

Abstract
Deregulated Myc expression drives many human cancers, including Burkitt's lymphoma and a highly aggressive subset of diffuse large cell lymphomas. Myc-driven tumors often display resistance to chemotherapeutics because of acquisition of mutations that impair the apoptosis pathway regulated by the Bcl-2 protein family. Given the need to identify new therapies for such lymphomas, we have evaluated the efficacy of ABT-737, a small molecule that mimics the action of the BH3-only proteins, natural antagonists of the prosurvival Bcl-2 proteins. ABT-737 selectively targets certain prosurvival proteins (Bcl-2, Bcl-xL, and Bcl-w) but not others (Mcl-1 and A1). We treated mice transplanted with lymphomas derived either from Eμ-myc transgenic mice or Eμ-myc mice that also expressed an Eμ-bcl-2 transgene. As a single agent, ABT-737 significantly prolonged the survival of mice transplanted with the myc/bcl-2 lymphomas but was ineffective for the myc lymphomas, probably because of the relatively higher Mcl-1 levels found in the latter. Strikingly, when combined with low-dose cyclophosphamide, ABT-737 produced sustained disease-free survival of all animals transplanted with two of three myc/bcl-2 lymphomas tested. The combination therapy was also more effective against some myc lymphomas than treatment with either agent alone. Our data suggest that antagonism of Bcl-2 with small organic compounds is an attractive approach to enhance the efficacy of conventional therapy for the treatment of Myc-driven lymphomas that over-express this prosurvival molecule.
The transcription factor Myc plays a pivotal role in controlling cell growth, proliferation, differentiation, and apoptosis. Up to 70% of human tumors exhibit deregulated expression of the Myc oncoprotein or one of its close relatives (1). Myc-driven lymphomas, typified by Burkitt's lymphoma in which the c-myc gene is deregulated by chromosome translocation to Ig gene loci (2), but which also include a subset of highly aggressive diffuse large cell lymphomas (3, 4), characteristically display high rates of proliferation and apoptosis. Whereas some are exquisitely sensitive to chemotherapeutics, others display intrinsic or acquired resistance (5).
It is now well recognized that treatment resistance can be acquired via perturbations in the p53-regulated pathway (6) and in the stress-induced apoptosis pathway that is regulated by the Bcl-2 protein family (7). In normal cells, stress signals activate BH3-only proteins, distant relatives of Bcl-2 that bind to and neutralize Bcl-2 and its anti-apoptotic relatives (Bcl-xL, Bcl-w, Mcl-1, and A1), thereby releasing restraints on proapoptotic proteins Bax and Bak (8). In tumor cells, the normal controls are often perturbed because Bcl-2 is elevated or activation of BH3-only genes is compromised. Mutations up-regulating Bcl-2 include the t (14, 18) chromosome translocation in follicular lymphoma (9) or the down-regulation of miR-15a and miR-16–1 microRNAs in chronic lymphocytic leukemias (10). Genetic lesions crippling BH3-only proteins include the following: for Noxa and Puma, functional inactivation of their key regulator p53, which occurs in >50% of human tumors (6); for Noxa, mutation and silencing (diffuse large B-cell lymphoma cell lines) (11); and for Bim, homozygous deletion (mantle cell lymphomas) (11, 12), promoter methylation (Burkitt's lymphoma and diffuse large B-cell lymphomas) (11), and amplification of the miR-17–92 locus (13).
Small molecules recently developed as BH3 mimetics by Abbott Laboratories have shown promising preclinical single agent activity in multiple hematopoietic tumor-derived cell lines or primary tumor samples, including leukemias, lymphomas, and myeloma (14–20). Like the BH3-only proteins, ABT-737 and an orally available compound in the same class, ABT-263 (21), induce apoptosis by antagonizing prosurvival Bcl-2 proteins. ABT-737 and ABT-263 directly bind to and neutralize Bcl-2, Bcl-xL, and Bcl-w but do not appreciably bind to Mcl-1 or A1, thereby mimicking the BH3-only protein Bad (14, 22, 23). Because these drugs act downstream of p53 and upstream of Bcl-2 and its closest homologs, they can potentially obviate the chemoresistance mediated by p53 mutations or overexpression of Bcl-2, Bcl-xL, or Bcl-w. However, because cell killing requires inactivation of most, if not all, prosurvival Bcl-2 relatives (22), ABT-737 and ABT-263 are not efficacious for cells harboring significant levels of Mcl-1 or A1 (15, 21, 23, 24).
Much has been learned about the biology of Myc-driven cancers through the study of lymphomas arising in Eμ-myc transgenic mice (25, 26), in which overexpression of c-myc in the B cell compartment is driven by the Ig heavy chain enhancer (Eμ), modeling human Burkitt's lymphoma (2). Eμ-myc transgenic mice are also invaluable for evaluating treatment strategies. Multiple primary Eμ-myc-driven lymphomas carrying either spontaneous or introduced mutations can be readily expanded by transplantation into syngeneic mice for in vitro and in vivo treatment studies (27). Given the need to identify new therapies for aggressive Myc-driven lymphomas that have failed standard therapies, and the knowledge that defective apoptosis in these tumors is commonly associated with chemoresistance (5), we have evaluated the impact of ABT-737 in immunocompetent mice bearing Eμ-myc lymphomas (25, 26) or Eμ-myc lymphomas overexpressing Bcl-2 (28).
Results
To evaluate the sensitivity of Myc-driven lymphomas to ABT-737, we collected primary tumors from C57BL/6 Eμ-myc transgenic mice (hereafter myc mice) (25, 26) and from bitransgenic C57BL/6 Eμ-myc/Eμ-bcl-2–22 mice (hereafter myc/bcl-2 mice) (28). All of the myc/bcl-2 tumors had a lymphomyeloid progenitor phenotype, as previously reported for tumors arising in a mixed genetic background (28, 29), whereas the myc lymphomas had a pre-B or IgM+ B cell phenotype (26). Each primary (T0) tumor was expanded by injection into multiple nonirradiated C57BL/6 recipients and the tumors arising in the transplant recipients (T1) were pooled and cryopreserved for analysis.
Mice were transplanted with (T1) lymphoma cells on day 0 and treatment was commenced on day 4 with a 14-day course of either ABT-737 (75 mg/kg/d) or the vehicle alone. Surprisingly, as a single agent, ABT-737 was ineffectual for treating myc lymphomas. It did not enhance survival of mice bearing any of the 6 pre-B or 6 B myc lymphomas tested (Fig. 1A) or reduce their tumor burden (supporting information (SI) Table S1). Consistent with this finding, these myc lymphomas were also resistant to ABT-737 in vitro (Fig. S1A).
Fig. 1.

Response of Myc-driven lymphomas to ABT-737 in vivo. (A) Kaplan–Meier survival curves of mice transplanted (day 0) with myc lymphomas and treated, from d4–17 (bar), with ABT-737 (75 mg/kg/d; n = 36, solid lines) or the vehicle (n = 36; dotted lines). Survival data were pooled from 6 pre-B (AH15, AH66, AI18, AI71, AG36, and AF44; Upper) or 6 B cell (AF47, AH29, AF40, AH54, AF52, and AH21; Lower) myc lymphomas. (B) Survival of mice bearing myc/bcl-2 lymphomas (#9, 12, and 16). Data were pooled from three lymphomas (Fig. S2); for each lymphoma, six mice were treated with either ABT-737 or the vehicle control. p values (log rank analysis) for vehicle vs. ABT-737 treatment are indicated.
In sharp contrast, ABT-737 treatment significantly prolonged the survival of mice transplanted with each of 3 independent myc/bcl-2 lymphomas (Fig. 1B and Fig. S2), despite the high levels of Bcl-2 they express (see below). All eventually succumbed to the disease, however, and autopsy revealed disseminated lymphoma, with elevated white blood cell counts, splenomegaly and lymphadenopathy (Table S1, data not shown).
To explore the basis for this marked difference in sensitivity to treatment, a panel of five myc/bcl-2 and eight myc tumors were analyzed for expression of Bcl-2 family proteins. Western blots (Fig. 2 and Fig. S3A, data not shown) revealed, as expected, high levels of transgenic human Bcl-2 protein in each of the myc/bcl-2 tumors. Of note, all of the myc tumors had higher levels of prosurvival Mcl-1 than the myc/bcl-2 tumors and most also expressed more Bcl-xL. Expression of most proapoptotic proteins, however, was comparable, with the exception of elevated Puma in the myc/bcl-2 lymphomas. Noxa protein was not assessable, because of the lack of a suitable antibody, but noxa transcripts were higher in myc/bcl-2 than myc tumors, as were puma transcripts (Fig. S4).
Fig. 2.
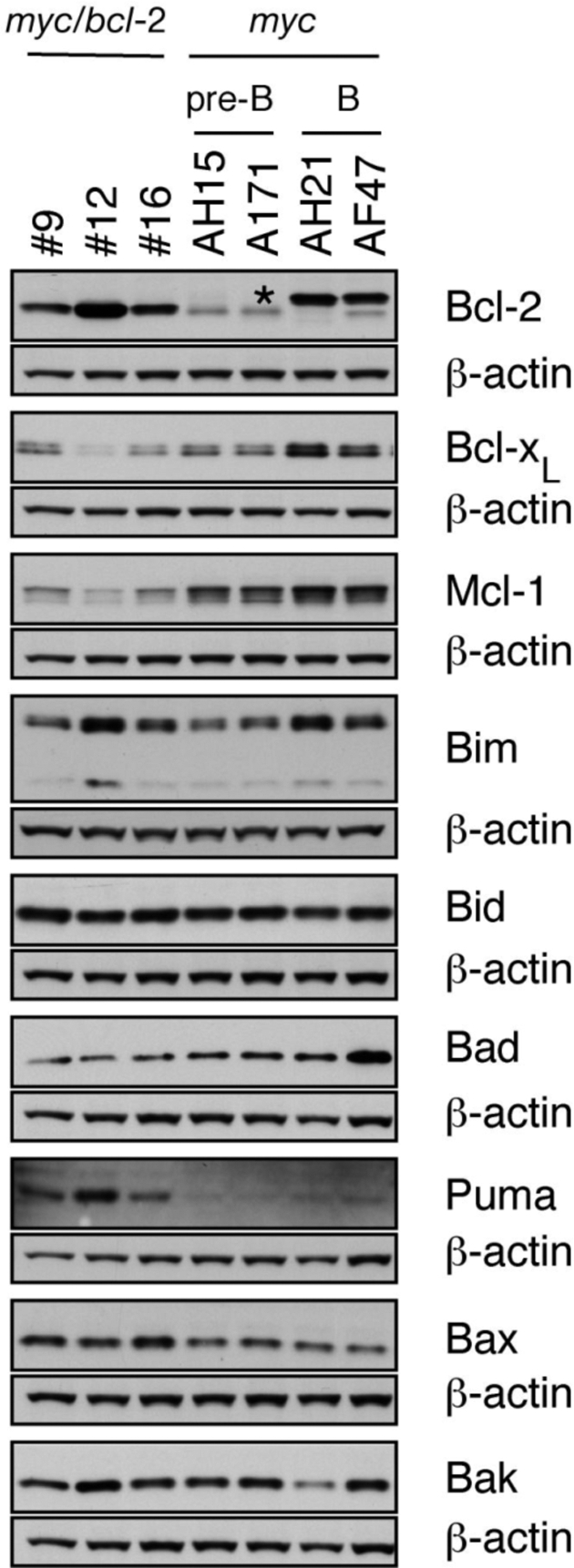
Expression of Bcl-2 family members in myc/bcl-2 and myc tumors. Western blot analysis of Bcl-2 family proteins in lymphomatous lymph nodes of transplant recipients. β-actin served as loading control. *, endogenous mouse Ig light chain present in B-cell tumors.
Thus, apart from high transgenic Bcl-2, the most striking differences in the expression of the Bcl-2 protein family were the lower level of prosurvival Mcl-1 and the higher level of proapoptotic Puma (and probably Noxa) in the myc/bcl-2 tumors. We surmise that the lower levels of Mcl-1 in the myc/bcl-2 tumors could account for their greater sensitivity to ABT-737 because reducing Mcl-1 enhances the responsiveness of several cell lines to ABT-737 (23, 30). Conversely, we found that increasing Mcl-1 increased the resistance of myc/bcl-2 lymphomas to ABT-737, but increasing Bcl-2 did not (23).
Response to Genotoxic Therapy.
Genotoxic agents activate p53 thereby inducing expression of Noxa (31) and Puma (32, 33). Because both of these BH3-only proteins bind to and inactivate Mcl-1 (22), we hypothesized that the combination of ABT-737 with a genotoxic agent should prove efficacious (23). We chose to test cyclophosphamide, an agent central for the treatment of patients with Burkitt's lymphoma (5).
We evaluated the efficacy of a single high dose of cyclophosphamide (200 or 300 mg/kg, equivalent to 667 mg/m2 and 1,000 mg/m2, respectively, close to the maximal tolerated dose) for treatment of 10 myc and 5 myc/bcl-2 lymphomas (Fig. 3 and Table S2). In this study, treatment of transplant recipients was initiated when lymphoma was detectable by palpation (enlarged spleen and/or lymph nodes), usually 14 days after tumor inoculation. Treatment with cyclophosphamide significantly prolonged the survival of most mice transplanted with myc lymphomas (Fig. 3A). Overall, the median survival was 98 days for treated mice, compared with only three days for the vehicle-treated control cohort (Table S2). Five of the 10 tumors were particularly responsive (Group 2, Fig. S5), with most tumor-inoculated mice still surviving when the experiment was terminated at 200 days.
Fig. 3.

Response of Myc-driven lymphomas to cyclophosphamide. (A) Kaplan–Meier plots indicating survival of mice bearing 10 independent myc lymphomas after treatment with a single (300 mg/kg ip) dose of cyclophosphamide (AF40, AG36, AH29, AH44, AH66, AF47, AH15, AH21, AI18, and AI71). P value (log rank analysis) for vehicle vs. cyclophosphamide is indicated. Treatment was initiated when tumors were palpable and survival is indicated from day of treatment (arrow, day 0). Overall median survival (time elapsed from day of treatment until 50% of the mice remained alive) with treatment was 98 days versus 3 days without (Table S2). (B) Survival of mice with five independent myc/bcl-2 lymphomas (#9, 10, 12, 16, and 30) treated similarly with cyclophosphamide. Median survival with treatment was 32 days and 12 days without (Table S2). P value as for A.
In contrast, the myc/bcl-2 lymphomas showed a uniformly poor response to cyclophosphamide; none of the treated mice survived beyond 60 days (Fig. 3B) and the median survival was only 32 days compared with 12 days for vehicle-treated controls (Table S2). These results suggest that high levels of Bcl-2 overwhelm the therapeutic potential of cyclophosphamide, as reported previously (7).
Promising Responses to Combination Therapy.
Given the poor response of lymphomas over-expressing Bcl-2 to cyclophosphamide as a single agent, we then embarked on combination studies with ABT-737 and cyclophosphamide. An initial dose-finding study (data not shown) established a safe regimen for in vivo treatment. For two myc/bcl-2 tumors (#9 and 16), the combination of ABT-737 and a low dose of cyclophosphamide (50 mg/kg, equivalent to 175 mg/m2) was highly efficacious; all tumor-transplanted mice receiving this treatment remained healthy when the experiment was terminated at day 150 (green lines in Fig. 4A). This was a significantly better outcome than for those treated with ABT-737 alone (blue lines) or cyclophosphamide alone (red line), even at a much higher dose of the latter (orange line). The third tumor, myc/bcl-2 #12, was more resistant to combination therapy although, even so, two of six mice survived out to day 150 (Fig. 4A). Of note, this tumor was also the most resistant to high-dose cyclophosphamide as a single agent.
Fig. 4.

Combination therapy with cyclophosphamide and ABT-737. Groups of mice transplanted with the myc/bcl-2 (A) or myc (B) lymphomas were treated with ABT-737 (75 mg/kg/d for 14 days; blue line) or vehicle (black), or with ABT-737 plus low-dose cyclophosphamide (50 mg/kg day 5 and day 9; green), or with low-dose cyclophosphamide alone (red) (n = 6 per arm). Another cohort received a single high dose of cyclophosphamide (300 mg/kg day 5; orange). Kaplan–Meier plots indicate survival; P values (log rank analysis) are indicated for low-dose cyclophosphamide (50 mg/kg) versus ABT-737 combined with low-dose cyclophosphamide. (C) noxa and puma are induced by DNA damage in sensitive lymphomas. Real-time qPCR for bim, puma, noxa, and p21 (positive control) using RNA from the indicated lymphomas 3-h after γ-irradiation (500 rad); negative control: p53-null myc lymphomas. Expression is normalized to that of β-actin and is expressed as the increase relative to untreated cells; results are means ± SEM of 2–4 independent experiments.
We also chose four myc lymphomas for combination studies from the cohort shown to be more resistant to high-dose cyclophosphamide as a single agent (Group 1 in Fig. S5). Mice transplanted with two of these tumors survived longer when treated with the combination of ABT-737 and a low dose of cyclophosphamide (green line) than with either agent alone (Fig. 4B) but in only one case (AH15) was the combination therapy significantly better than treatment with high dose cyclophosphamide.
We postulated that the failure of certain tumors to respond to combination therapy might be because of lesions in the DNA damage response and, consequently, failure to neutralize Mcl-1 by up-regulation of noxa and puma. To assess this, we performed real-time quantitative PCR on tumor cells treated in vitro with γ-irradiation (Fig. 4C) or etoposide (Fig. S6). Notably, noxa and puma transcripts increased more in the responsive myc/bcl-2 tumors (#9 and 16), than in the relatively resistant tumor (#12). Likewise, DNA damage provoked greater noxa and puma induction in the most sensitive myc lymphoma (AH15), than in the other three tumors (AI71, AH21, and AF47).
Long-Term Disease-Free Survival.
The survival data (Fig. 4A) suggested that treatment with ABT-737 plus low dose cyclophosphamide had very effectively reduced the tumor burden for most mice transplanted with myc/bcl-2 lymphomas (6/6 lymphoma # 9; 6/6 lymphoma #16; and 2/6 lymphoma #12). To test this directly, long-term survivors were killed at day 150 for autopsy and a full histologic analysis. Strikingly, none of the 14 survivors had any macroscopic or microscopic evidence of disease and their peripheral blood and bone marrow appeared normal (Fig. 5A and Table S3). Furthermore, no human Bcl-2 protein was detectable by flow cytometry in the bone marrow or blood (green curves in Fig. S7) and the human bcl-2 transgene was undetectable by PCR in 6/6 myc/bcl-2 #9 tumors analyzed (Fig. 5C).
Fig. 5.

Long-term disease-free survival. Mice autopsied at day 150 comprised 12 mice that had been transplanted with myc/bcl-2 lymphomas #9 and #16 and treated with ABT-737 plus low-dose cyclophosphamide, and the sole surviving myc/bcl-2 #9-transplanted mouse treated with high-dose cyclophosphamide. (A) All mice treated with combination therapy appeared macroscopically and microscopically normal (representative shown). (B) The mouse treated with high-dose cyclophosphamide had an enlarged spleen (S), enlarged liver (L), and peritoneal tumor mass (T) (Left); and prominent lymphoblasts in the blood (Center) and bone marrow (Right). Blood films were stained with Wrights' stain and sternal sections with H&E; magnification ×400. (C) Human bcl-2 transgene was not detectable by PCR in the spleen (S) of six survivors (#9) that received combination therapy but readily seen in the spleen (S) and lymph node from the survivor treated with high-dose cyclophosphamide. p53 - loading control.
In contrast, only four of the 18 mice treated with high dose cyclophosphamide survived out to day 150 and only three of these were disease-free (Table S3 and Fig. S7). The fourth, the sole myc/bcl-2 #9 survivor, displayed clear signs of recurrent disease. It had an enlarged spleen and liver, tumor masses in the peritoneum, lymphoblast infiltration of the blood and bone marrow (Fig. 5B), and readily detectable human Bcl-2 protein (Fig. S7A) and human bcl-2 DNA (Fig. 5C).
Discussion
For most adult patients with aggressive Myc-driven B cell lymphomas, intensive combination chemotherapy is curative (5). However, in the significant minority of patients who relapse, subsequent intensive salvage chemotherapy rarely results in durable survival (34), and new therapeutic approaches are therefore required. Our data in an immunocompetent mouse model of aggressive Myc-driven lymphoma provide clear evidence that BH3 mimetic drugs warrant clinical evaluation in this setting, particularly in combination with chemotherapy.
A surprising initial finding was that transplantable myc lymphoid tumors (both pre-B and B) were resistant to ABT-737 as a single agent (Fig. 1 A and Fig. S1). These tumors expressed Bcl-2 and Bcl-xL (Fig. 2) but inhibition of these prosurvival proteins by ABT-737 was insufficient to trigger apoptosis. Their significant expression of Mcl-1 (Fig. 2), which is not targeted by ABT-737 (21, 23), may well explain these findings. In sharp contrast, the myc/bcl-2 tumors, which expressed less Mcl-1 but high Bcl-2 displayed single agent sensitivity to ABT-737 (Fig. 1B and Fig. S2). Using a conditional bcl-2 transgene, Letai et al. (35) have previously reported that Bcl-2 is required for maintenance of myc/bcl-2 lymphoid tumors. The myc/bcl-2 tumors express more Puma than the myc lymphomas (Fig. 2 and Fig. S3A) and presumably this potent BH3-only protein (22) plays a role in triggering apoptosis once ABT-737 binds to and neutralizes Bcl-2 (Bcl-xL and Bcl-w). Interestingly, nonmalignant pre-B and B cells from healthy young myc/bcl-2 mice also have lower Mcl-1 and higher Puma levels than their counterparts from myc mice (Fig. S3B), suggesting that this expression signature correlates with high Bcl-2 expression rather than differentiation stage. In many different cell types, high Bcl-2 correlates with sensitivity to ABT-737 (e.g., 19, 21).
As a single agent, ABT-737 was unable to eradicate the myc/bcl-2 tumor cells, because all of the treated mice eventually succumbed to the disease (Fig. 1B, Fig. S2, and Table S1). Because genotoxic agents induce BH3-only proteins Noxa and Puma, which are natural antagonists for Mcl-1, we sought to enhance the response to ABT-737 by combining it with chemotherapy. As a single agent, cyclophosphamide provided only modest efficacy, even at a high dose (Figs. 3B and 4A). When combined with ABT-737, however, low dose cyclophosphamide was curative in all mice transplanted with 2 of 3 independent myc/bcl-2 lymphomas (Figs. 4A and 5). Whereas these responsive lymphomas displayed robust induction of noxa and puma after genotoxic stress, a more resistant tumor (#12) displayed minimal response (Fig. 4C and Fig. S6). Thus, we infer that cure of these highly resistant tumors requires neutralization of Mcl-1 as well as the ABT-737 targets.
Importantly, even though ABT-737 showed no activity against any of the myc pre-B and B lymphomas, it did synergise with low dose cyclophosphamide to produce a complete remission for 6/6 mice transplanted with one tumor (AH15) (Fig. 4B). Again, the most responsive tumor displayed robust induction of noxa and puma in response to genotoxic stress. Taken together, our results suggest that effective therapy for a Myc-driven tumor will require neutralization of most if not all prosurvival proteins expressed by the tumor.
Our data complement and extend a recent study of ABT-263, an orally available compound in the same class as ABT-737, for treating aggressive human B lymphoid tumor cell lines in a murine immuno-compromised xenograft model (21). In that study, two tumor-derived cell lines (DoHH2 to model diffuse large B cell lymphoma and GRANTA-519 to model mantle cell lymphoma) expressing high levels of Bcl-2 responded to ABT-263 and the response was significantly enhanced when combined with a standard therapy for non-Hodgkin's lymphoma. Our data in immunocompetent mice with myc/bcl-2 lymphomas not only confirm the benefits of combining Bad-like BH3 mimetics with chemotherapy, but also illustrate that these benefits may be achievable with lower doses of cytotoxic drugs and that high rates of complete tumor elimination are possible.
Tse et al. (21) also found that OPM-2 human myeloma cells, which express high levels of Mcl-1, did not respond in the xenograft model to treatment with ABT-263. Nevertheless, ABT-263 significantly enhanced the response to bortezomib, which was shown in vitro to induce Noxa, thereby inactivating Mcl-1. This finding is analogous to our data for myc tumors, which were unresponsive to ABT-737 as a single agent in vivo but could respond to ABT-737 plus cyclophosphamide, if able to induce noxa and puma in response to genotoxic stress.
From these findings we draw several conclusions regarding the possible utility of BH3 mimetics for treating aggressive myc driven lymphomas. First, for tumors that are dependent on Bcl-2 for survival, ABT-737 could well have significant single agent activity, even when genotoxic responses are impaired. Second, consistent with other observations (23, 24, 30), ABT-737 probably will be ineffective as a single agent for tumors in which Mcl-1 is expressed at significant levels. Third, the combination of chemotherapy and ABT-737 should be highly effective and potentially curative provided the DNA damage response is sufficiently intact to induce adequate levels of Noxa and Puma. For tumors with lesions in the DNA damage response, an alternative therapeutic that down-regulates and/or inactivates Mcl-1 in a p53-independent fashion could be used to synergise with ABT-737.
Materials and Methods
For studies with ABT-737, 6- to 8-week-old wild-type male mice were injected (i.p.) on day 0 with 1–3 × 106 T1 lymphoma cells and treatment was initiated on day 4 with i.p. (ip) injections of ABT-737 (Abbott Laboratories; 75 mg/kg in 30% propylene glycol, 5% Tween 80, 3.3% dextrose in water pH 4, 1% DMSO) administered daily for 14 days. For studies testing cyclophosphamide as a single agent (Fig. 3), mice were injected (i.v.) on day 0 with 1–3 × 106 T1 lymphoma cells and regularly checked until tumors were palpable (usually day 14), whereupon they were injected (ip) with water alone or with cyclophosphamide (Sigma; ≤ 300 mg/kg in water; 2–13 recipients per tumor). For combination studies (Fig. 4), mice were transplanted on day 0, then injected daily from day 4 with either vehicle alone or ABT-737 (75 mg/kg), each cohort also receiving either cyclophosphamide (ip, 50 mg/kg, equivalent to 175 mg/m2) or saline on days 5 and 9. A fifth cohort was given a dose of cyclophosphamide (300 mg/kg, equivalent to 1000 mg/m2) on day 5. The mice were culled when deemed unwell (lethargy, tremor, hindleg paralysis, >5% weight loss, palpable tumor) by animal husbandry staff who were blinded to the experiment. All mouse experiments were performed in accordance with guidelines administered by the Institutional Animal Ethics Committee. Details of mice, immunoblotting, real-time qPCR and genomic PCR analysis are provided within the SI Materials and Methods; see also Tables S4 and S5.
Supplementary Material
Acknowledgments.
We thank Abbott Laboratories for providing ABT-737; our colleagues for useful discussions; F. Battye, D. Cooper, H. Ierino, E. Jansen, A. Naughton, K. Pioch, G. Siciliano, A. Srikumar, A. Wiegmans for excellent technical assistance; and L. O'Reilly for reagents. This work was supported by the Australian National Health and Medical Research Council Program Grants 461221 and 461219 and Fellowships (to C.J.V., C.L.S., D.C.S.H., and A.W.R.), U.S. National Cancer Institute Grant CA43540, Leukemia and Lymphoma Society Specialized Center for Research Grant 7015–02 and a fellowship (to C.L.S.), Australian Cancer Research Foundation (Centre for Therapeutic Target Drug Discovery), Cancer Council of Victoria (K.D.M.), Leukemia Foundation (A.H.W.) and the Victorian Cancer Agency (K.D.M.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/0809957105/DCSupplemental.
References
- 1.Nilsson JA, Cleveland JL. Myc pathways provoking cell suicide and cancer. Oncogene. 2003;22:9007–9021. doi: 10.1038/sj.onc.1207261. [DOI] [PubMed] [Google Scholar]
- 2.Cory S. Activation of cellular oncogenes in hemopoietic cells by chromosome translocation. Adv Cancer Res. 1986;47:189–234. doi: 10.1016/s0065-230x(08)60200-6. [DOI] [PubMed] [Google Scholar]
- 3.Hummel M, et al. A biologic definition of Burkitt's lymphoma from transcriptional and genomic profiling. N Engl J Med. 2006;354:2419–2430. doi: 10.1056/NEJMoa055351. [DOI] [PubMed] [Google Scholar]
- 4.Dave SS, et al. Molecular diagnosis of Burkitt's lymphoma. N Engl J Med. 2006;354:2431–2442. doi: 10.1056/NEJMoa055759. [DOI] [PubMed] [Google Scholar]
- 5.Bishop PC, Rao VK, Wilson WH. Burkitt's lymphoma: Molecular pathogenesis and treatment. Cancer Invest. 2000;18:574–583. doi: 10.3109/07357900009012197. [DOI] [PubMed] [Google Scholar]
- 6.Vousden KH, Lu X. Live or let die: The cell's response to p53. Nat Rev Cancer. 2002;2:594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 7.Schmitt CA, Lowe SW. Bcl-2 mediates chemoresistance in matched pairs of primary Eμ-myc lymphomas in vivo. Blood Cells Mol Dis. 2001;27:206–216. doi: 10.1006/bcmd.2000.0372. [DOI] [PubMed] [Google Scholar]
- 8.Youle RJ, Strasser A. The Bcl-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 9.Tsujimoto Y, et al. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science. 1984;226:1097–1099. doi: 10.1126/science.6093263. [DOI] [PubMed] [Google Scholar]
- 10.Cimmino A, et al. miR-15 and miR-16 induce apoptosis by targeting Bcl-2. Proc Natl Acad Sci USA. 2005;102:13944–13949. doi: 10.1073/pnas.0506654102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mestre-Escorihuela C, et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007;109:271–280. doi: 10.1182/blood-2006-06-026500. [DOI] [PubMed] [Google Scholar]
- 12.Tagawa H, et al. Genome-wide array-based CGH for mantle cell lymphoma: Identification of homozygous deletions of the proapoptotic gene Bim. Oncogene. 2005;24:1348–1358. doi: 10.1038/sj.onc.1208300. [DOI] [PubMed] [Google Scholar]
- 13.Xiao C, et al. Lymphoproliferative disease and autoimmunity in mice with increased miR-17–92 expression in lymphocytes. Nat Immunol. 2008;9:404–414. doi: 10.1038/ni1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oltersdorf T, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 15.Konopleva M, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Tahir SK, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–1183. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 17.Kang MH, et al. Activity of vincristine, L-ASP, and dexamethasone against acute lymphoblastic leukemia is enhanced by the BH3-mimetic ABT-737 in vitro and in vivo. Blood. 2007;110:2057–2066. doi: 10.1182/blood-2007-03-080325. [DOI] [PubMed] [Google Scholar]
- 18.Chauhan D, et al. A novel Bcl-2/Bcl-xL/Bcl-w inhibitor ABT-737 as therapy in multiple myeloma. Oncogene. 2007;26:2374–2380. doi: 10.1038/sj.onc.1210028. [DOI] [PubMed] [Google Scholar]
- 19.Del Gaizo Moore V, et al. Chronic lymphocytic leukemia requires Bcl-2 to sequester prodeath Bim, explaining sensitivity to Bcl-2 antagonist ABT-737. J Clin Invest. 2007;117:112–121. doi: 10.1172/JCI28281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lock R, et al. Initial testing (stage 1) of the BH3 mimetic ABT-263 by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1181–1189. doi: 10.1002/pbc.21433. [DOI] [PubMed] [Google Scholar]
- 21.Tse C, et al. ABT-263: A potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res. 2008;68:3421–3428. doi: 10.1158/0008-5472.CAN-07-5836. [DOI] [PubMed] [Google Scholar]
- 22.Chen L, et al. Differential targeting of pro-survival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 23.van Delft MF, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin X, et al. ‘Seed’ analysis of off-target siRNAs reveals an essential role of Mcl-1 in resistance to the small-molecule Bcl-2/Bcl-xL inhibitor ABT-737. Oncogene. 2007;26:3972–3979. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]
- 25.Adams JM, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature. 1985;318:533–538. doi: 10.1038/318533a0. [DOI] [PubMed] [Google Scholar]
- 26.Harris AW, et al. The Eμ-myc transgenic mouse: A model for high-incidence spontaneous lymphoma and leukemia of early B cells. J Exp Med. 1988;167:353–371. doi: 10.1084/jem.167.2.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmitt CA, Rosenthal CT, Lowe SW. Genetic analysis of chemoresistance in primary murine lymphomas. Nat Med. 2000;6:1029–1035. doi: 10.1038/79542. [DOI] [PubMed] [Google Scholar]
- 28.Strasser A, Harris AW, Bath ML, Cory S. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature. 1990;348:331–333. doi: 10.1038/348331a0. [DOI] [PubMed] [Google Scholar]
- 29.Strasser A, Elefanty AG, Harris AW, Cory S. Progenitor tumours from Eμ-bcl-2-myc transgenic mice have lymphomyeloid differentiation potential and reveal developmental differences in cell survival. EMBO J. 1996;15:3823–3834. [PMC free article] [PubMed] [Google Scholar]
- 30.Chen S, et al. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 31.Oda E, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000;288:1053–1058. doi: 10.1126/science.288.5468.1053. [DOI] [PubMed] [Google Scholar]
- 32.Nakano K, Vousden KH. puma, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7:683–694. doi: 10.1016/s1097-2765(01)00214-3. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, et al. Puma induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- 34.Sweetenham JW, et al. Adult Burkitt's and Burkitt-like non-Hodgkin's lymphoma–outcome for patients treated with high-dose therapy and autologous stem-cell transplantation in first remission or at relapse: Results from the European Group for Blood and Marrow Transplantation. J Clin Oncol. 1996;14:2465–2472. doi: 10.1200/JCO.1996.14.9.2465. [DOI] [PubMed] [Google Scholar]
- 35.Letai A, Sorcinelli MD, Beard C, Korsmeyer SJ. Antiapoptotic Bcl-2 is required for maintenance of a model leukemia. Cancer Cell. 2004;6:241–249. doi: 10.1016/j.ccr.2004.07.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.