

Abstract
Light information reaches the suprachiasmatic nucleus (SCN) through a subpopulation of retinal ganglion cells. Previous work raised the possibility that brain-derived neurotrophic factor (BDNF) and its high-affinity tropomyosin-related receptor kinase may be important as modulators of this excitatory input into the SCN. In order to test this possibility, we used whole-cell patch-clamp methods to measure spontaneous excitatory currents in mouse SCN neurons. We found that the amplitude and frequency of these currents were increased by BDNF and decreased by the neurotrophin receptor inhibitor K252a. The neurotrophin also increased the magnitude of currents evoked by application of N-methyl-D-aspartate and amino-methyl proprionic acid. Next, we measured the rhythms in action potential discharge from the SCN brain slice preparation. We found that application of K252a dramatically reduced the magnitude of phase shifts of the electrical activity rhythm generated by the application of glutamate. By itself, BDNF caused phase shifts that resembled those produced by glutamate and were blocked by K252a. The results demonstrate that BDNF and neurotrophin receptors can enhance glutamatergic synaptic transmission within a subset of SCN neurons and potentiate glutamate-induced phase shifts of the circadian rhythm of neural activity in the SCN.
Keywords: circadian rhythms, mouse, neurotrophin, N-methyl-d-aspartate, suprachiasmatic nucleus
Introduction
In mammals, the neural structure responsible for most circadian behaviors can be localized to a bilaterally paired structure in the hypothalamus, the suprachiasmatic nucleus (SCN). Many SCN neurons function as intrinsic pacemakers that generate daily rhythms of gene expression and electrical activity with a frequency of close to 24 h (Herzog & Schwartz, 2002). In order to function adaptively, these neurons must be synchronized to each other as well as to the physical environment. This process of synchronization or entrainment occurs through daily light-induced phase advances and delays of the endogenous clock (Roenneberg et al., 2003). Cells in the SCN gain access to information about external lighting conditions, at least in part, through a specialized subpopulation of light-sensitive retinal ganglion cells that contain the photopigment melanopsin (Van Gelder, 2005). These ganglion cells express and utilize the neuropeptide pituitary adenylyl cyclase activating peptide and glutamate as cotransmitters to communicate with the SCN (Hannibal et al., 2000, 2002). Understanding the mechanisms underlying the photic regulation of SCN neurons is a critical problem in this research area.
Previous work has raised the possibility that brain-derived neurotrophic factor (BDNF) may be an important regulator of synaptic input into the SCN. Both BDNF and its high-affinity tropomyosin-related receptor kinase (TrkB) receptor are expressed in the SCN (Liang et al., 1998; Allen & Earnest, 2005). The levels of BDNF fluctuate rhythmically in this region with peak protein levels found during the night when exposure to light can alter the phase of circadian oscillations (Liang et al., 1998). Functionally, both BDNF- and TrkB-deficient mice exhibit a reduction in the phase-shifting effects of light on the circadian system (Liang et al., 2000; Allen et al., 2005). BDNF-expressing cells in the ventral SCN are well positioned to gate light input to the circadian system, i.e. BDNF secreted at night may be required for light-induced phase shifts. In the hippocampus and other brain regions, BDNF modulates glutamatergic synaptic transmission (e.g. Blum & Konnerth, 2005; Nagappan & Lu, 2005). Thus, we became interested in exploring the possible role of BDNF as a regulator of glutamatergic signaling within the SCN.
Materials and methods
Animals
The measurements of excitatory currents were carried out at the University of California, Los Angeles using an approved animal use protocol (Animal Research Committee no. 1997-027). C57Bl/6 mice were obtained from a breeding facility at the University of California, Los Angeles. The long-term recordings of neural activity rhythm were carried out at East Carolina University using an approved animal protocol (Animal Use and Care Committee no. Q201). Experiments were carried out in accordance with NIH Guidelines regarding the care and use of animals. For these studies, C57Bl/6 mice were obtained from Harlan (Indianapolis, IN, USA). At both locations, the mice were maintained on a daily 12-h light/dark cycle for at least 10 days prior to the experiment. The time of lights-off in the colony rooms prior to the preparation of the brain slices was defined as circadian time (CT) 12 for the physiological analysis. It has been well established that cells in the SCN continue to show circadian oscillations when isolated from the animal in a brain slice preparation and that the phase of rhythm is dependent upon the light/dark cycle to which the mouse was exposed.
Brain slice preparation
For whole-cell patch-clamp recordings, brain slices were prepared at the time of lights-off for recordings during the night (CT 14–18). Brain slices were prepared, using standard techniques, from mice between 4 and 6 weeks of age. For long-term recording of rhythms in neural activity, brain slices were prepared 2 h prior to the time of lights-off using mice between 6 and 8 weeks of age. In both cases, the animals were anesthetized with Isoflurane, killed by decapitation, and brains dissected and placed in cold oxygenated artificial cerebral spinal fluid containing (in mm): NaCl, 130; NaHCO3, 26; KCl, 3; MgCl2, 5; NaH2PO4, 1.25; CaCl2, 1.0 and glucose, 10 (pH 7.2–7.4). After cutting slices (Microslicer, DSK Model 1500E) from the areas to be analysed, coronal sections were placed in artificial cerebral spinal fluid (25–27 °C) for at least 1 h (in this solution CaCl2 is increased to 2mm and MgCl2 is decreased to 2 mm). Slices were constantly oxygenated with 95% O2/5% CO2 (pH 7.2—7.4, osmolality 290–300 mOsm).
Whole-cell patch-clamp electrophysiology
The methods used were similar to those described previously (Colwell, 2001; Michel et al., 2002). Briefly, electrodes were pulled on a multistage puller (P-97, Sutter, Novato, CA, USA). The electrode resistance in the bath was typically 4–6 MΩ, and the standard solution in the patch pipette contained (in mm): Cs-methanesulfonate, 125; HEPES, 8; MgATP, 5; NaCl, 4; KCl, 3; MgCl2, 1; GTP, 1; leupeptin, 0.1 and phosphocreatine, 10. The pH was adjusted to 7.25–7.3 using CsOH and the osmolarity to 280–290 mOsm using sucrose. Whole-cell recordings were obtained with a 200B amplifier and monitored on-line with pclamp (both Axon Instruments, Foster City, CA, USA). To minimize changes in offset potentials with changing ionic conditions, the ground path used an artificial cerebral spinal fluid agar bridge. Cells were approached with a slight positive pressure (2–3cm H2O) and offset potentials were corrected. The liquid junction potential was -14 mV. The pipette was lowered to the vicinity of the membrane keeping a positive pressure. After forming a high-resistance seal (2–10 GΩ) by applying negative pressure, a second pulse of negative pressure was used to break the membrane. While entering the whole-cell mode, a repetitive test pulse of 10 mV was delivered in a passive potential range (approx. -60 to -70 mV). The whole-cell capacitance and electrode resistance were neutralized and compensated (50–80%) using the test pulse. Data acquisition was then initiated. Series and input resistance were monitored repeatedly by checking the response to small pulses in a passive potential range. The series resistance was not compensated and the maximal voltage error due to this resistance was calculated to be 6 mV. The access resistance of these cells ranged from 20 to 40 MΩ whereas the cell capacitance was typically between 6 and 18 pF.
Under voltage clamp (Vm = -70 mV), the holding current was monitored throughout the experiment. In addition, the current—voltage relationship of the neuron was measured every 2–3 min by moving the cell’s membrane potential through a series of voltage steps. In these experiments, after the initial control ramps (no drug), each cell was exposed to amino-methyl proprionic acid (AMPA) or N-methyl-D-aspartate (NMDA) followed by a wash until the response returned to baseline. Current—voltage relationships were calculated as the response to the agonist minus the baseline response in the absence of the agonist. Current measurements were normalized to cell size by dividing peak inward current by cell capacitance.
Measurement of excitatory currents
Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded from SCN brain slices and analysed using the minianalysis program (Synaptosoft, Decatur, GA, USA) as previously described (Michel et al., 2002, 2006). The software analysed the number and peak amplitude of sEPSCs. Each automatically detected event was manually checked to ensure that the baseline and peak were accurately determined. The mean frequency and amplitude of the sEPSCs were calculated for each neuron during 360-s sampling periods before, during and after drug treatment.
Measurement of neural activity rhythms
Methods were similar to those described previously (Ding et al., 1994). Spontaneous action potentials were recorded from the SCN with single-unit extracellular electrodes (3–5 MΩ, filled with 5 m NaCl) under the visual guidance of a dissecting microscope for 4 min every 10-min interval. Action potentials generated from individual neurons were isolated on an oscilloscope with a window discriminator in real time based on amplitude, waveform, polarity and cadence, and counted by a customized program (labview, National Instruments, Austin, TX, USA). Under control conditions, the time average of spike train frequencies exhibited a stable phase relationship with the light/dark cycle, with the peak occurring at CT 7 for up to 3 days in vitro. Phase shifts were calculated as the time difference in hours between the peak of the circadian rhythm in spontaneous action potentials of treatment and control conditions.
Drug application
For acute electrophysiology experiments, solution exchanges within the slice were achieved by a rapid gravity-feed delivery system utilizing an electronically controlled valve. In this study, AMPA and NMDA were applied for 120 s with maximal responses typically observed approximately 90 s after the start of treatment. BDNF and K252a were applied for 240 s and, in some cases, these treatments were immediately followed by application of AMPA or NMDA. BDNF was applied focally (100 μm tip) using a pressurized application system (ALA Scientific Instruments, Westbury, NY, USA). For the experiments measuring neural activity rhythms, the drug treatments were achieved by stopping the perfusion pump (34 mL/h) in order to expose the surface of the brain slice. A syringe was then used to apply small drops (0.2 μL) of glutamate or BDNF bilaterally to the SCN (10 min application). 2-Amino-5-phosphonovalerate (AP5) and K252a were bath applied from 20 min prior to the indicated treatment time. Chemicals were purchased from Sigma (St Louis, MO, USA).
Statistical analyses
One-way anova and Scheffé means comparison procedures were used to evaluate the statistical significance of the phase-shifting experiments at an alpha level of 0.05 using origin 7.0 (OriginLab, Northampton, MA, USA). For measurements of excitatory currents, between-group differences were evaluated using t-tests or Mann—Whitney rank sum tests when appropriate. Values were considered significantly different if P < 0.05. These tests were performed using sigmastat (SPSS, Chicago, IL, USA). In the text, values are shown as mean ± SEM.
Results
Brain-derived neurotrophic factor enhances excitatory synaptic transmission measured in suprachiasmatic nucleus neurons
In order to test the hypothesis that BDNF regulates excitatory synaptic transmission within the mouse SCN, whole-cell voltage-clamp recording techniques were used to measure sEPSCs in ventral SCN neurons during the night (Fig. 1). The mean frequency and mean amplitude of sEPSCs recorded at a holding potential of -70 mV were 0.09 ± 0.06 events/s and -12.0 ± 2.4 pA, respectively (n = 16 neurons). These sEPSCs were recorded in the presence of tetrodotoxin and bicuculline (25 μm). The sEPSCs were completely abolished with 6-cyano-7-nitroquinoxaline-2,3-dione (25 μm, five of five neurons tested) and were stable over 20 min (data not shown, n = 5). Focal application of BDNF (100 ng/mL; 240 s) increased the sEPSC amplitude in a subset of the ventral SCN neurons examined (43 ± 11% increase in responding neurons; six of 12 neurons responded; P < 0.05). BDNF also increased the sEPSC frequency in some of these neurons (141 ± 49% increase in responding neurons; six of 12 neurons responded; P < 0.05). Although most of the responding neurons showed an increase in both amplitude and frequency, one neuron showed a BDNF-induced increase in sEPSC amplitude without a change in frequency and one neuron showed a BDNF-induced change in sEPSC frequency without a change in amplitude. The Trk-signaling pathway inhibitor K252a produced the opposite effects on the excitatory currents. K252a (100–200 nm; 240 s) decreased the sEPSC amplitude in about 30% of ventral SCN neurons (32 ± 5% decrease in responding neurons; five of 16 neurons responded). K252a also decreased the sEPSC frequency in most ventral SCN neurons (56 ± 7% decrease in responding neurons; 10 of 16 neurons; P < 0.05). Again, it was possible to dissociate the neurotrophin effects on amplitude and frequency as five neurons exhibited a K252a-induced decrease in frequency without a corresponding change in amplitude. Thus, BDNF can increase and conversely K252a can decrease both sEPSC frequency and amplitude.
Fig. 1.

Brain-derived neurotrophic factor (BDNF) enhances excitatory synaptic transmission in suprachiasmatic nucleus (SCN) neurons. Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded from the ventral SCN neuron during the night in the presence of tetrodotoxin (0.5 μM) and bicuculline (25 μM). (A) Examples of sEPSCs recorded from a neuron immediately before and after treatment with BDNF (100 ng/mL, 240 s). (B) Average sEPSC waveform recorded in this same neuron before (gray line) and after (black line) treatment with BDNF. (C) Application of BDNF increased the frequency and amplitude of the sEPSCs whereas K252a (100 nM, 240 s) decreased these same values. Neurons that did not respond to the BDNF treatment were not included in this analysis. Data are shown as means ± SEM. *Significance at P < 0.05.
Brain-derived neurotrophic factor-enhanced N-methyl-d-aspartate- and amino-methyl proprionic acid-evoked currents recorded in suprachiasmatic nucleus neurons
To directly test the hypothesis that BDNF modulates the postsynaptic response of SCN neurons to glutamatergic stimulation, whole-cell patch-clamp recording techniques were used to measure currents evoked by NMDA and AMPA in ventral SCN neurons. NMDA currents were blocked by AP5 (50 μm, 240 s) and were stable over 30 min (data not shown, n = 8). The bath application of NMDA (25 μm, 120 s) produced a normalized peak current of -6.4 ± 0.3 pA/pF (n = 17 neurons). Treatment with BDNF (100 ng/mL, 240 s) significantly enhanced the magnitude of NMDA-evoked currents in the SCN neurons examined (62 ± 19% increase in peak current in responding neurons; 14 of 17 neurons responded; P < 0.001; Fig. 2). AMPA currents were blocked by the AMPA/KA GluR antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (25 μm, 240 s) and were stable for the 30 min (data not shown, n = 6). The bath application of AMPA (25 μm, 120 s) produced a normalized peak current of -13.3 ± 1.4 pA/pF (n = 35). Treatment with BDNF (100 ng/mL, 240 s) significantly enhanced the magnitude of AMPA-evoked currents in most SCN neurons examined (43 ± 5% increase in responding neurons; 25 of 35 neurons responded; P < 0.001; Fig. 3). Pretreatment with K252a (100 nm, 240 s) prevented the stimulatory effect of BDNF on AMPA currents (2 ± 8% increase, n = 7). These data demonstrate that BDNF can modulate AMPA- and NMDA-evoked currents in the SCN through activation of neurotrophin receptors.
Fig. 2.
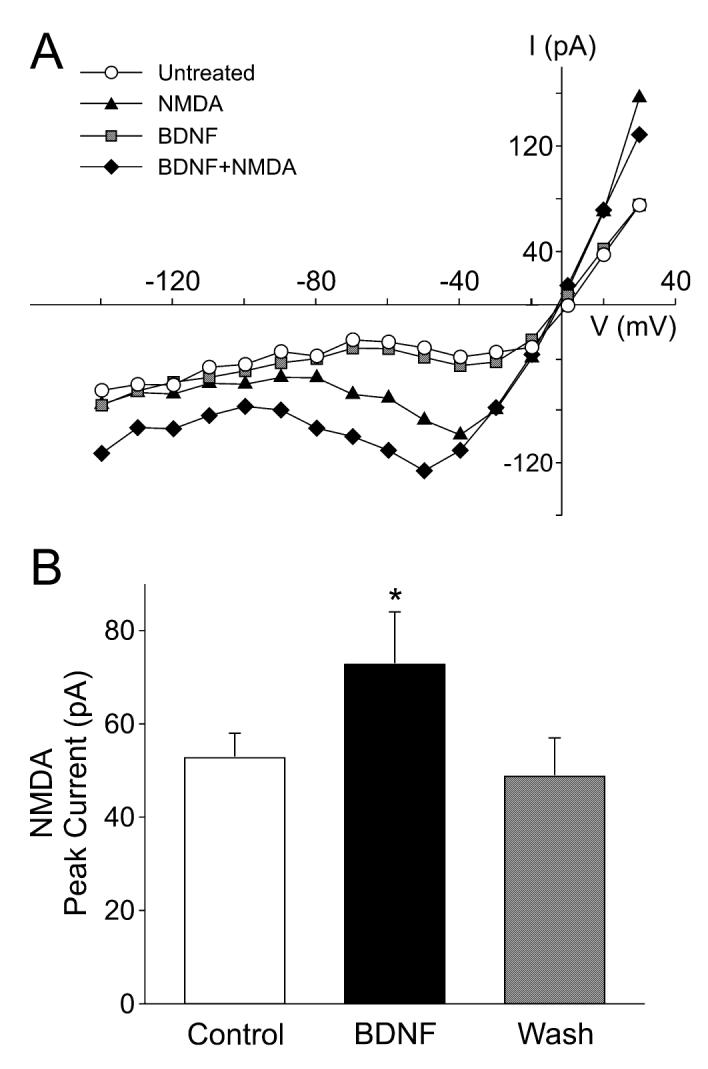
Brain-derived neurotrophic factor (BDNF) enhances the magnitude of N-methyl-D-aspartate (NMDA) currents in suprachiasmatic nucleus (SCN) neurons. Whole-cell patch-clamp recording techniques were used to measure currents evoked by NMDA in ventral SCN neurons during the night. The voltage dependence of the currents was measured by moving the membrane potential of the cell through a series of voltage steps before, during and after treatment with NMDA (25 μM, 120 s) or BDNF (100 ng/mL, 240 s) in the bath. (A) current—voltage relationship for peak NMDA currents recorded before (‘NMDA’) and after (‘BDNF + NMDA’) treatment. For comparison, the current—voltage curves obtained before any treatment (‘untreated’) and after BDNF alone (‘BDNF’) are shown. (B) Application of BDNF increased the magnitude of the peak NMDA currents. Control refers to NMDA-induced current before BDNF treatment. Neurons that did not respond to the BDNF treatment were not included in this analysis. Data are shown as means ± SEM. *Significance at P < 0.05.
Fig. 3.
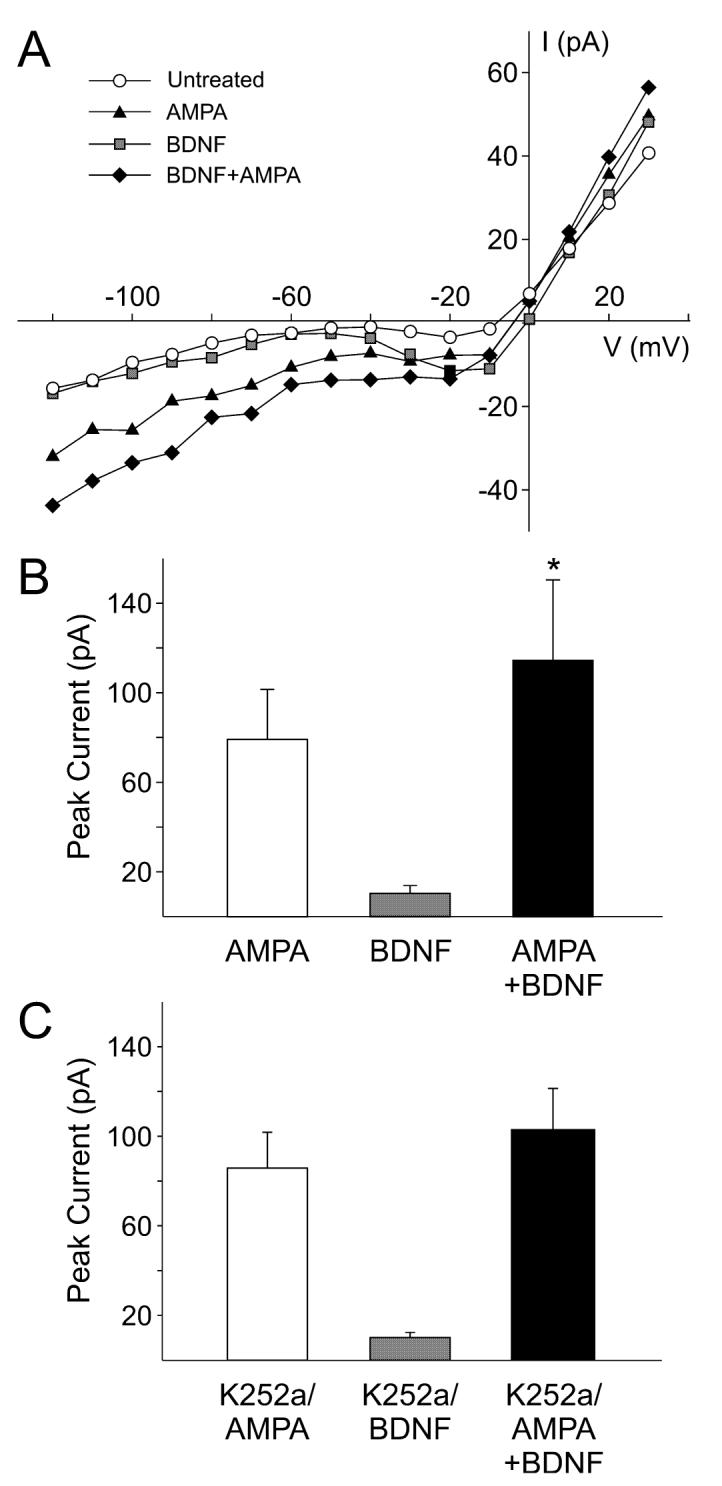
Brain-derived neurotrophic factor (BDNF) enhances the magnitude of amino-methyl proprionic acid (AMPA) currents in suprachiasmatic nucleus (SCN) neurons. Whole-cell patch-clamp recording techniques were used to measure currents evoked by AMPA in ventral SCN neurons during the night. The voltage dependence of the currents was measured by moving the membrane potential of the cell through a series of voltage steps before, during and after treatment with AMPA (25 μM, 120 s) or BDNF (100 ng/mL, 240 s) in the bath. (A) current—voltage relationship for peak AMPA currents recorded before (‘AMPA’) and after (‘BDNF + AMPA’) treatment. For comparison, the current—voltage curves obtained before any treatment (‘untreated’) and after BDNF alone (‘BDNF’) are shown. (B) Application of BDNF increased the magnitude of the peak AMPA currents. Neurons that did not respond to the BDNF treatment were not included in this analysis. (C) BDNF did not significantly increase AMPA currents in the presence of the neurotrophin receptor antagonist K252a (100 nm). Data are shown as means ± SEM. *Significance at P < 0.05.
Neurotrophin receptor antagonist K252a inhibits glutamate-induced phase shifts
In the next set of experiments, we sought to determine if neurotrophin receptors are required for glutamate-induced phase shifts of the neural activity rhythm. In the early subjective night (CT 16), glutamate alone (10 μm, 10 min) caused a 3.0 ± 0.2-h (n = 5) phase delay (Fig. 4), whereas K252a alone (100 nm, 30 min) had no effect on the timing of the peak (0.2 ± 0.2 h, n = 4). When the SCN slice was treated with glutamate in the presence of K252a, the magnitude of the glutamate-induced phase delay was significantly reduced to 1.3 ± 0.2 h (P = 0.00003, n = 3). Similarly, in the late subjective night (CT 22), glutamate alone (10 μm, 10 min) caused a 2.6 ± 0.01-h (n = 4) phase advance (Fig. 4), whereas K252a alone (100 nm, 30 min, n = 4) had no effect on the timing of the peak (0.2 ± 0.06 h). When the SCN slice was treated with glutamate in the presence of K252a, the magnitude of the glutamate-induced phase shift was significantly reduced to 1.25 ± 0.14 h (P = 0.00001, n = 3). These observations demonstrate that activation of neurotrophin receptors in the SCN is required for glutamate-induced phase shifts of normal magnitude.
Fig. 4.
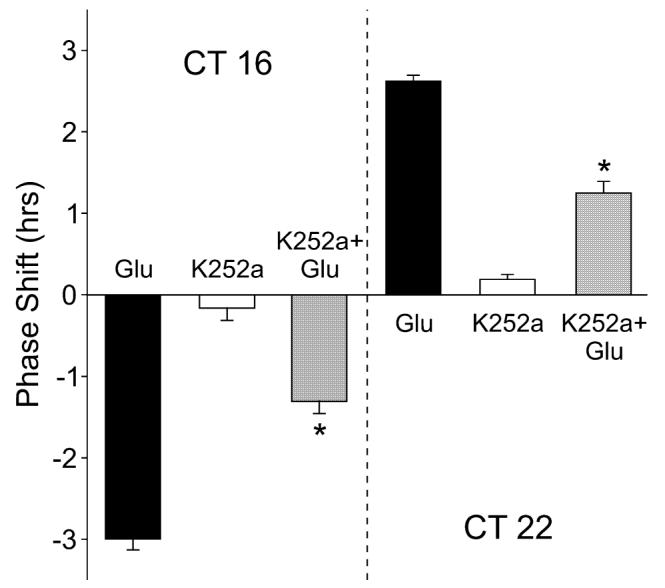
Neurotrophin receptor inhibitor reduces the magnitude of glutamate-induced phase shifts of the circadian rhythm of the suprachiasmatic nucleus (SCN) neuronal activity. Microdrops of glutamate (Glu) (10 μm, 10 min), K252a (100 nm, 10 min) or the combination of the two treatments were applied bilaterally to the SCN at circadian time (CT) 16 and CT 22. On the following day, the circadian rhythm in spontaneous action potentials was recorded. Application of K252a significantly reduced the magnitude of glutamate-induced phase delays and advances of the neural activity rhythm. By itself, K252a did not produce significant phase shifts. Data are shown as means ± SEM. *Significance at P < 0.05.
Brain-derived neurotrophic factor alone causes phase shifts of the neural activity rhythm recorded in the suprachiasmatic nucleus
In our final set of experiments, we sought to determine if BDNF itself could cause phase shifts of the neural activity rhythms recorded from the mouse SCN. Application of BDNF alone caused phase-dependent shifts in the time of peak activity measured from the SCN slices (Fig. 5A). Microdrop application of BDNF (10 μm, 0.2 μL, 10 min) during the early night (CT 16) caused a phase delay of the activity peak by 2.3 ± 0.3 h (n = 5) whereas the same treatment during the late night (CT 22) caused a phase advance of 2.3 ± 0.16 h (n = 3). No phase shift was observed when BDNF was applied during the day (CT 7). Both BDNF-induced phase advances (P = 0.0004, n = 3) and phase delay (P = 0.00001, n = 3) were fully blocked by the coadministration of the neurotrophin receptor inhibitor K252a (Fig. 5B). BDNF-induced phase shifts were also blocked by the coadministration of the NMDA receptor antagonist AP5 (P = 0.00004, n = 4). Application of K252a (100 nm, 30 min, n = 4) or AP5 (100 μm, 30 min, n = 4) did not cause phase by themselves. These data shifts demonstrate that BDNF alone can produce phase-dependent phase shifts and that these phase shifts require the activation of neurotrophin receptors as well as NMDA receptors.
Fig. 5.
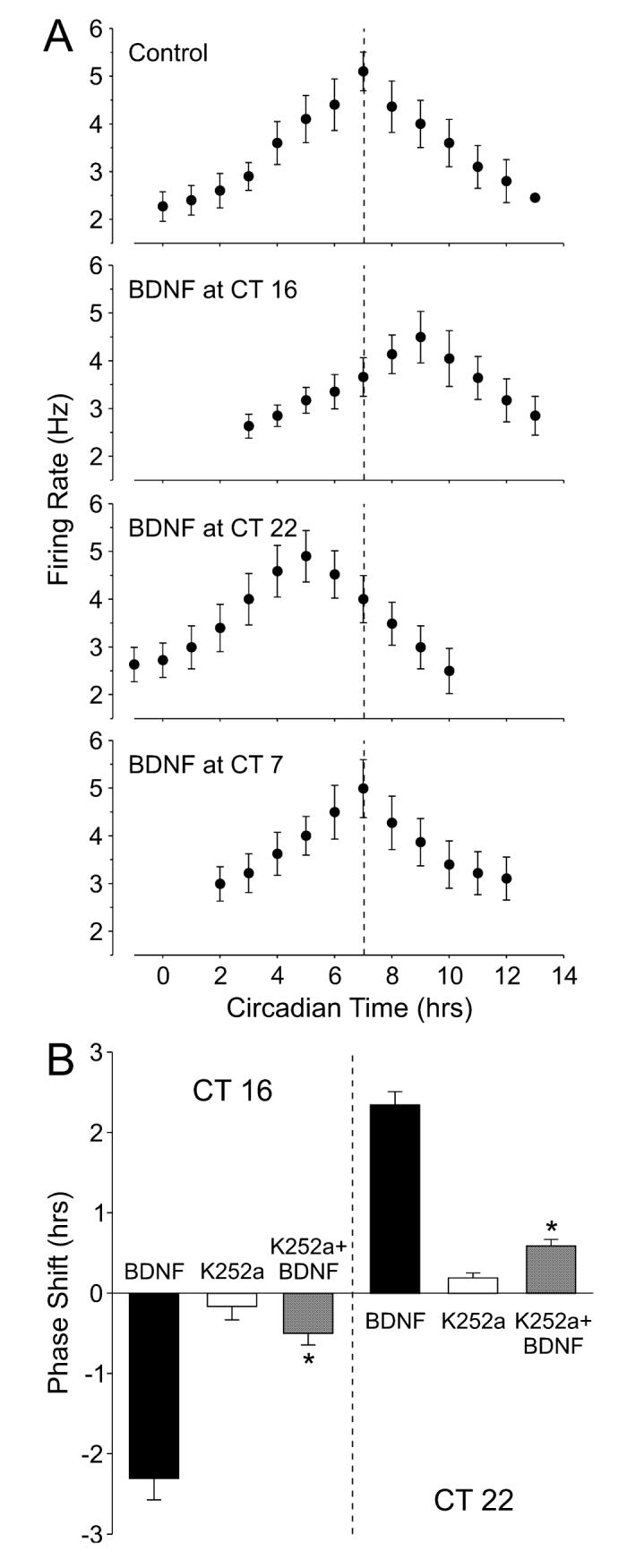
Brain-derived neurotrophic factor (BDNF) produces phase-dependent phase shifts of the circadian rhythm of the suprachiasmatic nucleus (SCN) neuronal activity. (A) Examples of neural activity rhythm recorded from the SCN. Microdrops of BDNF (10 μm, 10 min) were applied bilaterally to the SCN at circadian time (CT) 7, 16 and 22. On the following day, the circadian rhythms in spontaneous action potentials were recorded. The vertical dashed line indicates CT 7, which represents the peak of the rhythm in control slices (untreated). (B) Application of K252a (100 nm, 20 min) significantly reduced the magnitude of BDNF-induced phase delays and advances of the neural activity rhythm. By itself, K252a did not produce significant phase shifts. The phase shifts are plotted as the difference (h) in the time of peaks between the experimental and control slices. Data are shown as means ± SEM. *Significance at P < 0.05.
Discussion
Brain-derived neurotrophic factor regulates presynaptic release of glutamate
We found evidence that BDNF modulates two distinct aspects of glutamatergic synaptic transmission in the SCN. We found that BDNF increases the sEPSC frequency recorded in the SCN in the presence of tetrodotoxin. Each miniature synaptic current results from the spontaneous fusion of an individual synaptic vesicle with the presynaptic membrane subsequently resulting in quantal release of transmitter molecules. Changes in the frequency at which this process occurs are normally associated with alterations in the presynaptic release process. The finding that BDNF and the tyrosine kinase inhibitor K252a changed the sEPSC frequency demonstrates that this neurotrophin signaling system dynamically regulates the presynaptic release of glutamate. It is possible that some of the effects of K252a alone on presynaptic release are due to an inhibition of the effects of pituitary adenylyl cyclase activating peptide. This neuropeptide has emerged as a likely retinal messenger to the SCN and is expressed in terminals of retinal ganglion neurons innervating the SCN (Hannibal et al., 2000, 2002). There is some evidence that pituitary adenylyl cyclase activating peptide can activate Trk neurotrophin receptors (Lee et al., 2002; Rajagopal et al., 2004) and we have recently found physiological evidence that pituitary adenylyl cyclase activating peptide regulates the presynaptic release of glutamate onto SCN neurons (Michel et al., 2006).
Our physiological analysis does not allow us to identify the location of the glutamatergic cell bodies that are regulated by BDNF. Anatomical evidence suggests that a major source of glutamatergic input to the SCN region would be terminals of the retino-hypothalamic tract (Moore et al., 2002). A recent study found a substantial overlap in the distribution of TrkB immunoreactivity and retino-hypothalamic tract innervation in the rat SCN (Allen & Earnest, 2005). The authors’ suggestion that retino-hypothalamic tract fibers may express TrkB receptors complements the present physiological data demonstrating a presynaptic regulation of glutamate release. More broadly, these observations are consistent with previous reports of TrkB expression in retinal ganglion cells and the localization of TrkB receptors on axons in the optic nerve (Jelsma et al., 1993; Suzuki et al., 1998; Vecino et al., 2002). However, it is likely that there are other sources of glutamatergic input as most SCN neurons in both the dorsal and ventral regions exhibit AMPA-evoked currents and express glutamate receptors (Michel et al., 2002). In other regions, BDNF also presynaptically increases the frequency of excitatory currents (Lessmann, 1998; Schinder et al., 2000; Xu et al., 2000).
Brain-derived neurotrophic factor regulates the postsynaptic response to glutamate
Our physiological evidence also implicates AMPA and NMDA glutamate receptors as targets for BDNF modulation. We found that BDNF increased and K252a decreased the amplitude of sEPSCs recorded in the SCN in the presence of tetrodotoxin. Changes in the amplitude of the currents reflect postsynaptic changes in receptor sensitivity or ionic driving force and are consistent with a postsynaptic regulation of glutamate receptors. We then directly demonstrated that the application of BDNF enhanced the magnitude of the currents evoked by the application of AMPA and NMDA in SCN neurons. The stimulatory effects of BDNF were prevented by the application of K252a. The BDNF regulation of glutamate receptors has been explored in some detail in the hippocampus (Lessmann, 1998; Lin et al., 1998; Levine & Kolb, 2000). In this region, BDNF enhances the phosphorylation of the NR2B subunit (Lin et al., 1998) and increases the open probability of NMDA channels through this mechanism (Levine & Kolb, 2000). The BDNF- and tyrosine kinase-driven phosphorylation of AMPA receptors is a well-established mechanism for the regulation of receptor function (e.g. Hayashi & Huganir, 2004; Wu et al., 2004). These types of regulatory mechanisms are rapid and provide a likely explanation for the BDNF/neurotrophin receptor modulation of NMDA and AMPA currents observed in the present study. An alternative explanation is that BDNF could drive changes in the surface expression of AMPA and NMDA receptors. Dynamic changes in the number of postsynaptic AMPA receptors are a well-documented mechanism controlling the strength of excitatory synapses and there is growing evidence that at least some NMDA receptor subunits can also undergo rapid cycling in and out of the membrane (e.g. Lavezzari et al., 2004; Nong et al., 2004). The regulation of synaptic strength by phosphorylation of glutamate receptors and changes in subcellular localization are not mutually exclusive and both mechanisms could be at work in the SCN.
Brain-derived neurotrophic factor modulation occurs in a subset of suprachiasmatic nucleus neurons
A range of evidence is emerging for functionally distinct cell populations within the SCN (Antle & Silver, 2005). This anatomical evidence supports the broad division of the SCN into distinct ventral (core) and dorsal (shell) subdivisions (Morin et al., 2006). Neurons in the ventral region are innervated by visual inputs and, in many cases, express the neuropeptides vasoactive intestinal polypeptide or gastrin-releasing peptide. These retino-recipient cells must then communicate light information to the rest of the SCN. Although this study did not attempt to distinguish between BDNF responses among different SCN cell populations, we selectively targeted neurons in the ventral region using infrared differential interference contrast optics. About half of the neurons from which we recorded showed evidence of presynaptic regulation. For the subset of responding neurons, the regulation was dramatic with BDNF increasing the sEPSC frequency by about 141% and K252a reducing the frequency by 56%. When we examined the postsynaptic effects of BDNF on AMPA or NMDA currents, the percentage of responding neurons increased to 70%. Again, among the BDNF-responsive neurons, NMDA currents were increased by 62% and AMPA currents by 43%. Thus, it may well be that a distinct subset of neurons in the ventral SCN region is subject to regulation by BDNF/neurotrophin signaling. Importantly, this subset of cells is sufficient to generate phase shifts when BDNF is applied as well as prevent phase shifts when the Trk-signaling pathway is blocked. In the hamster, Antle & Silver (2005) identified a calbindin-positive cell population that is photically regulated but not electrically rhythmic (Jobst & Allen, 2002). These authors suggested that this distinct cell population may function to ‘gate’ the photic response of the SCN. Although the distribution of calbindin-positive cells varies between species, it would be interesting to determine whether these calbindin-positive neurons are the same cells that are regulated by BDNF.
Functional significance
Earnest and colleagues have previously shown that BDNF-deficient mice (BDNF+/-) exhibited a reduced magnitude of light-induced phase shifts and the infusion of exogenous BDNF resulted in mice responding to light during the day with large phase advances (Liang et al., 2000). Similar deficits in light-induced phase shifts were described in TrkB-deficient mice (TrkB+/-; Allen et al., 2005). In both cases, the mice homozygous for these mutations do not survive to adulthood so that the effects of the complete loss of BNDF/TrkB signaling could not be evaluated. In addition, the site of action of the loss of BDNF or TrkB receptors cannot be established with this type of transgenic approach. Nevertheless, this work strongly suggested a role for BDNF signaling in light- and glutamate-induced phase shifts of the circadian system. In the present study, using a brain slice preparation, we found that blocking the Trk-signaling pathway with K252a greatly reduced the magnitude of glutamate-induced phase shifts and that both advances and delays were affected. These data provide support for the behavioral studies and indicate that the site of action of BDNF is within the SCN. Together, the data indicate that neurotrophins are important modulators of the actions of light on the circadian system.
Our data also demonstrate that BDNF/Trk signaling is sufficient to generate phase shifts, a finding that could not have been predicted by the previous behavioral analysis. Using the well-established rhythms in neural activity to measure phase shifts, we found that BDNF alone mimicked the phase-shifting effects of glutamate and NMDA. Bath application of BDNF produced phase-dependent shifts in the neural activity rhythms with treatment in the early night resulting in phase delays. Treatment in the late night resulted in phase advances and treatment in the middle of the day produced no effect. These findings raise the possibility that BDNF mediates, not just modulates, light-induced phase shifts of the circadian system.
Mechanisms
The mechanisms by which BDNF and neurotrophin receptors regulate mature neurons has been the subject of extensive analysis (e.g. Chao, 2003; Blum & Konnerth, 2005; Nagappan & Lu, 2005). BDNF binds to Trk receptor tyrosine kinase and the p75 neurotrophin receptor at the cell membrane. BDNF and its receptors are then internalized and transported from the axon terminal to the cell body. The internalized receptors activate second messenger pathways including intracellular calcium, MAPK and CREB. The resulting changes in transcription are probably responsible for the BDNF-induced phase shifts of the circadian system. These same pathways are implicated in the photic regulation of the circadian system (Obrietan et al., 1998; Tischkau et al., 2003; Lundkvist et al., 2005). Whichever intracellular signaling pathways underlie the BDNF-induced phase shifts, there must be some contribution from synaptic processes within the SCN circuit. We found that the NMDA receptor antagonist AP5 blocked the BDNF-induced phase shifts. One explanation for this observation is that BDNF could stimulate the release of glutamate within the SCN circuit and the resulting activation of NMDA receptors mediates the phase shift. Our observation that BDNF treatment enhanced the presynaptic release of glutamate and the magnitude of the postsynaptic response to glutamate receptor agonists is consistent with this explanation. This BDNF-induced release of glutamate was not sufficiently large to evoke a measurable inward current in the presence of tetrodotoxin (see Fig. 2A). Alternatively, NMDA receptors can be expressed extrasynaptically and, in a few cases, result in a tonic NMDA current at the level of the cell body (e.g. Gottesman & Miller, 2003). BDNF could produce phase shifts through an increase in intracellular calcium mediated by these tonic NMDA currents. Clearly, more work will be required to develop a mechanistic explanation of BDNF-induced phase shifts as well as modulation of glutamatergic signaling within the SCN circuit.
Summary
In summary, BDNF and TrkB receptors are expressed in the SCN and perhaps the terminals at the retino-hypothalamic tract/SCN synaptic connection (Liang et al., 1998; Allen & Earnest, 2005). The BDNF content in the SCN fluctuates rhythmically such that protein levels peak during the night (Liang et al., 1998). Previous studies have shown that BDNF+/- and TrkB+/- mice exhibit deficits in the magnitude of light-induced phase shifts (Liang et al., 2000; Allen et al., 2005). The present data demonstrate that BDNF can cause phase shifts and neurotrophin receptors can modulate glutamate-induced phase shifts in the isolated SCN and thus provide an anatomical site of action for the effects of BDNF. Our cellular physiological findings demonstrate that BDNF acts both presynaptically to enhance the release of glutamate and postsynaptically to enhance the NMDA and AMPA receptor-mediated component of the SCN neurons to glutamate. This dual regulation would have the functional effect of increasing the coupling between the photic environment and the SCN during the night. Collectively, these results are consistent with the hypothesis that BDNF plays a critical role in the gating of the circadian system to light. We suggest that the presence of BDNF during the night may be a necessary requirement for glutamate to initiate a signal-transduction cascade that underlies light-induced phase shifts of the circadian system.
Acknowledgements
J.P.C. and J.M.D. were responsible for the neural activity data, and S.M. and C.S.C. were responsible for measurement of excitatory currents. We would like to thank Katherine Gniotczynski for technical assistance. Supported by NIH NS043169 to C.S.C. and NS47014 to J.M.D.
Abbreviations
- AMPA
amino-methyl proprionic acid
- AP5
2-amino-5-phosphonovalerate
- BDNF
brain-derived neurotrophic factor
- CT
circadian time
- NMDA
N-methyl-d-aspartate
- SCN
suprachiasmatic nucleus
- sEPSC
spontaneous excitatory postsynaptic current
- TrkB
tropomyosin-related receptor kinase
References
- Allen GC, Earnest DJ. Overlap in the distribution of TrkB immunoreactivity and retinohypothalamic tract innvervation of the rat suprachiasmatic nucleus. Neurosci. Lett. 2005;376:200–204. doi: 10.1016/j.neulet.2004.11.076. [DOI] [PubMed] [Google Scholar]
- Allen GC, Qu X, Earnest DJ. TrkB-deficient mice show diminished phase shifts to the circadian activity rhythm in response to light. Neurosci. Lett. 2005;378:150–155. doi: 10.1016/j.neulet.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Antle MC, Silver R. Orchestrating time: arrangements of the brain circadian clock. Trends Neurosci. 2005;28:145–151. doi: 10.1016/j.tins.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Blum R, Konnerth A. Neurotrophin-mediated rapid signaling in the central nervous system: mechanisms and functions. Physiology (Bethesda) 2005;20:70–78. doi: 10.1152/physiol.00042.2004. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Colwell CS. NMDA-evoked calcium transients and currents in the suprachiasmatic nucleus: Gating by the circadian system. Eur. J. Neurosci. 2001;13:1420–1428. doi: 10.1046/j.0953-816x.2001.01517.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JM, Chen D, Weber ET, Faiman LE, Rea MA, Gillette MU. Resetting the biological clock: Mediation of nocturnal circadian shifts by glutamate and NO. Science. 1994;266:1713–1717. doi: 10.1126/science.7527589. [DOI] [PubMed] [Google Scholar]
- Gottesman J, Miller RF. N-methyl-D-aspartate receptors contribute to the baseline noise of retinal ganglion cells. Vis. Neurosci. 2003;20:329–333. doi: 10.1017/s0952523803203114. [DOI] [PubMed] [Google Scholar]
- Hannibal J, Moller M, Ottersen OP, Fahrenkrug J. PACAP and glutamate are co-stored in the retinohypothalamic tract. J. Comp. Neurol. 2000;418:147–155. [PubMed] [Google Scholar]
- Hannibal J, Hindersson P, Knudsen SM, Georgm B, Fahrenkrug J. The photopigment melanopsin is exclusively present in pituitary adenylate cyclase-activating polypeptide-containing retinal ganglion cells of the retinohypothalamic tract. J. Neurosci. 2002;22:RC191. doi: 10.1523/JNEUROSCI.22-01-j0002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Huganir RL. Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J. Neurosci. 2004;24:6152–6160. doi: 10.1523/JNEUROSCI.0799-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog ED, Schwartz WJ. A neural clockwork for encoding circadian time. J. Appl. Physiol. 2002;92:401–408. doi: 10.1152/japplphysiol.00836.2001. [DOI] [PubMed] [Google Scholar]
- Jelsma TN, Friedman HH, Berkelaar M, Bray GM, Aguayo AJ. Different forms of the neurotrophin receptor TrkB mRNA predominate in rat retina and optic nerve. J. Neurobiol. 1993;24:1207–1214. doi: 10.1002/neu.480240907. [DOI] [PubMed] [Google Scholar]
- Jobst EE, Allen CN. Calbindin neurons in the hamster suprachiasmatic nucleus do not exhibit a circadian variation in spontaneous firing rate. Eur. J. Neurosci. 2002;16:2469–2474. doi: 10.1046/j.1460-9568.2002.02309.x. [DOI] [PubMed] [Google Scholar]
- Lavezzari G, McCallum J, Dewey CM, Roche KW. Subunit-specific regulation of NMDA receptor endocytosis. J. Neurosci. 2004;24:6383–6391. doi: 10.1523/JNEUROSCI.1890-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee FS, Rajagopal R, Kim AH, Chang PC, Chao MV. Activation of Trk neurotrophin receptor signaling by pituitary adenylate cyclase-activating polypeptides. J. Biol. Chem. 2002;277:9096–9102. doi: 10.1074/jbc.M107421200. [DOI] [PubMed] [Google Scholar]
- Lessmann V. Neurotrophin-dependent modulation of glutamatergic synaptic transmission in the mammalian CNS. Gen. Pharmacol. 1998;31:667–674. doi: 10.1016/s0306-3623(98)00190-6. [DOI] [PubMed] [Google Scholar]
- Levine ES, Kolb JE. Brain-derived neurotrophic factor increases activity of NR2B-containing N-methyl-D-aspartate receptors in excised patches from hippocampal neurons. J. Neurosci. Res. 2000;62:357–362. doi: 10.1002/1097-4547(20001101)62:3<357::AID-JNR5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Liang FQ, Walline R, Earnest DJ. Circadian rhythm of brain-derived neurotrophic factor in the rat suprachiasmatic nucleus. Neurosci. Lett. 1998;242:89–92. doi: 10.1016/s0304-3940(98)00062-7. [DOI] [PubMed] [Google Scholar]
- Liang FQ, Allen G, Earnest D. Role of brain-derived neurotrophic factor in the circadian regulation of the suprachiasmatic pacemaker by light. J. Neurosci. 2000;20:2978–2987. doi: 10.1523/JNEUROSCI.20-08-02978.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, Black IB. BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res. Mol. Brain Res. 1998;55:20–27. doi: 10.1016/s0169-328x(97)00349-5. [DOI] [PubMed] [Google Scholar]
- Lundkvist GB, Kwak Y, Davis EK, Tei H, Block GD. A calcium flux is required for circadian rhythm generation in mammalian pacemaker neurons. J. Neurosci. 2005;25:7682–7686. doi: 10.1523/JNEUROSCI.2211-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel S, Itri J, Colwell CS. Excitatory mechanisms in the suprachiasmatic nucleus: The role of AMPA/KA glutamate receptors. J. Neurophysiol. 2002;88:817–828. doi: 10.1152/jn.2002.88.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel S, Itri J, Han JH, Gniotczynski K, Colwell CS. Regulation of glutamatergic signalling by PACAP in the mammalian suprachiasmatic nucleus. BMC Neurosci. 2006;7:15. doi: 10.1186/1471-2202-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY, Speh JC, Leak RK. Suprachiasmatic nucleus organization. Cell Tissue Res. 2002;309:89–98. doi: 10.1007/s00441-002-0575-2. [DOI] [PubMed] [Google Scholar]
- Morin LP, Shivers KY, Blanchard JH, Muscat L. Complex organization of mouse and rat suprachiasmatic nucleus. Neuroscience. 2006;137:1285–1297. doi: 10.1016/j.neuroscience.2005.10.030. [DOI] [PubMed] [Google Scholar]
- Nagappan G, Lu B. Activity-dependent modulation of the BDNF receptor TrkB: mechanisms and implications. Trends Neurosci. 2005;28:464–471. doi: 10.1016/j.tins.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Nong Y, Huang YQ, Salter MW. NMDA receptors are movin’ in. Curr. Opin. Neurobiol. 2004;14:353–361. doi: 10.1016/j.conb.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Impey S, Storm DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat. Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- Rajagopal R, Chen ZY, Lee FS, Chao MV. Transactivation of Trk neurotrophin receptors by G-protein-coupled receptor ligands occurs on intracellular membranes. J. Neurosci. 2004;24:6650–6658. doi: 10.1523/JNEUROSCI.0010-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roenneberg T, Daan S, Merrow M. The art of entrainment. J. Biol. Rhythms. 2003;18:183–194. doi: 10.1177/0748730403018003001. [DOI] [PubMed] [Google Scholar]
- Schinder AF, Berninger B, Poo M. Postsynaptic target specificity of neurotrophin-induced presynaptic potentiation. Neuron. 2000;25:151–163. doi: 10.1016/s0896-6273(00)80879-x. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Nomura S, Morii E, Fukuda Y, Kosaka J. Localization of mRNAs for trkB isoforms and p75 in rat retinal ganglion cells. J. Neurosci. Res. 1998;54:27–37. doi: 10.1002/(SICI)1097-4547(19981001)54:1<27::AID-JNR4>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Tischkau SA, Mitchell JW, Tyan SH, Buchanan GF, Gillette MU. Ca2+/cAMP response element-binding protein (CREB)-dependent activation of Per1 is required for light-induced signaling in the suprachiasmatic nucleus circadian clock. J. Biol. Chem. 2003;278:718–723. doi: 10.1074/jbc.M209241200. [DOI] [PubMed] [Google Scholar]
- Van Gelder RN. Nonvisual ocular photoreception in the mammal. Meth. Enzymol. 2005;393:746–755. doi: 10.1016/S0076-6879(05)93039-5. [DOI] [PubMed] [Google Scholar]
- Vecino E, Garcia-Grespo D, Garcia M, Martinez-Millan L, Sharma SC, Carrascal E. Rat retinal ganglion cells co-express brain derived neurotrophic factor (BDNF) and its receptor TrkB. Vis. Res. 2002;42:151–157. doi: 10.1016/s0042-6989(01)00251-6. [DOI] [PubMed] [Google Scholar]
- Wu K, Len GW, McAuliffe G, Ma C, Tai JP, Xu F, Black IB. Brain-derived neurotrophic factor acutely enhances tyrosine phosphorylation of the AMPA receptor subunit GluR1 via NMDA receptor-dependent mechanisms. Brain Res. Mol. Brain Res. 2004;130:178–186. doi: 10.1016/j.molbrainres.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving TrkB. J. Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]