

Abstract
Inherited retinal diseases are a common cause of visual impairment in children and young adults, often resulting in severe loss of vision in later life. The most frequent form of inherited retinopathy is retinitis pigmentosa (RP), with an approximate incidence of 1 in 3,500 individuals worldwide1,2. RP is characterized by night blindness and progressive degeneration of the midperipheral retina, accompanied by bone spicule-like pigmentary deposits and a reduced or absent electroretinogram (ERG). The disease process culminates in severe reduction of visual fields or blindness. RP is genetically heterogeneous, with autosomal dominant, autosomal recessive and X-linked forms. Here we have identified two mutations in a novel retina-specific gene from chromosome 8q that cause the RP1 form of autosomal dominant RP in three unrelated families. The protein encoded by this gene is 2,156 amino acids and its function is currently unknown, although the amino terminus has similarity to that of the doublecortin protein, whose gene (DCX) has been implicated in lissencephaly in humans17. Two families have a nonsense mutation in codon 677 of this gene (Arg677stop), whereas the third family has a nonsense mutation in codon 679 (Gln679stop). In one family, two individuals homozygous for the mutant gene have more severe retinal disease compared with heterozygotes.
Several genes have been identified for each of the forms of RP (refs 3-16), but most RP genes are still uncloned and known only through linkage studies (http://www.sph.uth.tmc.edu/RetNet/). The disease loci in three families with adRP have been mapped to the 8q11–q12 region. This locus was designated RP1, as it was the first well-characterized adRP locus18. Linkage data from the first RP1 family, UCLA-RP01, were combined with data from a second family, Australian family D, to narrow the disease locus to a 4-cM region between markers D8S601 and D8S285, with a maximum combined 2-point lod score of 16.9 (ref. 19). The disease locus in a third adRP family, family UK-RP1 from southwest England, also maps to this region, with a maximum multipoint lod score of 3.0 (ref. 20). All three families have similar clinical characteristics, with relatively late onset of night blindness and slow progression18,20,21. Clinical findings in UCLA-RP01 include diffuse retinal pigmentation, progressive decrease in recordable ERGs and concentric visual field loss. Fundus findings include retinal atrophy, pigment deposits and vascular attenuation.
Consanguinity is present in several segments of the extensive UCLA-RP01 pedigree and, in one case, two affected third cousins, once-removed, married and had several affected children. Two of these children have unusually severe clinical manifestations of RP relative to the rest of the family. Specifically, the eldest daughter had noticeable night blindness by age 6, noticeable visual field loss by 8 and severe retinal atrophy and non-recordable ERGs by age 18. Her youngest brother, examined at age seven, had experienced night blindness since early childhood and already had severe visual field constriction. These clinical findings suggested that both were homozygous for the RP1 mutation and linkage mapping confirmed they were homozygous for all tested markers across the RP1 critical region22.
The RP1 region spans approximately 4 Mb and is partly syntenic with mouse chromosome 4, where a dominant retinal degeneration locus (Rd4) caused by a chromosomal inversion has also been mapped23. In our search for RP1 candidate genes, we gave preference to ESTs that mapped to both RP1 and Rd4 regions. We identified two candidate ESTs, AA018812 and stSG29859, and mapped them to the RP1 YAC contig.
We determined the complete sequence of the cDNA clone from which EST AA018812 was derived and found a large 2,024-bp ORF with no apparent stop codon. Database searches revealed no sequence similarity with known proteins, but led to a BAC end sequence containing part of the 5′ end of the gene. Sequencing of this clone provided the intron/exon junctions for the first three exons. Continuing database searches revealed a recently sequenced BAC clone that completely spans the gene.
The ORF identified in the original cDNA clone extends for an additional 4,000 bp with a total length of 6,471 bp. We determined the polyadenylation site and the 3′ end of the transcript using 3′-RACE with a retinal cDNA library. The gene consists of 4 exons with the start codon located 12 bp into exon 2 (Fig. 1a). The complete mRNA size is approximately 7 kb and the protein is predicted to be 2,156 amino acids (Fig. 1c) with a calculated molecular weight of 240 kD.
Fig. 1.

Gene and protein structure of RP1. a, RP1 consists of four exons, three of which (exons 2–4) contain coding sequence. All disease-causing mutations and polymorphic sites are in exon 4. b, Protein sequence of RP1 (2,156 aa). c, Protein alignment of RP1. The partial alignment demonstrates a conserved region in the first 370 residues of RP1 that shares similarity with several eukaryotic proteins, including: a unique protein fragment deduced from a retina-specific EST cluster consensus (STACK accession number eye2931), with 50% identity over 86 residues; human DCX, with 25% identity over 328 residues; a human protein of unknown function (KIAA0369), with 20% identity over 707 residues; mouse Dcx, with 25% identity over 328 residues; a C. elegans predicted gene product (W07G1.5), with 25% identity over 332 residues; and Schizosaccharomyces pombe predicted protein product CAB9768.1, with 17% identity over 195 residues. Colouring represents degree of physiochemical conservation of residues as follows: purple, proline/glycine; yellow, cysteine; blue, hydrophilic; light blue, aromatic; green, negative; red, positive; pink, aliphatic/hydrophobic. A continuous line over the RP1 sequence represents matches to Prosite nuclear localization signal PS50079.
DNA from members of family UCLA-RP01 was screened for mutations by direct DNA sequencing using primer pairs that amplified all exons and intron/exon junctions. A nonsense mutation was identified in exon 4 at codon 677 (Arg677 stop; CGA→TGA) in affected members (Fig. 2a) and predicts a protein one-third the size of wild type. We used restriction analysis to screen all family members and additional normal controls. The mutation was found in all affected individuals (69 tested), but not in 150 normal chromosomes. The two severely affected family members in UCLA-RP01 were homozygous for the mutation (Fig. 3).
Fig. 2.
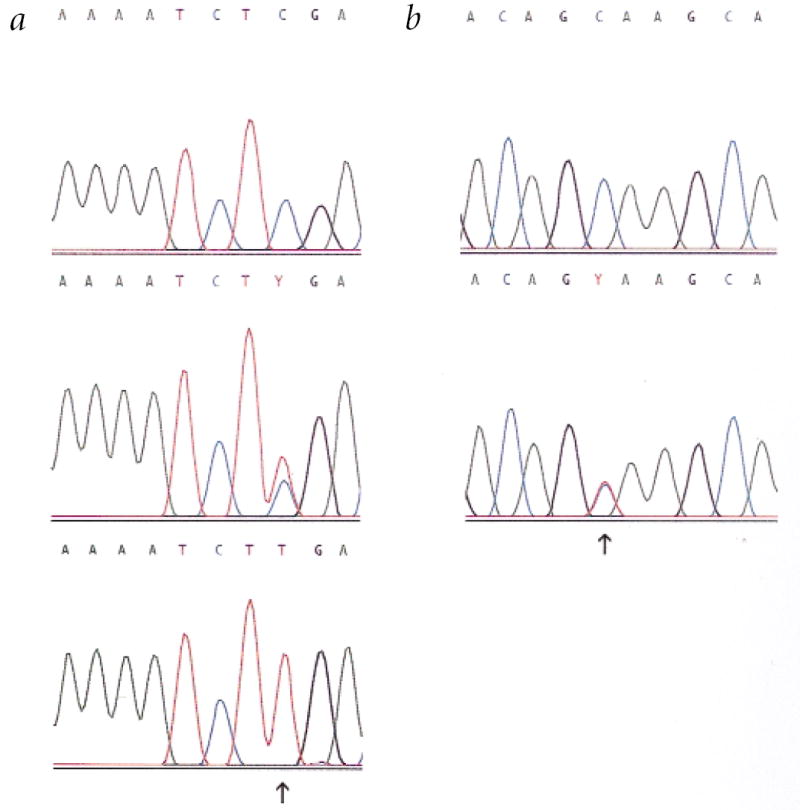
Sequence electropherograms of mutations found in three adRP families. a, The Arg677stop mutation found in American family UCLA-RP01 and Australian family D. Top, an unaffected control (CGA/CGA). Middle, heterozygous C/T mutation at codon 677 (CGA/TGA). Bottom, DNA sequence of a homozygous, affected member of UCLA-RP01 (TGA/TGA). This mutation destroys a TaqI site. b, The Gln679stop mutation in English family UK-RP1. Normal sequence (top; CAA/CAA) and the heterozygous C/T mutation (bottom; CAA/TAA) are shown. This mutation destroys a Cac8I site.
Fig. 3.
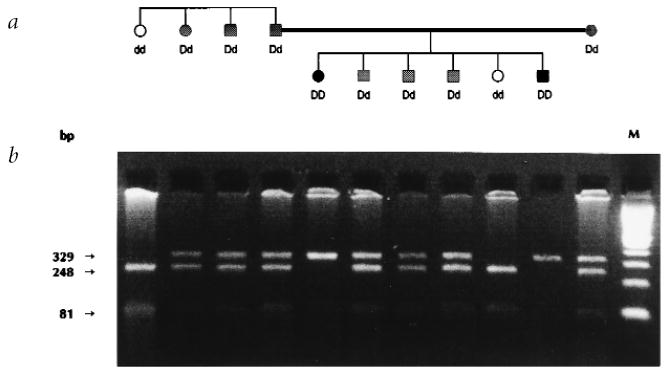
Pedigree and mutation screen of a consanguineous nuclear family in UCLA-RP01. a, Pedigree. Disease phenotypes in this portion of the family suggest that two individuals are homozygous for the disease mutation. Open symbols are unaffected individuals (dd), hatched symbols are affected heterozygotes (Dd) and filled symbols are severely affected homozygotes (DD). The parents of the two severely affected individuals are third cousins once removed. b, Segregation of the Arg677 stop mutation. Digestion of amplimer F with TaqI in an unaffected individual produces a 248-bp fragment and an 81-bp fragment. The CGA→TGA change at codon 677 eliminates the TaqI site, producing an undigested band of 329 bp. Unaffected individuals have the two lower bands, heterozygous affected individuals have three bands and homozygous affected individuals have one large, undigested fragment. M, molecular weight marker.
We screened DNA from an affected individual from Australian family D by sequencing and found the same nonsense mutation identified in UCLA-RP01, Arg677stop (CGA→TGA). The rest of family D was screened by digestion at the TaqI site and the mutation was found to co-segregate with the disease in all cases (5 affected and 11 unaffected.) Microsatellite markers D8S165, D8S593 and D8S591, all within 1 Mb of RP1, were tested in UCLA-RP01 and family D. Two distinct chromosomal haplotypes segregate with disease in the two families, suggesting that the families are not related or that the mutation is very old (data not shown).
DNA sequencing in the third family, UK-RP1, revealed a nonsense mutation (Fig. 2b) at codon 679 (Gln679stop; CAA→TAA). This mutation predicts a truncated protein only two amino acids longer than that found in the other families. We sequenced all members of the family through this region and found the mutation to segregate with the disease phenotype (eight affected and ten unaffected). The codon 679 mutation causes a change in a Cac8I site and restriction digestion with Cac8I was used to confirm that the mutation was not present in 180 normal chromosomes. In addition to the disease-causing mutations found in affected individuals, we found several polymorphic amino acid substitutions in unaffected controls (Table 1).
Table 1.
Polymorphic amino acid substitutions in RP1 protein
| Amino acid no. | Exon/amplimer | Amino acid (codon) | Frequency | Amino acid (codon) | Frequency |
|---|---|---|---|---|---|
| 872 | 4/H | Arg (CGT) | 0.75 | His (CAT) | 0.25 |
| 985 | 4/I | Asn (AAT) | 0.54 | Tyr (TAT) | 0.46 |
| 1,670 | 4/P | Ala (GCA) | 0.78 | Thr (ACA) | 0.22 |
| 1,691 | 4/P | Ser (TCT) | 0.77 | Pro (CCT) | 0.23 |
| 1,725 | 4/P | Gln (CAA) | 0.80 | Gln (CAG) | 0.20 |
| 2,033 | 4/S | Cys (TGT) | 0.46 | Tyr (TAT) | 0.54 |
PCR primers from RP1 were used to amplify portions of the mouse homologue from a mouse retinal cDNA library. Several amplimers were sequenced and were more than 90% identical to RP1 sequence. We used these primers to screen a mouse BAC library and identified two BACs containing the gene. These BACs were then used for fluorescent in situ hybridization of mouse fibroblasts to determine the chromosomal location of mouse Rp1. Both BACs map to mouse chromosome 1A4-5 (data not shown) and exclude Rp1 as the causative gene in the Rd4 mouse. Additionally, SSCP analysis in the Jackson Laboratory BSS mapping panel also placed the mouse homologue of RP1 on chromosome 1, near the centromere.
Database searches suggest the RP1 gene product may be a protein kinase, with developmental-regulatory activity, sharing functional similarity with human doublecortin24, KIAA0369 (DCAMKL1; ref. 25) and EYE2931 (encoded by a gene fragment assembled from ESTs). Specifically, the protein (2,156 residues) has significant similarity over the first 310 residues of its sequence to human and mouse doublecortin, processed EST assembly EYE2931, human KIAA0369 and the predicted, uncharacterized Caenorhabditis elegans protein 4008399 (Fig. 1b). The greatest similarity is within the doublecortin (DC) domain itself, implying the existence of a functionally conserved structure.
RP1 may be a member of the same family as EYE2931 and KIAA0369, as it shares 15–50% identity over the DC domain, but it has fewer detectable protein kinase signatures throughout its extended length. The KIAA0369 gene product is expressed in alternatively spliced forms and has a calmodulin-dependent kinase (CaM kinase)-like domain following the DC domain25,26. By contrast, the RP1 protein bears weak similarity to known serine/threonine protein kinases, suggesting there may be a generalized similarity in regulatory function with KIAA0369.
A nucleoside diphosphate kinase motif found at residues 1,820–1,828 and 3 nuclear localization profiles found in the first 700 residues of the RP1 protein imply that the protein may be transported to the nucleus. Also, many potential phosphorylation sites were matched with Prosite motifs throughout the length of the protein (data not shown).
Northern-blot analysis (Fig. 4) indicates that expression of RP1 message is limited to retina, with no detectable expression in heart, brain, placenta, lung, liver, skeletal muscle, kidney or pancreas. Searches of EST databases indicate that the RP1 sequence appears only twice, as ESTs from both ends of the retinal cDNA clone sequenced originally (AA018811/AA018812). ESTs for KIAA0369 and DCX, with which RP1 shares sequence similarity, are found predominantly in brain27. EYE2931, another related sequence, appears to be exclusively expressed in retina, with ten retinal ESTs currently identified. The products encoded by RP1, DCX and the sequence-related ESTs may represent a novel family of retinal proteins.
Fig. 4.
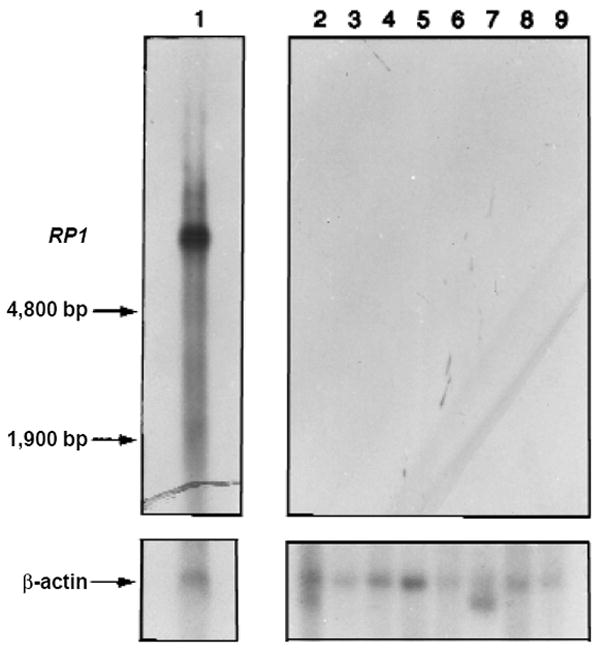
Expression of RP1 transcript in human tissues. We incubated northern blots containing poly(A)+ RNA from human adult tissues with an RP1 probe. Lane 1, adult retina; lane 2, heart; lane 3, whole brain; lane 4, placenta; lane 5, lung; lane 6, liver; lane 7, skeletal muscle; lane 8, kidney; lane 9, pancreas. A 2-h exposure is shown. A 42-h exposure reveals nothing over background in lanes 2–9. As control, the same blots were incubated with a human β-actin probe.
The RP1 form of adRP was one of the earliest well-characterized forms of autosomal retinopathy and identification of RP1 concludes a long-standing effort to clone the causative gene. Although little is known about the RP1 protein at present, its similarity to doublecortin suggests it may have a role in development or maintenance of the neural retina or neuronal components of photoreceptor cells.
Methods
YAC mapping
We tested published primer pairs for ESTs AA018812 and stSG 29859 in the following CEPH YACs (Research Genetics): 786E11, 938A9, 936B9, 784C8, 962A10, 834A9, 902B4, 935E9, 958A5, 898G12, 911H2 and 967D2. AA018812 amplified only from YACs 958A5 and 898G12, whereas stSG29859 amplified from 935E9, 967D2, 911H2 and 958A5.
cDNA and BAC sequencing
A partial cDNA clone (IMAGE 363092) corresponding to EST AA018812 and a BAC containing the 5′ end of the RP1 coding region (137I22) were obtained (Research Genetics) and sequenced by primer walking using an ABI 310 Prism Genetic Analyzer (Perkin Elmer) and BigDye terminator chemistry (Applied Biosystems). We confirmed intron/exon junctions by comparison of the cDNA sequence with the published genomic sequence of BAC clone 18L18.
3′-RACE
To identify the 3′ end of RP1 we performed 3′-RACE using human retina Marathon-Ready cDNA (Clontech) and the Marathon RACE kit (Clontech). A gene-specific primer from the 3′ end of the RP1 coding region (5′–CCAGGGCTCAAGAACAAATCTCAACC–3′) was used to amplify retinal cDNA in conjunction with the AP1 RACE primer (Clontech). We sequenced the resulting product using the same primer and an additional nested primer (5′–CTCAGGGCATCAAAATGTG–3′).
Northern-blot analysis
A human multiple tissue northern (MTN) blot (Clontech) and a human retinal northern blot were simultaneously probed with radioactively labelled amplimers from exon 4, generated using the Strip-EZ PCR kit (Ambion). Blots were incubated in ULTRA-hyb hybridization solution (Ambion) and washed according to the manufacturer’s protocol. As a positive control, both blots were also incubated with human β-actin probe using the same hybridization conditions.
Mutation/polymorphism screening
We chose 25 primer pairs to PCR amplify the entire coding region in overlapping segments. Genomic DNA was amplified and sequenced using the ABI BigDye cycle sequencing dye terminator kit (Applied Biosystems) and an ABI Prism 310 Genetic Analyzer (Perkin Elmer). To detect the Arg677stop mutation, we amplified a 330-bp fragment from exon 4 containing the site from all available family members and a panel of 75 unaffected controls using primers 4FF/4FR. After amplification, the PCR product was digested with TaqI (Boehringer) and visualized on a 2.5% agarose gel. To detect the Gln679stop mutation, we used primers mut2F (5′–TTCAGCAGATGCAACCCAT–3′) and 4FR to amplify a 453-bp genomic fragment. PCR products were radiolabelled, digested with Cac8I (New England Biolabs) and separated on 6% Long Ranger (FMC Bioproducts) gels. To determine the frequencies of the polymorphic amino acid substitutions in a panel of 90 unrelated normal controls, amplimers containing the polymorphic site were amplified and typed by single strand conformational analysis (SSCA) as described16.
Mapping of mouse homologues
When DNA sequence was available from a mouse homologue, we designed PCR primers to amplify a portion of the 3′ UTR. These primers were then used on two mouse mapping panels: the Jackson Laboratory BSS mapping panel and the whole-genome radiation hybrid panel (WG-RH T31).
Database searches and sequence comparisons
Database comparisons were performed against all available non-redundant protein data sets at the National Center for Biotechnology Information (NCBI) as of 29 March 1999 using BLAST (ref. 28) and Smith-Waterman via the BCM search launcher (http://dot.imgen.bcm.tmc.edu:9331/). We performed electronic analysis of the expression localization of RP1 using the Sequence Tag Alignment Consensus Knowledgebase search launcher at SANBI (http://ziggy.sanbi.ac.za/stack/stacksearch.htm). STACK accession EYE2931 was processed using the STACK_PACK toolset to produce a predicted protein sequence from a series of assembled ESTs (R.T. Miller et al., manuscript submitted). Sequences were aligned using Pileup (Wisconsin Genetics Package) and presented using GeneDoc (http://www.cris.com/~Ketchup/genedoc.html). We performed multiple alignment and automated prediction using the GeneQuiz server (http://www.sander.ebi.ac.uk).
GenBank accession numbers
RP1 cDNA, AF143222; human BAC, AF128525; human exons and primer sequences, AF143223–AF143226; BAC clone 18L18, AF128525; ESTs, AA059469, H85579, AA059256, AA019117, R84770, H85580, AA058855, AA019118, R85514 and AA059222; DCX, AF034634; Dcx, AF045547; C. elegans W07G1.5, EMBL Z82076; S. pombe CAB9768.1, EMBL Z97052.1.
Acknowledgments
We thank family members of UCLA-RP01 for participation, R. McInnes for retinal northern blots and P. Forsythe for technical assistance. This work was supported by grants from the Foundation Fighting Blindness and the George Gund Foundation, the William Stamps Farish Fund, the M.D. Anderson Foundation, the John S. Dunn Research Foundation, grant EY07142 from the National Eye Institute–National Institutes of Health (L.S.S., S.J.B. and S.P.D.), grant 5-FY98-0725 from the March of Dimes Birth Defects Foundation (J.Z.), a grant from the British Retinitis Pigmentosa Society and grant 035535/Z/96 from the Wellcome Trust (C.F.I.).
References
- 1.Heckenlively JR. Retinitis Pigmentosa. J.B. Lippincott; Philadelphia: 1988. [Google Scholar]
- 2.Heckenlively JR, Daiger SP. Hereditary retinal and choroidal degenerations. In: Rimon DL, Conner JM, Pyeritz RE, editors. Principals and Practices of Medical Genetics. Churchill Livingstone; New York: 1997. pp. 2555–2576. [Google Scholar]
- 3.Banerjee P, et al. TULP1 mutation in two recessive extended Dominican kindreds with autosomal recessive retinitis pigmentosa. Nature Genet. 1998;18:177–179. doi: 10.1038/ng0298-177. [DOI] [PubMed] [Google Scholar]
- 4.Cremers FPM, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet. 1998;7:355–362. doi: 10.1093/hmg/7.3.355. [DOI] [PubMed] [Google Scholar]
- 5.Dryja TP, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- 6.Dryja TP, et al. Mutations in the gene encoding the α subunit of the rod cGMP-gated channel in autosomal recessive retinitis pigmentosa. Proc Natl Acad Sci USA. 1995;92:10177–10181. doi: 10.1073/pnas.92.22.10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hagstrom SA, North MA, Nishina PM, Berson EL, Dryja TP. Recessive mutations in the gene encoding the tubby-like protein TULP1 in patients with retinitis pigmentosa. Nature Genet. 1998;18:174–176. doi: 10.1038/ng0298-174. [DOI] [PubMed] [Google Scholar]
- 8.Huang SH, et al. Autosomal recessive retinitis pigmentosa caused by mutations in the α subunit of rod cGMP phosphodiesterase. Nature Genet. 1995;11:468–471. doi: 10.1038/ng1295-468. [DOI] [PubMed] [Google Scholar]
- 9.Kajiwara K, et al. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature. 1991;354:480–483. doi: 10.1038/354480a0. [DOI] [PubMed] [Google Scholar]
- 10.Kajiwara K, Berson EL, Dryja TP. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science. 1994;264:1604–1608. doi: 10.1126/science.8202715. [DOI] [PubMed] [Google Scholar]
- 11.Martinez-Mir A, et al. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nature Genet. 1998;18:11–12. doi: 10.1038/ng0198-11. [DOI] [PubMed] [Google Scholar]
- 12.Maw MA, et al. Mutations in the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nature Genet. 1997;17:198–200. doi: 10.1038/ng1097-198. [DOI] [PubMed] [Google Scholar]
- 13.McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the β-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nature Genet. 1993;4:130–134. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 14.Meindl A, et al. A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3) Nature Genet. 1996;13:35–42. doi: 10.1038/ng0596-35. [DOI] [PubMed] [Google Scholar]
- 15.Schwahn U, et al. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nature Genet. 1998;19:327–332. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- 16.Sohocki MM, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription factor gene. Am J Hum Genet. 1998;63:1307–1315. doi: 10.1086/302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sossey-Alaoui K, et al. Human doublecortin (DCX) and the homologous gene in mouse encode a putative Ca2+-dependent signaling protein which is mutated in human X-linked neuronal migration defects. Hum Mol Genet. 1998;7:1327–1332. doi: 10.1093/hmg/7.8.1327. [DOI] [PubMed] [Google Scholar]
- 18.Blanton SH, et al. Linkage mapping of autosomal dominant retinitis pigmentosa (RP1) to the pericentric region of human chromosome 8. Genomics. 1991;11:857–869. doi: 10.1016/0888-7543(91)90008-3. [DOI] [PubMed] [Google Scholar]
- 19.Xu S-Y, Denton M, Sullivan LS, Daiger SP, Gal A. Genetic mapping of RP1 on 8q11–q21 in an Australian family with autosomal dominant retinitis pigmentosa reduces the critical region to 4 cM between D8S601 and D8S285. Hum Genet. 1996;98:741–743. doi: 10.1007/s004390050296. [DOI] [PubMed] [Google Scholar]
- 20.Inglehearn CF, et al. A new family linked to the RP1 dominant retinitis pigmentosa locus on 8q. J Med Genet. in press. [PMC free article] [PubMed] [Google Scholar]
- 21.Jimenez JB, et al. No evidence of linkage between the locus for autosomal dominant retinitis pigmentosa and D3S47 (C17) in three Australian families. Hum Genet. 1991;86:265–267. doi: 10.1007/BF00202406. [DOI] [PubMed] [Google Scholar]
- 22.Daiger SP, et al. Progress in positional cloning of RP10 (7q31.3), RP1 (8q11–q21) and VMD1 (8q24) In: LaVail M, Hollyfield JG, Anderson RE, editors. Degenerative Retinal Diseases. Plenum Publishing; New York: 1997. pp. 277–289. [Google Scholar]
- 23.Roderick TH, Chang B, Hawes NL, Heckenlively JR. A new dominant retinal degeneration (Rd4) associated with a chromosomal inversion in the mouse. Genomics. 1997;42:393–396. doi: 10.1006/geno.1997.4717. [DOI] [PubMed] [Google Scholar]
- 24.Hannan AJ, et al. Expression of doublecortin correlates with neuronal migration and pattern formation in diverse regions of the developing chick brain. J Neurosci Res. 1999;55:650–657. doi: 10.1002/(SICI)1097-4547(19990301)55:5<650::AID-JNR12>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 25.Omori Y, et al. Expression and chromosomal localization of KIAA0369, a putative kinase structurally related to doublecortin. J Hum Genet. 1998;43:169–177. doi: 10.1007/s100380050063. [DOI] [PubMed] [Google Scholar]
- 26.Matsumoto N, Pilz DT, Ledbetter DH. Genomic structure, chromosomal mapping, and expression pattern of human DCAMKL1 (KIAA0369), a homologue of DCX (XLIS) Genomics. 1999;56:179–183. doi: 10.1006/geno.1998.5673. [DOI] [PubMed] [Google Scholar]
- 27.Nagase T, et al. Prediction of the coding sequences of unidentified human genes. VII. The complete sequences of 100 new cDNA clones from brain which can code for large proteins in vitro. DNA Res. 1997;4:141–150. doi: 10.1093/dnares/4.2.141. [DOI] [PubMed] [Google Scholar]
- 28.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]