

Abstract
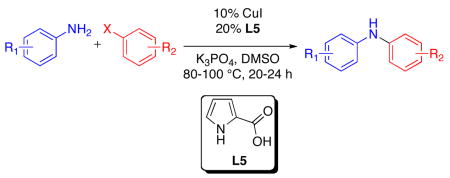
Pyrrole 2-carboxylic acid (L5) was found to be an effective ligand for the Cu-catalyzed mono-arylation of anilines with aryl iodides and bromides. Under the reported conditions (10% CuI/20% L5/DMSO/K3PO4/80–100 °C/20–24 h), a variety of useful functional groups were tolerated, and moderate to good yields of the diaryl amine products were obtained.
The diaryl amine moiety can be found in a variety of biologically active pharmaceuticals, natural products, and materials.1 Metal-catalyzed cross-coupling reactions of anilines with aryl halides are among the foremost methods for assembling this substructure.1,2 For reactions of poorly nucleophilic primary anilines with aryl halides, Pd-based catalyst systems are highly efficient.1 This is due, in great part, to the rapid transmetallation of anilines to Pd(II), a phenomenon that arises from the large increase in acidity of the nucleophile when coordinated to Pd(II).3 The complementary nature of Pd- and Cu-catalyzed C–N bond-forming processes, and the issues involving removal of the trace Pd from the products encourage the development of Cu-based catalyst systems for the preparation of diaryl amines.
In recent years, Cu-catalyzed C–N bond-forming reactions have evolved as reliable alternatives to Pd-catalyzed reactions.2 However, Cu-based catalyst systems for the synthesis of diaryl amines are less general and useful than the Pd-based protocols.4 The Cu-catalyzed reactions of anilines with aryl halides are slow enough that a wide variety of N–H and O–H nucleophiles, including amides, nitrogen heterocycles, aliphatic, benzylic and allylic amines, as well as aliphatic and benzylic alcohols are selectively arylated in the presence of an anilino-NH2 group.5 In the absence of a competing reactant, the poor nucleophilicity of the aniline, when employing Cu-based catalysts, further manifests itself in the need to use high catalyst loadings (> 20% Cu),4a,f long reaction times (> 30 h),4b–c,e strong bases that preclude the presence of many common functional groups,4g and/or anilines with strong electron-withdrawing groups in the para-position.4b Further, the few examples of Cu-catalyzed reactions of anilines with ortho-substituted aryl halides require even higher catalyst loadings (35–50% Cu).4f Finally, when employing Cu-based catalysts, the propensity of the diaryl amine product to undergo further N-arylation to form a triarylamine provides an added level of complexity to developing a suitable catalyst system.6
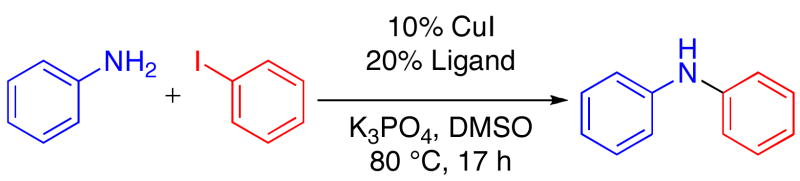
We began our investigation into the Cu-catalyzed reactions of aromatic amines with aryl iodides, by evaluating previously reported catalysts for this transformation.4 Since the more successful systems employed proline-type ligands,4b–e we sought to evaluate the use of new ligands that would provide a more active and generally applicable catalyst for the reaction of aniline with an aryl iodide (Table 1). While several heterocyclic-2-carboxylic acids, including some previously reported as ligands for Cu-catalyzed and -mediated nucleophilic substitution reactions of aryl halides,7 provided poor results for this transformation (entries 1–4), pyrrole-2-carboxylic acid, L5, manifested good catalytic activity (entry 5). Both the N–H and carboxylate functional groups of this ligand are important to the activity of the catalysts derived from it. This can be seen as modification of these groups provided less-active catalysts (entries 6–8). Benzannulated analogs L9 and L10 also provided less-active catalyst (entries 9–10); presumably because they are too hindered. Finally, L5 provided a more active catalyst system than those derived from commercially-available ligands previously reported for this transformation (entries 11–14).4a–b,d,f
Table 1.
Cu-Catalyzed Reaction of Aniline with Iodobenzene
 | |||
|---|---|---|---|
| Entry | Ligand | GC Conversion (%) | GC Yield (%) |
| 1 | L1 | 58 | 35 |
| 2 | L2 | 46 | 27 |
| 3 | L3 | 47 | 27 |
| 4 | L4 | 0 | 0 |
| 5 | L5 | 94 | 68 |
| 6 | L6 | 57 | 37 |
| 7 | L7 | 55 | 22 |
| 8 | L8 | 66 | 0 |
| 9 | L9 | 62 | 26 |
| 10 | L10 | 60 | 22 |
| 11 | L11 | 64 | 34 |
| 12 | L12 | 64 | 29 |
| 13 | L13 | 48 | 15 |
| 14 | L14 | 64 | 19 |
| 15 | L15 | 51 | 30 |
Reactions Conditions: 1.0 mmol ArNH2, 0.5 mmol ArI, 1.0 mmol K3PO4, 0.050 mmol CuI, 0.10 mmol ligand, 0.25 mL DMSO, at 80 °C in a sealed tube under an N2 atmosphere for 17 h.
Further optimization of the reaction conditions using L5 revealed that the base/solvent combination of K3PO4/DMSO typically provided a superior system than combinations involving K2CO3, Cs2CO3, KOH, and NaOt-Bu in DMF, 1,4-dioxane, toluene, and acetonitrile. Since diaryl ether and phenolic products (up to 20% of ArX consumption) were frequently produced under the reaction conditions and observed by GC/MS,8 the base was flame-dried under reduced pressure then cooled under a positive pressure of N2 prior to use.
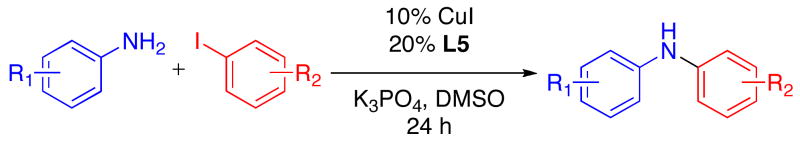
The reaction conditions we developed (1.0 equiv ArX/2.0 equiv ArNH2/10% CuI/20% L5/K3PO4/DMSO/80–90 °C) could be used to couple aryl iodides with anilines in moderate to good yields (Table 2). For the reaction of p-anisidine with 4-chloroiodobenzene, the catalyst loading and reaction temperature could be reduced to 5% CuI and 70 °C, respectively (entry 1). Attempts to reduce the catalyst loading down to 2 mol%, however, were unsuccessful. For this example, the lower catalyst loading employed can be attributed to the increased nucleophilicity of p-anisidine relative to anilines that lack a strong electron-donating substituent at the para position. Substrates containing base-sensitive functional groups such as benzoic esters and benzonitriles, which do not tolerate heating in the presence of hydroxide,5c were transformed to the desired product in respectable yields (entries 2–3). In addition, the presence of an ortho substituent on the aryl halide was tolerated (entry 4). Using the standard conditions, reactions of anilines containing strongly electron-withdrawing substituents at the 4-position provided the diaryl amine product in lower yields (entries 5–7). In these reactions, significant quantities of triarylamine byproducts were observed.4d The reaction of 4-nitroaniline with an aryl iodide provided the undesired triarylamine as the major product. However, an aryl halide could be coupled with N-(4-aminophenyl)acetamide to provide a product with a similar substitution pattern (entry 8). As previously noted, the Cu-catalyzed coupling of an anilino-NH2 group in the presence of an amide is unusual for a Cu-catalyzed reaction of this type.6a–b In this case, the observed chemoselectivity is likely due to the slow reaction of secondary amides.9 In contrast to this result, the reaction of 4-aminobenzamide with 4-iodoanisole provided a complex mixture of products. Lastly, the Cu/L5-catalyzed reaction of 2-aminobenzothiazole with 4-iodoanisole arylated the heterocyclic nitrogen as opposed to the anilino-NH2 (entry 9).10 This result is noteworthy, since the Pd-catalyzed reactions of this nucleophile with aryl bromides selectively react at the anilino-NH2 position.11
Table 2.
Cu-Catalyzed Reactions of Anilines with Aryl Iodides.a
 | ||||
|---|---|---|---|---|
| entry | product | temperature (°C) | yield (%)b | |
| 1 |
|
70 | 82c | |
| 2 |
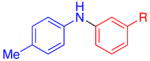
|
R=CO2Et | 80 | 78 |
| 3 | CN | 80 | 73 | |
| 4 |
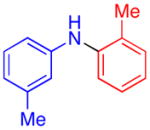
|
80 | 71 | |
| 5 |
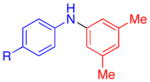
|
R=Ac | 90 | 60 |
| 6 | CO2Et | 90 | 50 | |
| 7 | CN | 90 | 52 | |
| 8 |
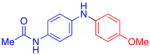
|
80 | 82 | |
| 9 |
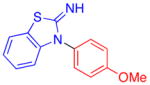
|
90 | 68 | |
Reactions Conditions: 2.0 mmol ArNH2, 1.0 mmol ArI, 2.0 mmol K3PO4, 0.10 mmol CuI, 0.20 mmol L5, 0.5 mL DMSO, in a sealed tube under an N2 atmosphere for 24 h.
Yields reported are the average of at least two runs determined to be > 95% pure by elemental analysis or 1H NMR.
5% CuI, 10% L5, 1.5 mmol ArNH2
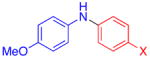
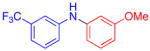
Aryl bromides were also successfully coupled using the CuI/L catalyst system (Table 3), although higher temperatures were required (100 °C). Increasing the reaction temperature to 110 °C provided significant quantities of N-arylated and decarboxylated pyrrole, and low yields of the diarylamine products. Electron-donating and -withdrawing substituents were tolerated on both the nucleophile and electrophile (entries 1–4). In addition, anilines and aryl bromides containing ortho-substituents were effectively combined (entries 5–8). When employing 3-bromoquinoline as a substrate, a significant quantity of reduced heteroarene was observed (entry 9). The formation of this byproduct is common for Cu-catalyzed reactions of heteroaryl halides with amines.5c
Table 3.
Cu-Catalyzed Reactions of Anilines with Aryl Bromides.a
 | |||
|---|---|---|---|
| entry | product | yield (%)b | |
| 1 |
|
X = Cl | 76 |
| 2 | F | 72 | |
| 3 |
|
55 | |
| 4 |
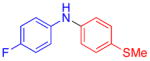
|
71 | |
| 5 |
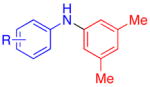
|
R = 2,5-Me2 | 75c |
| 6 | 2-OMe | 70 | |
| 7 |
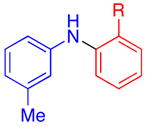
|
R = Me | 74d |
| 8 | OMe | 67d | |
| 9 |
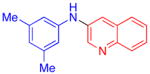
|
51 | |
Reactions Conditions: 2.0 mmol ArNH2, 1.0 mmol ArBr, 2.0 mmol K3PO4, 0.10 mmol CuI, 0.20 mmol L5, 0.5 mL DMSO, in a sealed tube under an N2 atmosphere for 24 h.
Yields reported are the average of at least two runs determined to be > 95% pure by elemental analysis or 1H NMR.
20% L15 employed as a ligand in DMF at 110 °C.
30 h.
In conclusion, pyrrole 2-carboxylic acid was employed as a suitable ligand for the Cu-catalyzed monoarylation of anilines with aryl iodides and bromides. Anilines and aryl halides possessing diverse electronic properties and useful functional groups were all tolerated. In many cases, the relatively low catalyst loading (10% Cu), the breadth of functional groups tolerated by the catalyst system, and the cost and commercial availability of the metal and ligand, might offset the required use of two equivalents of amine, and the moderate yields obtained. We are continuing our investigations to develop newer and more active Cu-based catalyst systems for this transformation.
Experimental Section
General procedure for the Cu-catalyzed cross-coupling of anilines with aryl halides
An oven-dried screw-cap test tube was charged with K3PO4 (424 mg, 2.0 mmol). The tube was sealed and the base was flame-dried under vacuum, and cooled under a purge of N2. CuI (19 mg, 0.10 mmol), Pyrrole-2-carboxylic acid (22 mg, 0.20 mmol), aryl halide (1.0 mmol, if solid), amine (2.0 mmol, if solid) and a magnetic stir bar were added to the cooled vessel. The tube was then evacuated and back-filled with nitrogen. The evacuation/backfill sequence was repeated two additional times. Aryl halide (1.0 mmol, if liquid), amine (2.0 mmol, if liquid) and DMSO (0.50 mL) were then added by syringe. The vessel was immersed in a preheated oil bath and the reaction mixture was stirred vigorously until TLC and/or GC analysis of the crude reaction mixture indicated that the aryl halide had been completely consumed. The reaction mixture was cooled to room temperature. Ethyl acetate (15 mL), NH4Cl(aq) (2 mL), and H2O (1mL) were added and the mixture was stirred. The organic layer was separated, and filtered through a plug of silica. The aqueous layer was extracted twice more with ethyl acetate (10 mL), and each extract was sequentially filtered through the pad of silica gel. The filtrate was concentrated and the resulting residue was purified by flash chromatography (hexanes/ethyl acetate, gradient elution) to provide the desired product.
Supplementary Material
Experimental procedures and characterization data for all new and known compounds, kinetic and computational data for complexes. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.

Ligands Examined for N-Arylation Reactions of Aniline.
Acknowledgments
We thank the National Institutes of Health (GM58160 and GM46059) for financial support for this project. RAA is grateful to the National Institutes of Health (F31GM081905) for a predoctoral fellowship. We thank Merck for additional unrestricted funding. The NMR instruments used for this publication were furnished by funds from the National Science Foundation (CHE 9808061 and DBI 9729592).
References
- 1.(a) Surry DS, Buchwald SL. Angew Chem Int Ed. 2008 doi: 10.1002/anie.200800497. Manuscript Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiang L, Buchwald SL. Palladium-Catalyzed Aromatic Carbon-Nitrogen Bond Formation. In: de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2. Wiley-VCH; Weinheim: 2004. pp. 699–760. [Google Scholar]
- 2.(a) Beletskaya IP, Cheprakov AV. Coord Chem Rev. 2004:2337. doi: 10.1021/cr9903048. [DOI] [PubMed] [Google Scholar]; (b) Ley SV, Thomas AW. Angew Chem Int Ed. 2003;42:5400. doi: 10.1002/anie.200300594. [DOI] [PubMed] [Google Scholar]; (c) Kunz K, Scholz U, Ganzer D. Synlett. 2003:2428. [Google Scholar]
- 3.Biscoe MB, Barder TE, Bucwhald SL. Angew Chem Int Ed. 2007;46:7232. doi: 10.1002/anie.200702122. [DOI] [PubMed] [Google Scholar]
- 4.(a) Gujadhur R, Venkataraman D, Kintigh JT. Tetrahedron Lett. 2001;42:4791. [Google Scholar]; (b) Zhang H, Cai Q, Ma D. J Org Chem. 2005;70:5164. doi: 10.1021/jo0504464. [DOI] [PubMed] [Google Scholar]; (c) Rao H, Fu H, Jiang Y, Zhao Y. J Org Chem. 2005;70:8107. doi: 10.1021/jo051221w. [DOI] [PubMed] [Google Scholar]; (d) Guo X, Rao H, Fu H, Jiang Y, Zhao Y. Adv Synth Catal. 2006;348:2197. [Google Scholar]; (e) Rao H, Jin Y, Fu H, Jiang Y, Zhao Y. Chem Eur J. 2006;12:3636. doi: 10.1002/chem.200501473. [DOI] [PubMed] [Google Scholar]; (f) Liu Y, Bai Y, Zhang J, Li Y, Jiao J, Qi X. Eur J Org Chem. 2007:6084. [Google Scholar]; (g) Rout L, Jammi S, Punniyamurthy T. Org Lett. 2007;9:3397. doi: 10.1021/ol0713887. [DOI] [PubMed] [Google Scholar]
- 5.(a) Huang X, Anderson KW, Zim D, Jiang L, Klapars A, Buchwald SL. J Am Chem Soc. 2003;125:6653. doi: 10.1021/ja035483w. [DOI] [PubMed] [Google Scholar]; (b) Klapars A, Huang X, Buchwald SL. J Am Chem Soc. 2002;124:7421. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]; (c) Altman RA, Koval ED, Buchwald SL. J Org Chem. 2007;72:6190. doi: 10.1021/jo070807a. [DOI] [PubMed] [Google Scholar]; (d) Antilla JC, Klapars A, Buchwald SL. J Am Chem Soc. 2002;124:11684. doi: 10.1021/ja027433h. [DOI] [PubMed] [Google Scholar]; (e) Shafir A, Buchwald SL. J Am Chem Soc. 2006;128:8742. doi: 10.1021/ja063063b. [DOI] [PubMed] [Google Scholar]; (f) Shafir A, Lichtor PA, Buchwald SL. J Am Chem Soc. 2007;129:3490. doi: 10.1021/ja068926f. [DOI] [PubMed] [Google Scholar]; (g) Altman RA, Shafir A, Choi A, Lichtor PA, Buchwald SL. J Org Chem. 2008;73:284. doi: 10.1021/jo702024p. [DOI] [PubMed] [Google Scholar]
- 6.(a) Goodbrand HB, Hu NX. J Org Chem. 1999;64:670. [Google Scholar]; (b) Kelkar AA, Patil NM, Chaudhari RV. Tetrahedron Lett. 2002;43:7143. [Google Scholar]; (c) Patil NM, Kelkar AA, Chaudhari RV. J Mol Catal A: Chem. 2004;223:45. [Google Scholar]
- 7.(a) Allred GD, Liebeskind LS. J Am Chem Soc. 1996;118:2748. [Google Scholar]; (b) Yip SF, Cheung HY, Zhou Z, Kwong FY. Org Lett. 2007;9:3469. doi: 10.1021/ol701473p. [DOI] [PubMed] [Google Scholar]
- 8.Cristau HJ, Cellier PP, Hamada S, Spindler JF, Taillefer M. Org Lett. 2004;6:913. doi: 10.1021/ol036290g. [DOI] [PubMed] [Google Scholar]
- 9.Klapars A, Huang X, Buchwald SL. J Am Chem Soc. 2002;124:7421. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]
- 10.Authentic N-(4-methoxyphenyl)benzo[d]thiazol-2-amine was independently prepared from the reaction of 2-chlorobenzothiazole and p-anisidine according to the procedure in Cossey HD, Judd J, Stephens FF. J Chem Soc. 1965:954. The product of this uncatalyzed reaction was found to be different than the product obtained from the reaction described in Table 2, entry 9.
- 11.Yin J, Zhao MM, Huffman MA, McNamara JM. Org Lett. 2002;4:3481. doi: 10.1021/ol0265923. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and characterization data for all new and known compounds, kinetic and computational data for complexes. This material is available free of charge via the Internet at http://pubs.acs.org.