated with 50 μl of goat anti-mouse Ig (IgG + IgM + H + L)-alkaline phosphatase conjugate at a 1:1000 dilution in blocking buffer at room temperature for 1 h. Color was developed for 30 min with 100 μl of 1 mg/ml p-nitrophenyl phosphate solution in diethanolamine buffer (consisting of 1 m diethanolamine and 0.5 mm MgCl2). Reaction was quenched by the addition of 100 μl of 3 n NaOH. Optical density at 405 nm was monitored using a plate reader. The plate-specific parameter (PSP) was used to normalize the plate to plate variability. The PSP for each plate was obtained as follows. A linear standard curve of known ESH8 antibody concentrations was obtained for each plate and used to calculate the maximum and minimum predicted absorbance. Half of the difference between the maximum and minimum predicted absorbance was calculated as the PSP. A linear regression of the plot of absorbance of various dilutions (1:100 to 1:40,000) versus log of dilution was used to calculate the dilution that gave an optical density equal to the PSP. The dilution so obtained was the antibody titer for the sample.
Inhibitory antibody titers were determined using the Nijmegen modification of the Bethesda assay as described previously (32). Briefly, 100 μl of various dilutions of mouse plasma diluted in human FVIII-deficient plasma were mixed with an equal volume of normal human plasma and incubated at 37 °C for 2 h. Residual activity at the end of the incubation was determined using an activated partial thromboplastin time assay. A linear regression of the plot of residual activity of various dilutions (1:2 to 1:32,000) versus log of dilution was performed to calculate the dilution with 50% reduction in activity and was expressed in Bethesda units.
Statistical analysis was conducted using Minitab Statistical Software, Minitab Release 14 (Minitab Inc., State College, PA).
RESULTS AND DISCUSSION
Effect of OPLS and Phosphatidylserine Analogs on the Tertiary Structure of rFVIII
In order to determine the effect of OPLS on the tertiary structure of the protein, the steady state fluorescence spectrum of the protein was monitored in the presence of various concentrations of OPLS. A concentration-dependent decrease in F/Fo of rFVIII at 335 nm was observed in the presence of OPLS (Fig. 2a) without any change in emission λmax. Similar spectral changes have been interpreted as conformational fluctuations for factor Va upon interaction with short chain phospholipids (33). The data were fit using Equation 3 (described under “Experimental Procedures”) to estimate the binding parameters. A high apparent KD value (5.71 μm) indicates that OPLS interaction with rFVIII is weak. Phosphatidylserine analogues with a hydrophobic acyl chain such as dihexyl phosphatidylserine showed a lower apparent KD value (3.31 μm), and the observed higher affinity is probably due to both hydrophobic and electrostatic interactions (data not shown). It is appropriate to mention that the apparent KD is a phenomenological parameter and may not represent the actual binding affinity of OPLS to rFVIII.
Fig. 2. Effect of OPLS on structure of rFVIII.
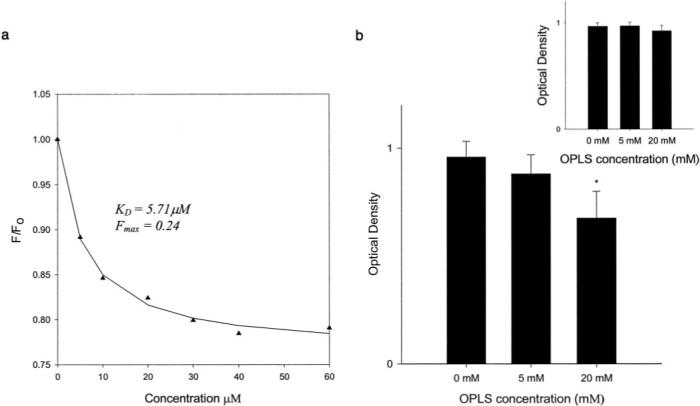
a, changes in the intrinsic fluorescence (F/Fo) of rFVIII as a function of OPLS concentration. The excitation was set at 285 nm, and emission was monitored at 335 nm with a 4-nm slit width. The protein concentration typically used was 5 μg/ml. Raw data were fitted using Equation 3 to obtain estimates for the binding parameters. b, effect of OPLS on the binding of rFVIII to ESH4 antibody as determined by sandwich ELISA. Inset, effect of preincubation of OPLS on the binding of rFVIII with ESH4. Statistical analysis was by analysis of variance followed by post hoc Dunnet's test (*, p < 0.05).
Sandwich ELISA: Interaction of OPLS with rFVIII
Sandwich ELISA studies were carried out to determine whether OPLS binds to the lipid-binding region (2303−2332) of the C2 domain. The monoclonal antibody, ESH4, that binds to the lipid-binding region was used as a stationary antibody, whereas biotinylated ESH8 that recognizes a nonoverlapping region in the C2 domain was used as probe antibody. If OPLS binds to the lipid-binding region of the protein, it would compete with ESH4, leading to a reduction in rFVIII binding to ESH4 (Fig. 2b). As can be seen from the figure, the binding of rFVIII to ESH4 antibody reduces with increasing concentrations of OPLS, suggesting that the binding site of OPLS and ESH4 overlap. In order to evaluate the possible interference from OPLS (a charged molecule) on the amounts of ESH4 coated on the plates, a control experiment was conducted where OPLS alone was incubated with ESH4-coated wells prior to the addition of rFVIII. As can be seen from the inset of Fig. 2b, the optical densities were unaltered and are independent of the concentration of OPLS. The result indicates that the reduction of rFVIII binding to ESH4 observed with OPLS is mainly due to competition between ESH4 and OPLS for binding to rFVIII.
Effect of OPLS on Thermal Denaturation of rFVIII
Thermal stress has been used to evaluate the folding and stability relationship of several proteins (34, 35). It has also been used to mimic pharmaceutical development conditions (36) and to evaluate excipients for protein formulations (37). The propensity of the protein to aggregate is an indicator of the expected stability, efficacy and immunogenicity of a protein formulation. rFVIII has been shown to be susceptible to aggregation following subtle conformational changes (38), which may at least involve the C2 domain (encompassing the lipid-binding region) (39). In order to determine whether the binding of OPLS to this region interferes with aggregation of rFVIII, thermal denaturation studies of rFVIII in the presence and in the absence of OPLS were carried out.
Circular Dichroism Spectroscopy
The CD spectrum of the rFVIII taken at 20 °C, in the absence of OPLS, showed a negative band at 215 nm, suggesting that the protein is composed predominantly of β-sheets (38). The ellipticity values monitored at 215 nm over a temperature range of 20−80 °C at a heating rate of 60 °C/h were used to generate the melting profile (Fig. 3a). No significant change in the ellipticity values was observed between 20 and 50 °C, suggesting that the protein undergoes very little change in the secondary structure. Beyond 50 °C, the negative ellipticity increased with the increase in temperature, initiating thermal transition. Size exclusion chromatography studies conducted at various temperatures along the thermal denaturation profile confirmed the coexistence of the native and aggregated forms of the protein during the transition. A representative chromatogram obtained at 55 °C is shown in Fig. 3d. At temperatures greater than 70 °C, the CD spectral characteristics displayed a positive band below 210 nm, and the negative band reduced in intensity and was red-shifted, consistent with the formation of intermolecular β strands (38, 40). This spectral change resulted in decrease in ellipticity values at 215 nm and distorted the melting profile, causing a reverse trend (indicated as an asterisk in Fig. 3a). When thermal denaturation was carried out in the presence of 5 mm OPLS, the loss of negative ellipticity beyond 70 °C was not observed. The spectral analysis of rFVIII in the presence of 5 mm OPLS and at temperatures beyond 70 °C showed that the characteristic features of intermolecular β-strands were absent (Fig. 3b), indicating that the presence of OPLS reduced the aggregate formation.
Fig. 3. Effect of OPLS on Stability of rFVIII.
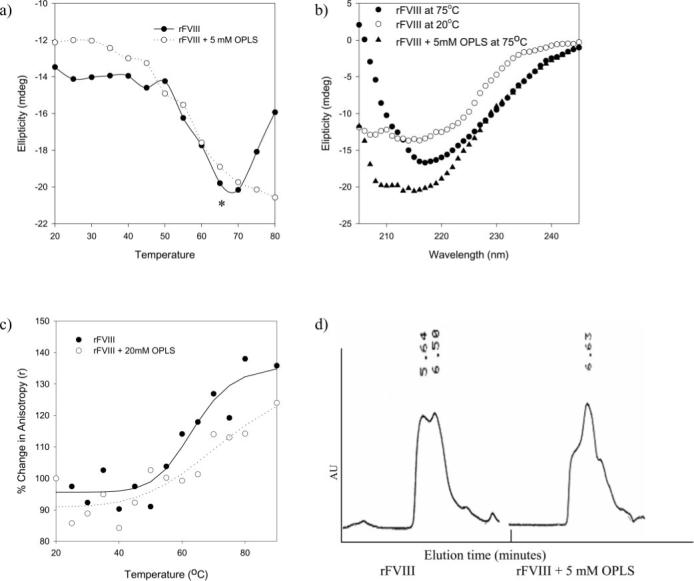
a, temperature-dependant ellipticity changes of rFVIII in the presence (○) and in the absence (●) of OPLS monitored at 215 nm. Secondary structural transition of rFVIII was monitored over the temperature range of 20−80 °C at a heating rate of 60 °C/h. The concentration of the protein used was ∼25 μg/100 μl, and the path length of the quartz cuvette was 0.1 cm. b, CD spectra of rFVIII in the presence and in the absence of OPLS acquired at various temperatures. Open symbols, 20 °C; closed symbols, 75 °C. c, temperature-dependent changes in the steady state fluorescence anisotropy of rFVIII in the presence (○) and in the absence (●) of 20 mm OPLS. Changes in anisotropy were monitored over the temperature range of 20−80 °C at a heating rate of 60 °C/h. The protein concentration was ∼5 μg/ml. The excitation wavelength was set at 280 nm, and the emission was monitored at 335 nm. d, a representative size exclusion chromatography profile of rFVIII obtained at 55 °C in the presence and in the absence of OPLS. rFVIII was subjected to thermal stress over the temperature range of 25−75 °C at 60 °C/h, and elution was monitored using a fluorescence detector with excitation and emission wavelengths set at 285 and 335 nm, respectively. The elution time is shown in minutes. Gel filtration was carried out under isocratic conditions at a flow rate of 0.4 ml/min. Samples were cooled down and stored for not more than 60 min at 4 °C prior to analysis. The column temperature was maintained at 20 °C. AU, arbitrary units.
Steady State Fluorescence Anisotropy Studies
Thermal denaturation and aggregation of rFVIII was further investigated by fluorescence anisotropy studies. The anisotropy values of aggregated protein would be higher than that of the native protein due to increase in the molecular volume (24). In order to determine whether OPLS interferes with the formation of aggregates, the anisotropy values of rFVIII in the presence and in the absence of OPLS were measured in the temperature range of 20−80 °C. For rFVIII alone, the anisotropy values did not change in the temperature range of 20−50 °C, but a further increase in temperature resulted in an increase in anisotropy values, resembling the melting profile generated by CD studies (Fig. 3c). Since the anisotropy value represents an average of several populations (native and aggregated forms; dimers, trim-ers, and multimers) of rFVIII, an increase in anisotropy values indicated that the relative fraction of aggregates and/or aggregate size increased with temperature. In the presence of OPLS, the magnitude of anisotropy changes is reduced, indicating that OPLS reduces the fraction and/or size of aggregates.
Size Exclusion Chromatography
The effect of OPLS on aggregation behavior of rFVIII was further confirmed by SEC studies. Native rFVIII appears as a single broad peak at ∼6.5−7 min. The samples collected during the thermal transition were subjected to SEC analysis. Similar to the observations with CD spectroscopy, the SEC profiles of rFVIII show very little change over the temperature range of 20−50 °C. At 55 °C, formation of aggregates was detected as a peak at 5.6 min, which progressively increases with temperature (Fig. 3d). The peak height ratio (PHR) of the aggregated species to native protein represents the relative populations of the two. At 55 °C, a substantial fraction of aggregated protein was detected when rFVIII alone was subjected to thermal stress (PHR = 0.987). In the presence of OPLS, the relative height of the native protein peak is greater than the aggregated peak (PHR = 0.605), suggesting that OPLS decreases the formation of aggregates.
Aggregation of rFVIII is a principle mechanism responsible for the inactivation rFVIII (41). rFVIII is susceptible to aggregation that may be initiated by subtle conformational changes in the tertiary structure (38) possibly occurring in the C2 domain (39). Based on three-dimensional structure and mutational analysis, it has been proposed that the C2 domain contains 2−4 hydrophobic loops that are susceptible to intermolecular interactions (15-17). Further, the C2 domain also encompasses several unstructured loops, having high flexibility (Fig. 1) (13). OPLS binding may stabilize this sticky region, prevent nonspecific binding, and interfere with the aggregation of rFVIII. We speculate that these stabilizing effects may be beneficial during storage and administration of rFVIII preparations, since they may reduce aggregation and surface adsorption to vials and syringes.
Determination of rFVIII·OPLS Complex Activity in Vivo
In order to investigate the activity and systemic effect of rFVIII and rFVIII·OPLS complex upon subcutaneous administration, survival studies were carried out in hemophilia A mice. These mice do not produce any active murine FVIII. Hence, bleeding caused by the tail clipping would result in the death of the animal in about 17 h, as observed for animals given Tris buffer alone. Animals given rFVIII and the rFVIII·OPLS complex survived beyond 18 h; therefore, rFVIII·OPLS and rFVIII were equally effective in promoting coagulation and, hence, survival of the animals after tail clipping. Upon visual examination, all of the animals receiving rFVIII or rFVIII·OPLS ceased to bleed within 2 h of the tail clip. This suggests that complexation of rFVIII with OPLS did not result in inactivation of rFVIII, and sufficient protein reached the systemic circulation after subcutaneous administration, possibly via lymphatics, to exert a pharmacological effect. Due to the qualitative nature of the in vivo activity measurement, a meaningful dose-response relationship could not be elucidated. A more quantitative pharmacokinetic and pharmacodynamic analysis aimed at determining the concentration and activity of rFVIII in blood, bioavailability, and half-life is in progress.
Immunogenicity of rFVIII·OPLS Complex
The relative immunogenicity of rFVIII·OPLS complex and rFVIII was evaluated in the hemophilia A mice. Hemophilia A mice have been routinely employed for investigating the immunogenicity of FVIII preparations, since their immune response is qualitatively similar to that observed in humans (28, 42). Further, murine models are valuable to measure relative immunogenicity of protein preparations (43, 44).
The total antibody titers and inhibitory titers in immunized mice were determined by antibody capture ELISA and the Nijmegen modification of the Bethesda assay (Fig. 4), respectively. Studies conducted in hemophilia A mice after intravenous administration resulted in mean total antibody titer of 1532.3 ± 646 (S.E.; n = 5) for rFVIII and 449.93 ± 397 (S.E.; n = 6) for rFVIII·OPLS at 6 weeks. Further, four of the six animals administered with rFVIII·OPLS did not develop any detectable antibody titers. However, due to the lower magnitude of immune response after intravenous administration (43) and high variability, a meaningful statistical analysis could not be performed.
Fig. 4. Effect of OPLS on the Immunogenicity of rFVIII.
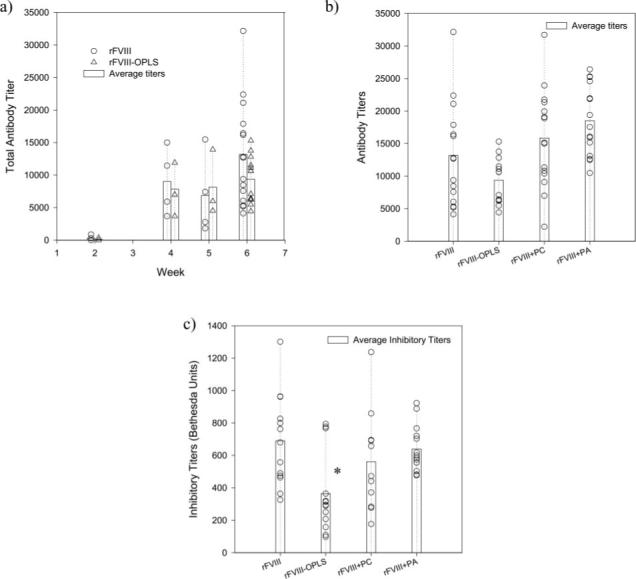
a, total mean (bars) and individual (open symbols) antibody titers against rFVIII as determined by antibody capture ELISA in hemophilia A mice immunized with rFVIII (○) or rFVIII·OPLS (△). b, total mean (bars) and individual (open symbols) antibody titers at 6 weeks as determined by antibody capture ELISA in hemophilia A mice immunized with rFVIII, rFVIII·OPLS, rFVIII + PC, or rFVIII + PA. c, mean (bars) and individual (open symbols) inhibitory titers at 6 weeks as determined by the Nijmegen modification of the Bethesda assay in hemophilia A mice immunized with rFVIII, rFVIII·OPLS, rFVIII + PC, or rFVIII + PA. Statistical analysis was by analysis of variance followed by post hoc Dunnet's test (*, p < 0.05).
In subcutaneously administered animals, antibodies were detected as early as 2 weeks with rFVIII and the rFVIII·OPLS complex. Immune responses were higher at 4 weeks for both preparations relative to the second week (Fig. 4a). Statistical analysis to determine differences in immune response between groups (rFVIII and rFVIII·OPLS) in the earlier weeks (2, 4, and 5 weeks; n = 4) was not attempted, since the number of animals per group was small. At the end of 6 weeks, for mice immunized with rFVIII, the mean total antibody titer was 13,167 ± 2042.0 (S.E.; n = 15), and the inhibitory antibody titer was 689.7 ± 78.1 Bethesda units (S.E.; n = 13). The high inhibitory antibody titers observed may be due to the administration of human protein in a nontolerized mice model. The administration of the rFVIII·OPLS complex resulted in a lower immune response at the end of 6 weeks compared with rFVIII. The mean total antibody titer and the inhibitory antibody titer for mice immunized with the rFVIII·OPLS complex was 9355.0 ± 979.00 (S.E.; n = 13) and 364.7 ± 69.25 Bethesda units/ml (S.E.; n = 13, p < 0.05), respectively. The presence of OPLS in the rFVIII preparation lowered anti-rFVIII inhibitory antibody formation by almost 50% compared with rFVIII alone (Fig. 4c). Further, the immunogenicity of rFVIII administered with PC or PA as control was also investigated to demonstrate the specificity of OPLS effect. The total antibody titers and inhibitory titers (Fig. 4, b and c) in the presence of PC or PA are comparable with rFVIII alone (p value < 0.05) and are higher than that observed for the rFVIII·OPLS complex. Overall, rFVIII·OPLS appears to have lower immunogenicity compared with rFVIII in hemophilia A mice due to the specific interaction of OPLS with rFVIII.
The precise mechanism responsible for the lower immune response observed with rFVIII·OPLS complex compared with rFVIII is not clear. However, we speculate that the reduction in immunogenicity could be due to inefficient processing of the FVIII·OPLS complex by the immune system. Based on structural characteristics, the C2 domain (epitope region 2291−2330) has several immunodominant epitopes for efficient processing by antigen-presenting cells (13). The binding of OPLS to this immunodominant epitope may induce subtle conformational changes that may result in inefficient processing of rFVIII by the immune system compared with rFVIII alone.2 This, in turn, may lead to changes in the clonal expansion of Th2 cells and cytokine (e.g. interleukin-10 (47)) levels necessary for antibody production. Similar subtle changes in protein conformation have been linked to the lower immunogenicity of the freeze-dried form of the anti-idiotypic antibody MMA-383 (48).
Overall, the findings suggest that OPLS may be a beneficial excipient for the development of less immunogenic rFVIII preparations. Considering that OPLS is used as a supplement in polyamino acid formulations to treat amino acid deficiencies, any toxicity associated with OPLS as an excipient is unlikely (49). The present study clearly demonstrates a difference in the overall magnitude of the immune response between rFVIII and rFVIII·OPLS. We believe that this may lead to rational development of low immunogenic rFVIII preparations.
Acknowledgments
We thank the Pharmaceutical Sciences Instrumentation Facility (University at Buffalo) for the use of the circular dichroism spectropolarimeter and spectrofluorometer. We thank Renee Tetreault and Melinda Mucci for technical support. We thank Dr. Joseph P. Balthasar for suggestions, proofreading of the manuscript, and the use of the plate reader for antibody titer determinations using ELISA.
Footnotes
This work was supported by National Institutes of Health Grant RO1 HL-70227 (to S. V. B.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The abbreviations used are: rFVIII, recombinant human factor VIII; ELISA, enzyme-linked immunosorbent assay; FVIII, factor VIII; OPLS, O-phospho-l-serine; PA, phosphatidic acid; PBT, phosphate buffer containing Tween; PC, phosphocholine; PHR, peak height ratio; PSP, plate-specific parameter; SEC, size exclusion chromatography; ITT, immune tolerance therapy.
The complexation between rFVIII and OPLS is very weak; hence, it is difficult to envisage that rFVIII would circulate as a complex with OPLS. Further, OPLS and von Willebrand factor bind to overlapping epitopes (lipid-binding region) in the C2 domain (45, 46). OPLS would hence be displaced by von Willebrand factor in circulation. OPLS may provide protection to rFVIII over a very brief window of time (i.e. after administration but before association with von Willebrand factor). This window of time may be critical, since rFVIII may be exposed to processing by potent antigen-presenting cells.
REFERENCES
- 1.Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orr EC, Amphlett GW, Foster BW, Coe ML, Knutson GJ, Fass DN, Hewick RN. Nature. 1984;312:342–347. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- 2.Kaufman RJ, Wasley LC, Dorner AJ. J. Biol. Chem. 1988;263:6352–6362. [PubMed] [Google Scholar]
- 3.Vehar GA, Keyt B, Eaton D, Rodriguez H, O'Brien DP, Rotblat F, Oppermann H, Keck R, Wood WI, Harkins RN, Tuddenham EGD, Lawn RD, Capon DJ. Nature. 1984;312:337–342. doi: 10.1038/312337a0. [DOI] [PubMed] [Google Scholar]
- 4.Fay PJ. Arch. Biochem. Biophys. 1988;262:525–531. doi: 10.1016/0003-9861(88)90404-3. [DOI] [PubMed] [Google Scholar]
- 5.VanAken WG. Transfus. Med. Rev. 1997;11:6–14. doi: 10.1016/s0887-7963(97)80005-3. [DOI] [PubMed] [Google Scholar]
- 6.Jacquemin MG, Saint-Remy JM. Haemophilia. 1998;4:552–557. doi: 10.1046/j.1365-2516.1998.440552.x. [DOI] [PubMed] [Google Scholar]
- 7.Lollar P, Healey JF, Barrow RT, Parker ET. Adv. Exp. Med. Biol. 2001;489:65–73. doi: 10.1007/978-1-4615-1277-6_6. [DOI] [PubMed] [Google Scholar]
- 8.Ingerslev J. Haematologica. 2000;85:15–20. [PubMed] [Google Scholar]
- 9.Ho AY, Height SE, Smith MP. Drugs. 2000;60:547–554. doi: 10.2165/00003495-200060030-00003. [DOI] [PubMed] [Google Scholar]
- 10.Gensana M, Altisent C, Aznar JA, Casana P, Hernandez F, Jorquera JI, Magallon M, Massot M, Puig L. Haemophilia. 2001;7:369–374. doi: 10.1046/j.1365-2516.2001.00526.x. [DOI] [PubMed] [Google Scholar]
- 11.Ananyeva NM, Lacroix-Desmazes S, Hauser CA, Shima M, Ovanesov MV, Khrenov AV, Saenko EL. Blood Coagul. Fibrinolysis. 2004;15:109–124. doi: 10.1097/00001721-200403000-00001. [DOI] [PubMed] [Google Scholar]
- 12.Scandella DH, Nakai H, Felch M, Mondorf W, Scharrer I, Hoyer LW, Saenko EL. Thromb. Res. 2001;101:377–385. doi: 10.1016/s0049-3848(00)00418-7. [DOI] [PubMed] [Google Scholar]
- 13.Reding MT, Okita DK, Diethelm-Okita BM, Anderson TA, Conti-Fine BM. J. Thromb. Haemost. 2003;1:1777–1784. doi: 10.1046/j.1538-7836.2003.00251.x. [DOI] [PubMed] [Google Scholar]
- 14.Pratt KP, Qian J, Ellaban E, Okita DK, Diethelm-Okita BM, Conti-Fine B, Scott DW. Thromb. Haemost. 2004;92:522–528. doi: 10.1160/TH03-12-0755. [DOI] [PubMed] [Google Scholar]
- 15.Pratt KP, Shen BW, Takeshima K, Davie EW, Fujikawa K, Stoddard BL. Nature. 1999;402:439–442. doi: 10.1038/46601. [DOI] [PubMed] [Google Scholar]
- 16.Gilbert GE, Kaufman RJ, Arena AA, Miao H, Pipe SW. J. Biol. Chem. 2002;277:6374–6381. doi: 10.1074/jbc.M104732200. [DOI] [PubMed] [Google Scholar]
- 17.Stoilova-McPhie S, Villoutreix BO, Mertens K, Kemball-Cook G, Holzenburg A. Blood. 2002;99:1215–1223. doi: 10.1182/blood.v99.4.1215. [DOI] [PubMed] [Google Scholar]
- 18.Raju R, Navaneetham D, Okita D, Diethelm-Okita B, McCormick D, Conti-Fine BM. Eur. J. Immunol. 1995;25:3207–3214. doi: 10.1002/eji.1830251202. [DOI] [PubMed] [Google Scholar]
- 19.Kemball-Cook G, Barrowcliffe TW. Thromb. Res. 1992;67:57–71. doi: 10.1016/0049-3848(92)90258-c. [DOI] [PubMed] [Google Scholar]
- 20.Gilbert GE, Furie BC, Furie B. J. Biol. Chem. 1990;265:815–822. [PubMed] [Google Scholar]
- 21.Gilbert GE, Drinkwater D. Biochemistry. 1993;32:9577–9585. doi: 10.1021/bi00088a009. [DOI] [PubMed] [Google Scholar]
- 22.Doering C, Parker ET, Healey JF, Craddock HN, Barrow RT, Lollar P. Thromb. Haemost. 2002;88:450–458. [PubMed] [Google Scholar]
- 23.Purohit VS, Ramani K, Kashi RS, Durrani MJ, Kreiger TJ, Balasubramanian SV. Biochim. Biophys. Acta. 2003;1617:31–38. doi: 10.1016/j.bbamem.2003.08.012. [DOI] [PubMed] [Google Scholar]
- 24.Lakowicz JR. Principles of Fluorescence Spectroscopy. 2nd Ed. Kluwer Academic/Plenum Publishers; New York: 1999. pp. 291–318. [Google Scholar]
- 25.Saenko EL, Scandella D. J. Biol. Chem. 1997;272:18007–18014. doi: 10.1074/jbc.272.29.18007. [DOI] [PubMed] [Google Scholar]
- 26.Derrick TS, Kashi RS, Durrani M, Jhingan A, Middaugh CR. J. Pharm. Sci. 2004;93:2549–2557. doi: 10.1002/jps.20167. [DOI] [PubMed] [Google Scholar]
- 27.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH., Jr. Nat. Genet. 1995;10:119–121. doi: 10.1038/ng0595-119. [DOI] [PubMed] [Google Scholar]
- 28.Qian J, Borovok M, Bi L, Kazazian HH, Jr., Hoyer LW. Thromb. Haemost. 1999;81:240–244. [PubMed] [Google Scholar]
- 29.Sarkar R, Xiao W, Kazazian HH., Jr. J. Thromb. Haemost. 2003;1:220–226. doi: 10.1046/j.1538-7836.2003.00096.x. [DOI] [PubMed] [Google Scholar]
- 30.Reipert BM, Ahmad RU, Turecek PL, Schwarz HP. Thromb. Haemost. 2000;84:826–832. [PubMed] [Google Scholar]
- 31.Crowther JR. Methods in Molecular Biology. Vol. 149. Humana Press; Totowa, NJ: 2000. [Google Scholar]
- 32.Verbruggen B, Novakova I, Wessels H, Boezeman J, van den Berg M, Mauser-Bunschoten E. Thromb. Haemost. 1995;73:247–251. [PubMed] [Google Scholar]
- 33.Zhai X, Srivastava A, Drummond DC, Daleke D, Lentz BR. Biochemistry. 2002;41:5675–5684. doi: 10.1021/bi011844d. [DOI] [PubMed] [Google Scholar]
- 34.Pace CN, Hebert EJ, Shaw KL, Schell D, Both V, Krajcikova D, Sevcik J, Wilson KS, Dauter Z, Hartley RW, Grimsley GR. J. Mol. Biol. 1998;279:271–286. doi: 10.1006/jmbi.1998.1760. [DOI] [PubMed] [Google Scholar]
- 35.Vermeer AW, Norde W. Biophys. J. 2000;78:394–404. doi: 10.1016/S0006-3495(00)76602-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Remmele RL, Jr., Gombotz WR. BioPharm. 2000;13:36–46. [Google Scholar]
- 37.Tsai PK, Volkin DB, Dabora JM, Thompson KC, Bruner MW, Gress JO, Matuszewska B, Keogan M, Bondi JV, Middaugh CR. Pharm. Res. (N.Y.) 1993;10:649–659. doi: 10.1023/a:1018939228201. [DOI] [PubMed] [Google Scholar]
- 38.Grillo AO, Edwards KL, Kashi RS, Shipley KM, Hu L, Besman MJ, Middaugh CR. Biochemistry. 2001;40:586–595. doi: 10.1021/bi001547t. [DOI] [PubMed] [Google Scholar]
- 39.Ramani K, Purohit VS, Miclea R, Lakhman SS, Balasubramanian SV. AAPS Pharm. Sci. 2003;5:T2091. [Google Scholar]
- 40.Kendrick BS, Cleland JL, Lam X, Nguyen T, Randolph TW, Manning MC, Carpenter JF. J. Pharm. Sci. 1998;87:1069–1076. doi: 10.1021/js9801384. [DOI] [PubMed] [Google Scholar]
- 41.Wang W, Kelner DN. Pharm. Res. (N. Y.) 2003;20:693–700. doi: 10.1023/a:1023271405005. [DOI] [PubMed] [Google Scholar]
- 42.Behrmann M, Pasi J, Saint-Remy JM, Kotitschke R, Kloft M. Thromb. Haemost. 2002;88:221–229. [PubMed] [Google Scholar]
- 43.Braun A, Kwee L, Labow MA, Alsenz J. Pharm. Res. (N. Y.) 1997;14:1472–1478. doi: 10.1023/a:1012193326789. [DOI] [PubMed] [Google Scholar]
- 44.Chirino AJ, Ary ML, Marshall SA. Drug Discov. Today. 2004;9:82–90. doi: 10.1016/S1359-6446(03)02953-2. [DOI] [PubMed] [Google Scholar]
- 45.Saenko EL, Shima M, Rajalakshmi KJ, Scandella D. J. Biol. Chem. 1994;269:11601–11605. [PubMed] [Google Scholar]
- 46.Saenko EL, Scandella D. J. Biol. Chem. 1995;270:13826–13833. doi: 10.1074/jbc.270.23.13826. [DOI] [PubMed] [Google Scholar]
- 47.Wu H, Reding M, Qian J, Okita DK, Parker E, Lollar P, Hoyer LW, Conti-Fine BM. Thromb. Haemost. 2001;85:125–133. [PubMed] [Google Scholar]
- 48.Taschner N, Muller SA, Alumella VR, Goldie KN, Drake AF, Aebi U, Arvinte T. J. Mol. Biol. 2001;310:169–179. doi: 10.1006/jmbi.2001.4736. [DOI] [PubMed] [Google Scholar]
- 49.Matera M, Castana R, Insirello L, Leonardi G. Int. J. Clin. Pharmacol. Res. 1993;13:93–105. [PubMed] [Google Scholar]