

Abstract
Breathing (especially deep breathing) antagonizes development and persistence of airflow obstruction during bronchoconstrictor stimulation. Force fluctuations imposed on contracted airway smooth muscle (ASM) in vitro result in its relengthening, a phenomenon called force fluctuation-induced relengthening (FFIR). Because breathing imposes similar force fluctuations on contracted ASM within intact lungs, FFIR represents a likely mechanism by which breathing antagonizes bronchoconstriction. While this bronchoprotective effect appears to be impaired in asthma, corticosteroid treatment can restore the ability of deep breaths to reverse artificially induced bronchoconstriction in asthmatic subjects. We previously demonstrated that FFIR is physiologically regulated through the p38 MAPK signaling pathway. While the beneficial effects of corticosteroids have been attributed to suppression of airway inflammation, we hypothesized that alternatively they might exert their action directly on ASM by augmenting FFIR as a result of inhibiting p38 MAPK signaling.
We tested this possibility in the present study by measuring relengthening in contracted canine tracheal smooth muscle (TSM) strips.
Our results indicate that dexamethasone treatment significantly augmented FFIR of contracted canine TSM. Canine tracheal ASM cells treated with dexamethasone demonstrated increased MAP kinase phosphatase (MKP)-1 expression and decreased p38 MAPK activity, as reflected in reduced phosphorylation of the p38 MAPK downstream target, HSP27.
These results suggest that corticosteroids may exert part of their therapeutic effect through direct action on ASM, by decreasing p38 MAPK activity and thus increasing FFIR.
Keywords: asthma, bronchoprotection, bronchoconstriction, deep breaths, steroids, tidal breathing
INTRODUCTION
Tidal breathing, especially with larger tidal volumes, antagonizes the development and persistence of airflow obstruction during bronchoconstrictor stimulation in normal animals and people [1–6]. However, this bronchoprotective effect is impaired in individuals suffering from asthma [5, 7, 8]. Corticosteroids have long been a mainstay in asthma therapy. These agents reduce airway constrictor hyperresponsiveness in mice with experimental allergen-induced airway inflammation [9, 10] and improve or restore the bronchodilatory effect of deep inspiration that is typically impaired in asthmatic patients [11–13]. The mechanisms by which breathing confers this beneficial effect in normal individuals and how corticosteroids restore this effect in asthmatic individuals are not clear. Here we propose a potential mechanism by which breathing antagonizes bronchoconstriction and suggest a novel hypothesis of how corticosteroids may restore this effect.
It is known that superimposing load fluctuations (that mimic those generated by breathing) upon isotonically contracted tracheal smooth muscle (TSM) strips causes them to relengthen [14–16], a phenomenon we’ve termed force fluctuation-induced relengthening (FFIR). We have proposed that FFIR may be one mechanism by which breathing antagonizes bronchoconstriction. Importantly, FFIR can be physiologically regulated, for pharmacological inhibition of actin polymerization [17] or of p38 mitogen-activated protein kinase (MAPK) signaling [14] augments FFIR in vitro.
In general, the beneficial effects of corticosteroids have been attributed to their anti-inflammatory actions, as proinflammatory cytokines can modulate ASM contractile and relaxant function. Grunstein and colleagues [18] have shown that IL-1β and TNF-α increase ASM contractility to acetylcholine (ACh) and impair ASM relaxation with isoproterenol, changes prevented when ASM is pretreated with dexamethasone. However, dexamethasone also increases relaxation with KCl [19] and isoproterenol [20] in contracted TSM even in the absence of a proinflammatory environment suggesting that corticosteroids might exert direct effects on ASM contraction independent of their anti-inflammatory effect. However, since these studies were conducted on rabbit tracheal and bronchial ring segments with intact epithelium, corticosteroids may have exerted their effect indirectly by acting on the epithelium or mucosa rather than on the smooth muscle itself. Corticosteroids have also been reported to induce expression of MAPK phosphatase-1 (MKP-1), which dephosphorylates and inactivates p38 MAPK [21], as well as other MAP kinases [22, 23]. Since pharmacological inhibition of p38 MAPK enhances FFIR [14], we reasoned that corticosteroid treatment might augment FFIR as well. In the present study, we found that dexamethasone treatment does indeed increase FFIR of contracted canine trachealis strips (epithelium removed) in vitro, and that such treatment increases MKP-1 expression in cultured canine tracheal myocytes. The latter was accompanied by decreased p38 MAPK activity, as reflected in diminished phosphorylation of HSP27 (a well-established downstream target of p38 MAPK [24]). Together, these results suggest that FFIR contributes to the bronchoprotective effect of breathing and that corticosteroid treatment may restore this effect in asthma by augmenting FFIR via reduction of p38 MAPK activation.
METHODS
Assessment of Force-Fluctuation-Induced Relengthening of ACh-Contracted Canine TSM Strips
In accordance with Institutional Animal Care and Use Committee (IACUC) approved protocols, random source dogs were anesthetized and killed by overdose with pentobarbital sodium (30 mg/kg iv). Tracheas were excised and rinsed several times in Krebs-Henseleit (K-H) solution (in mM: 115 NaCl, 25 NaCO3, 1.38 KH2PO4, 2.51 KCl, 2.46 MgCl2, 2.5 CaCl2, and 11.2 dextrose). K-H was gassed with 95% O2/5% CO2 to maintain a pH between 7.3 and 7.5. Some tissues were stored for up to 4 days at 4ºC prior to study, without apparent effect on results. All studies were conducted at 37ºC in K-H solution. As described previously [17], parallel fibered bundles of canine TSM (CTSM) were dissected free of all overlying connective tissue and epithelium and fastened at either end in aluminum foil clips (Laser Services Inc., Westford, MA). The strips were then placed in a horizontal dip-tray style of organ bath and connected to a 300B lever arm/force transducer (Aurora Scientific, Aurora, Canada); the 300B lever arm measures both force output and length changes. All force and length changes of the TSM strips were monitored using ADInstruments Powerlab Chart software.
As described previously [17], after equilibration, reference length (Lref) of the tissues measured between 3.5 and 6.0 mm and maximal response (Fmax) to 100 μM acetylcholine (ACh) was determined. Lref and Fmax in response to ACh were then used as base parameters for force oscillation contraction sequences. Muscles were allowed to relax by reperfusing with K-H alone. Twenty min after force reached relaxed baseline, tissues were re-exposed to 10−4 M ACh, and allowed to shorten isotonically against an afterload of 32% Fmax for 20 min and then without delay and during continued ACh exposure, force oscillations were superimposed (0.2 Hz and amplitude ± 16% Fmax) for 20 min; thereafter, TSM strips were allowed to relax by switching to ACh-free K-H solution. All length changes were noted. Next, tissues were incubated for ~2 hr in K-H solution containing 4 μM dexamethasone or vehicle control. This concentration was chosen based on studies done on a separate cohort of TSM strips that showed it to have minimal effects on isometric force (data not shown). After this equilibration period, the entire isotonic contraction sequence was repeated in the continued presence of dexamethasone or vehicle; length changes during contractions before and after inhibitor treatment were expressed as %Lref. FFIR was calculated as the extent of relengthening from the end of the isotonic shortening period until the end of the oscillation period. Differences between the first and second isotonic/force oscillation sequence (i.e., before and after dexamethasone or vehicle) were expressed as ΔFFIR.
Assessment of MKP-1 Expression and HSP27 Phosphorylation in Cultured Canine Tracheal Smooth Muscle Cells
Because we previously found that the inhibition of p38 MAPK augmented FFIR [14] and others have found that corticosteroids induce the expression of dual-specific phosphatases, especially MKP-1 [21–23], which in turn dephosphorylates and inactivates p38 MAPK [21], we measured MKP-1 expression and HSP27 phosphorylation (downstream target of p38 MAPK) in canine TSM cells. Airway myocytes were dissociated from adult canine trachealis and cultured using previously described methods [25, 26]. Briefly, TSM cells were enzymatically digested from dissected trachealis using 10 U/ml elastase, 600 U/ml collagenase, and 2 U/ml Nagarse protease. Myocytes were grown on uncoated plastic culture plates in Dulbecco’s modified Eagle’s medium/F12 (DMEM/F12) supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin, 50 μg/ml streptomycin, and 50 μg/ml gentamicin. Low passage (1–3) myocytes from 5 different primary cell lines were treated with 4 μM dexamethasone (or in media alone as untreated control) for 1 or 2 hours. Protein lysates from treated and untreated myocytes were collected using CelLytic lysis/extraction buffer (Sigma-Aldrich). Following the manufacturer’s protocol, the cells were washed with PBS, lysed for 15 minutes with 0.5ml lysis buffer supplemented with Complete (Roche) protease inhibitor cocktail mix, then centrifuged to pellet the cellular debris. The protein containing supernatant was then used for western blots.
Proteins from dexamethasone- and vehicle-treated canine TSM cells were extracted as described previously [17, 27]. All lanes in all gels were loaded with equal volumes and concentrations of total protein extracts. Denatured proteins were separated by SDS-polyacrylamide gel electrophoresis (Invitrogen, NuPage 4–12% gels), transferred to Immobilin-P PVDF membranes, and probed for phosphorylated and non-phosphorylated HSP27, MKP-1 and β-actin. Phosphorylated and non-phosphorylated HSP27 were detected on the same gels using Pierce SuperSignal West Pico chemiluminescent substrate. Membranes initially probed for phosphorylated HSP27 were stripped for 30 min at 50ºC (0.76% Tris base, 2% SDS, 0.7% β-mecaptoethanol, pH 6.7) and re-probed for total HSP27. Blot intensities (volumes) were calculated using a BioRad S710 densitometer and software. The ratios of phosphorylated to total HSP27 and MKP-1 to β-actin expression were assessed relative to data derived for vehicle-treated cells, on the same western blot. All primary antibodies were raised in rabbits, except for β-actin which was raised in mouse, and were from the following sources: HSP27, Dr. W.T. Gerthoffer; phosphorylated HSP27, Stressgen Bioreagents; anti-MKP-1, Santa Cruz Biotechnology; anti-β-actin, Sigma-Aldrich.
Data Analysis
All data were expressed as means ± standard errors. Results from the control and dexamethasone-treated groups were compared with student t-tests. Significant differences were defined when p < 0.05.
RESULTS
FFIR in CTSM strips
Superimposition of force fluctuations upon a steady load against which ACh-stimulated CTSM strips had shortened caused them to relengthen (Figure 1). After dexamethasone treatment, CTSM strips demonstrated significantly increased FFIR, whereas no change in FFIR was observed in vehicle treated CTSM strips; the increase in FFIR was significantly larger in dexamethasone-treated trachealis strips than in control strips (Figure 2, ΔFFIR = 6.6 % ± 2.70 % versus 0.52 % ± 0.72 % respectively, p = 0.029). Isotonic shortening post-treatment was not different in dexamethasone and vehicle-treated tissues (Figure 3), so this parameter could not account for the differences observed in ΔFFIR in dexamethasone-treated TSM.
Figure 1. Experimental protocol to assess drug effect on FFIR.

Tracheal smooth muscle (TSM) strips were attached to a force/length transducer and equilibrated allowing for determination of reference length (Lref) and isometric maximal force [Fmax - upon exposure to 100 μM acetylcholine (ACh)]. For the next exposure to ACh, TSM strips were contracted against a load equal to 32% of established Fmax (32% Fmax), thus producing isotonic shortening. At 20 min into the isotonic contraction, sinusoidal force fluctuations (0.2 Hz to simulate tidal breathing) were superimposed on 32% Fmax that were ± 16% Fmax. Force fluctuation-induced relengthening (FFIR) was noted at the end of 20 min. ACh was again washed out and baseline force and Lref re-established. TSM was then incubated for ~2 h in 4 μM dexamethasone (DEX) or vehicle (K-H solution alone). Differences in FFIR (ΔFFIR) post-vs pre-treatment were compared.
Figure 2. Cumulative data for FFIR in control and dexamethasone-treated canine TSM strips.
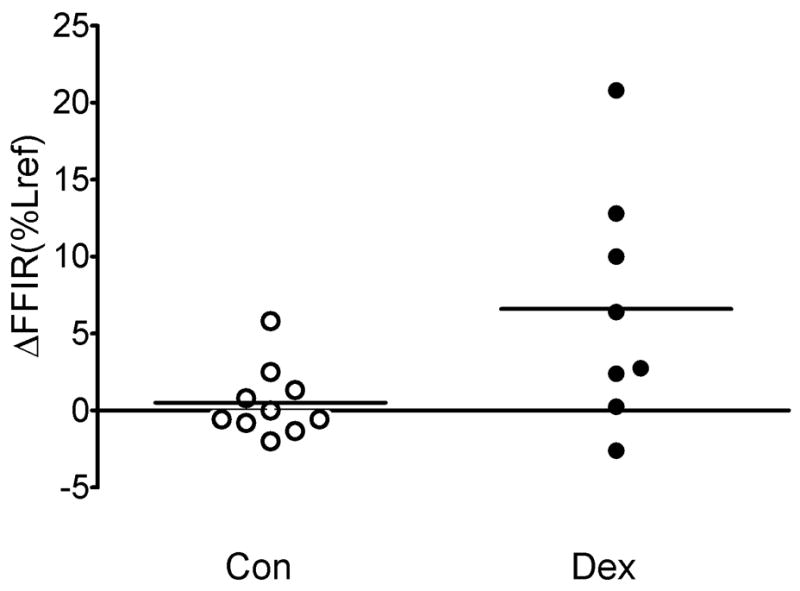
Dexamethasone-treated strips demonstrated a greater increase in FFIR (Δ FFIR) than did control strips when comparing FFIR posttreatment to pretreatment (Δ FFIR = 6.6 % ± 2.70 % versus 0.52 % ± 0.72 % respectively, p = 0.029).
Figure 3. Cumulative data for isotonic shortening control and dexamethasone-treated canine TSM strips.
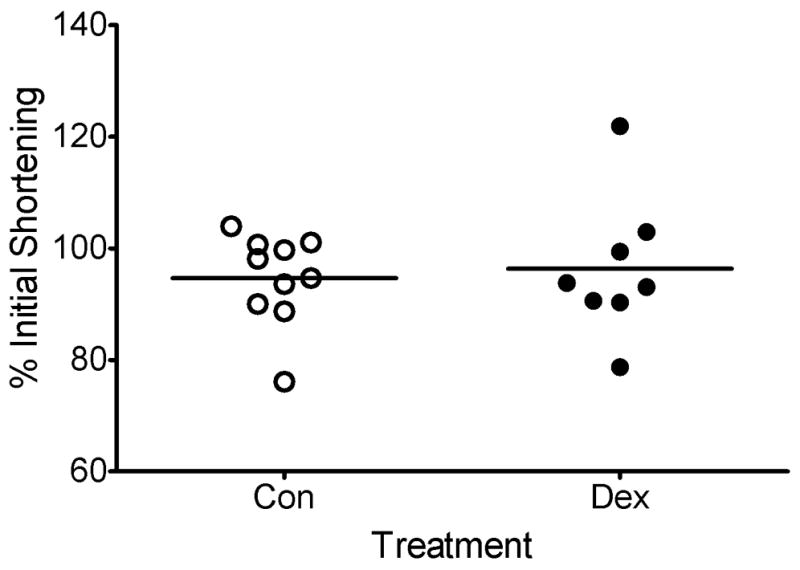
There was no significant difference in the change in isotonic (no oscillations) shortening after treatment between control and dexamethasone-treated tissues (94.65 ± 2.59% versus 96.32 ± 4.44% of initial shortening, respectively, p = 0.737).
Canine TSM cell culture experiments
Canine TSM cells incubated in 4 μM dexamethasone demonstrated significantly increased MKP-1 expression within 1 h as compared to control cells (Figure 4). Although at 1 h there was no difference in HSP27 phosphorylation between dexamethasone-treated cells and control cells, by 2 h, cells treated with dexamethasone demonstrated a significant decrease in HSP27 phosphorylation, as compared to control cells.
Figure 4. MKP-1 and HSP27 expression in dexamethasone-treated canine TSM cells.
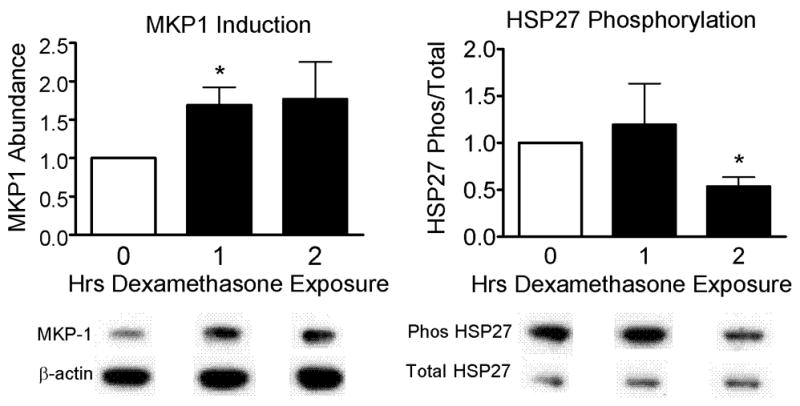
After 1 h of incubation in 4 μM dexamethasone (Dex) TSM cells demonstrated a significant increase in MKP-1 expression (relative to β-actin), as compared to control (Con) cells (1.69 ± 0.23 versus Con = 1.00, p=0.040). Although HSP27 phosphorylation (relative to total HSP27) was unchanged at 1 h incubation, by 2 h there was significantly less HSP27 phosphorylation in cells incubated in dexamethasone compared to control cells (0.54 ± 0.10 versus Con = 1.00, p=0.041). Molecular weights: HSP27, total and phosphorylated ~ 27 kD; MKP-1, β-actin ~ 40 kD. * p<0.05
DISCUSSION
We demonstrated enhancement of force fluctuation-induced relengthening of ACh-contracted tracheal smooth muscle strips by treatment with corticosteroids, a class of drugs that also augments the ability of deep breaths to reverse bronchoconstriction in people with asthma. Application of large force-fluctuations resulted in significant relengthening of isotonically shortened smooth muscle strips, and dexamethasone treatment further enhanced this effect. It therefore seems possible that corticosteroids might help restore the bronchodilatory effect of deep inspiration in asthmatic patients [11–13] in part by enhancing FFIR in their airway smooth muscle.
Dexamethasone might conceivably affect smooth muscle FFIR by a number of mechanisms. Corticosteroids are potent anti-inflammatory agents. Dexamethasone decreases bronchoconstriction in sensitized animals [9, 10] and enhances the bronchodilatory effect of deep inspiration in asthmatic individuals [11–13]. In our experiments, however, it seems less likely that dexamethasone influenced FFIR through an anti-inflammatory action, as the TSM strips used were obtained from non-sensitized, healthy dogs. However, length oscillations applied to bovine smooth muscle strips have been found to induce the expression of IL-6 and IL-8 genes, and this expression is reduced when the frequency of oscillations is reduced [28]. Other studies using cultured cells and mechanical stretch/strain have shown similar results [29, 30]. In cultured human ASM cells, corticosteroids reduce TNF-α-induced IL-6 release from cultured ASM cells by upregulation of MKP-1 [31]. These data raise the possibility that the reduction of cytokine release from ASM itself by corticosteroids may be involved in their enhancement of FFIR.
Dexamethasone has multiple effects on smooth muscle function [32], that include reducing intracellular calcium [33], uncoupling of H1 histamine receptors [34], and reducing muscarinic receptor expression [35]. Together, these effects could act in concert to reduce smooth muscle contractile activation in response to a variety of stimuli. However, in our experiments, dexamethasone treated TSM demonstrated similar isotonic shortening as did control tissues upon acetylcholine exposure and the 4 μM concentration of dexamethasone was chosen because it did not significantly affect isometric force in a separate cohort of TSM strips, suggesting that smooth muscle contractile activation was likely not impaired by dexamethasone. Glucocorticoids also can enhance smooth muscle relaxation by increasing adenylate cyclase activity [36], reducing β2 receptor desensitization [37], increasing the number of β2 receptors [38], and increasing Na+/K+ pump activity [19]. The latter effect, in particular, may be relevant because it occurs in less than 1 h. Corticosteroids can also reduce smooth muscle proliferation [39–41], though one might not expect change in cell number to be relevant within the short time course of our experiments.
Numerous studies have demonstrated that corticosteroids increase the expression of dual-specific phosphatases, especially MKP-1 [21–23], and by doing so decrease p38 MAPK activity, which is dephosphorylated and inactivated by MKP-1 [21]. We have previously demonstrated that when p38 MAPK activity is inhibited pharmacologically with SB203580, force fluctuation-induced smooth muscle relengthening is enhanced [14]. In the experiments presented here, canine TSM cells incubated with dexamethasone demonstrated increased MKP-1 expression within 1 h and decreased HSP27 phosphorylation by 2h. HSP27 is a well-established downstream phosphorylation target of p38 MAPK signaling [42, 43], and thus these data strongly suggest that dexamethasone treatment suppresses p38 MAPK activity. HSP27 is also an actin capping protein that, when phosphorylated, promotes actin polymerization [43–45]. Inhibition of HSP27 phosphorylation could be expected to decrease actin polymerization; we previously demonstrated that inhibition of actin polymerization with latrunculin B increases FFIR [17, 46]. Thus, we propose that dexamethasone may enhance FFIR of contracted tracheal smooth muscle by inducing MKP-1 expression, which in turn reduces p38 MAPK activation and HSP27 phosphorylation.
It is noteworthy that the effect of dexamethasone on the ability of contracted airway smooth muscle to maintain shortening was revealed in our experiments through a loading protocol that simulates physiological conditions. This effect would otherwise have been inapparent, had we measured only isotonic shortening or isometric force. Thus most traditional studies of muscle contractility have not considered the physiologic pathways and mechanisms that are evoked here
In conclusion, we have demonstrated that corticosteroids enhance FFIR in contracted airway smooth muscle and inhibit the p38 MAPK pathway. Others have demonstrated the importance of deep breaths in reversing bronchoconstriction and that this phenomenon is impaired in asthma, but restored by corticosteroid treatment. Our studies suggest that FFIR is a mechanism by which deep inspirations protect against bronchoconstriction and that corticosteroids may restore this effect that is impaired in asthma, through inhibition of the p38 MAPK pathway and augmentation of FFIR. These results suggest that novel therapies that enhance FFIR, perhaps by targeting p38 MAPK, may have a beneficial effect in asthma.
Acknowledgments
NIH Grants HL 79368, AI 56352 and HD 043387
References
- 1.Shen X, Gunst SJ, Tepper RS. Effect of tidal volume and frequency on airway responsiveness in mechanically ventilated rabbits. J Appl Physiol. 1997;83:1202–8. doi: 10.1152/jappl.1997.83.4.1202. [DOI] [PubMed] [Google Scholar]
- 2.Salerno FG, et al. Tidal volume amplitude affects the degree of induced bronchoconstriction in dogs. J Appl Physiol. 1999;87:1674–7. doi: 10.1152/jappl.1999.87.5.1674. [DOI] [PubMed] [Google Scholar]
- 3.Murphy TM, et al. Ontogeny of dry gas hyperpnea-induced bronchoconstriction in guinea pigs. J Appl Physiol. 1994;76:1150–5. doi: 10.1152/jappl.1994.76.3.1150. [DOI] [PubMed] [Google Scholar]
- 4.Freedman S, et al. Abolition of methacholine induced bronchoconstriction by the hyperventilation of exercise or volition. Thorax. 1988;43:631–6. doi: 10.1136/thx.43.8.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown R, Mitzner W. Effects of tidal volume stretch on airway constriction in vivo. J Appl Physiol. 2001;91(5):1995–8. doi: 10.1152/jappl.2001.91.5.1995. [DOI] [PubMed] [Google Scholar]
- 6.Sly PD, et al. Volume dependence of airway and tissue impedances in mice. J Appl Physiol. 2003;94(4):1460–6. doi: 10.1152/japplphysiol.00596.2002. [DOI] [PubMed] [Google Scholar]
- 7.Scichilone N, Permutt S, Togias A. The lack of the bronchoprotective and not the bronchodilatory ability of deep inspiration is associated with airway hyperresponsiveness. Am J Respir Crit Care Med. 2001;163(2):413–9. doi: 10.1164/ajrccm.163.2.2003119. [DOI] [PubMed] [Google Scholar]
- 8.Skloot G, Togias A. Bronchodilation and bronchoprotection by deep inspiration and their relationship to bronchial hyperresponsiveness. Clin Rev Allergy Immunol. 2003;24(1):55–72. doi: 10.1385/CRIAI:24:1:55. [DOI] [PubMed] [Google Scholar]
- 9.Kim J, et al. Prevention and reversal of pulmonary inflammation and airway hyperresponsiveness by dexamethasone treatment in a murine model of asthma induced by house dust. Am J Physiol Lung Cell Mol Physiol. 2004;287(3):L503–9. doi: 10.1152/ajplung.00433.2003. [DOI] [PubMed] [Google Scholar]
- 10.Trifilieff A, El-Hashim A, Bertrand C. Time course of inflammatory and remodeling events in a murine model of asthma: effect of steroid treatment. Am J Physiol Lung Cell Mol Physiol. 2000;279(6):L1120–8. doi: 10.1152/ajplung.2000.279.6.L1120. [DOI] [PubMed] [Google Scholar]
- 11.Slats AM, et al. Improvement in bronchodilation following deep inspiration after a course of high-dose oral prednisone in asthma. Chest. 2006;130(1):58–65. doi: 10.1378/chest.130.1.58. [DOI] [PubMed] [Google Scholar]
- 12.Lim TK, et al. The effects of deep inhalation on maximal expiratory flow during intensive treatment of spontaneous asthmatic episodes. Am Rev Respir Dis. 1989;140(2):340–3. doi: 10.1164/ajrccm/140.2.340. [DOI] [PubMed] [Google Scholar]
- 13.Corsico A, et al. Effects of inhaled steroids on methacholine-induced bronchoconstriction and gas trapping in mild asthma. Eur Respir J. 2000;15(4):687–92. doi: 10.1034/j.1399-3003.2000.15d11.x. [DOI] [PubMed] [Google Scholar]
- 14.Lakser OJ, Lindeman RP, Fredberg JJ. Inhibition of the p38 MAP kinase pathway destabilizes smooth muscle length during physiological loading. Am J Physiol Lung Cell Mol Physiol. 2002;282(5):L1117–21. doi: 10.1152/ajplung.00230.2000. [DOI] [PubMed] [Google Scholar]
- 15.Fredberg JJ, et al. Airway smooth muscle, tidal stretches, and dynamically determined contractile states. Am J Respir Crit Care Med. 1997;156:1752–9. doi: 10.1164/ajrccm.156.6.9611016. [DOI] [PubMed] [Google Scholar]
- 16.Fredberg JJ, et al. Perturbed equilibrium of myosin binding in airway smooth muscle and its implications in bronchospasm. Am J Respir Crit Care Med. 1999;159:959–67. doi: 10.1164/ajrccm.159.3.9804060. [DOI] [PubMed] [Google Scholar]
- 17.Dowell ML, et al. Latrunculin B increases force fluctuation-induced relengthening of ACh-contracted, isotonically shortened canine tracheal smooth muscle. J Appl Physiol. 2005;98(2):489–97. doi: 10.1152/japplphysiol.01378.2003. [DOI] [PubMed] [Google Scholar]
- 18.Hakonarson H, et al. Association between IL-1beta/TNF-alpha-induced glucocortico0id-sensitive changes in multiple gene expression and altered responsiveness in airway smooth muscle. Am J Respir Cell Mol Biol. 2001;25(6):761–71. doi: 10.1165/ajrcmb.25.6.4628. [DOI] [PubMed] [Google Scholar]
- 19.Schramm CM, Grunstein MM. Corticosteroid modulation of Na(+)-K+ pump-mediated relaxation in maturing airway smooth muscle. Br J Pharmacol. 1996;119(5):807–12. doi: 10.1111/j.1476-5381.1996.tb15744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schramm CM. beta-adrenergic relaxation of rabbit tracheal smooth muscle: a receptor deficit that improves with corticosteroid administration. J Pharmacol Exp Ther. 2000;292(1):280–7. [PubMed] [Google Scholar]
- 21.Lasa M, et al. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol Cell Biol. 2002;22(22):7802–11. doi: 10.1128/MCB.22.22.7802-7811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Engelbrecht Y, et al. Glucocorticoids induce rapid up-regulation of mitogen-activated protein kinase phosphatase-1 and dephosphorylation of extracellular signal-regulated kinase and impair proliferation in human and mouse osteoblast cell lines. Endocrinology. 2003;144(2):412–22. doi: 10.1210/en.2002-220769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kassel O, et al. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. Embo J. 2001;20(24):7108–16. doi: 10.1093/emboj/20.24.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hedges JC, et al. A role for p38(MAPK)/HSP27 pathway in smooth muscle cell migration. J Biol Chem. 1999;274(34):24211–9. doi: 10.1074/jbc.274.34.24211. [DOI] [PubMed] [Google Scholar]
- 25.Halayko AJ, et al. Divergent differentiation paths in airway smooth muscle culture: induction of functionally contractile myocytes. Am J Physiol. 1999;276(1 Pt 1):L197–206. doi: 10.1152/ajplung.1999.276.1.L197. [DOI] [PubMed] [Google Scholar]
- 26.Halayko AJ, et al. Markers of airway smooth muscle cell phenotype. Am J Physiol. 1996;270(6 Pt 1):L1040–51. doi: 10.1152/ajplung.1996.270.6.L1040. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell RW, et al. Selective restoration of calcium coupling to muscarinic M(3) receptors in contractile cultured airway myocytes. Am J Physiol Lung Cell Mol Physiol. 2000;278(5):L1091–100. doi: 10.1152/ajplung.2000.278.5.L1091. [DOI] [PubMed] [Google Scholar]
- 28.Kanefsky J, Lenburg M, Hai CM. Cholinergic receptor and cyclic stretch-mediated inflammatory gene expression in intact ASM. Am J Respir Cell Mol Biol. 2006;34(4):417–25. doi: 10.1165/rcmb.2005-0326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hasaneen NA, et al. Cyclic mechanical strain-induced proliferation and migration of human airway smooth muscle cells: role of EMMPRIN and MMPs. FASEB J. 2005;19(11):1507–9. doi: 10.1096/fj.04-3350fje. [DOI] [PubMed] [Google Scholar]
- 30.Kumar A, Knox AJ, Boriek AM. CCAAT/enhancer-binding protein and activator protein-1 transcription factors regulate the expression of interleukin-8 through the mitogen-activated protein kinase pathways in response to mechanical stretch of human airway smooth muscle cells. J Biol Chem. 2003;278(21):18868–76. doi: 10.1074/jbc.M212777200. [DOI] [PubMed] [Google Scholar]
- 31.Quante T, et al. Corticosteroids Reduce IL-6 in ASM Cells Via Upregulation of MKP-1. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2007-0014OC. [DOI] [PubMed] [Google Scholar]
- 32.Hirst SJ, Lee TH. Airway smooth muscle as a target of glucocorticoid action in the treatment of asthma. Am J Respir Crit Care Med. 1998;158(5 Pt 3):S201–6. doi: 10.1164/ajrccm.158.supplement_2.13tac190. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka H, et al. Arachidonic acid metabolites and glucocorticoid regulatory mechanism in cultured porcine tracheal smooth muscle cells. Lung. 1995;173(6):347–61. doi: 10.1007/BF00172142. [DOI] [PubMed] [Google Scholar]
- 34.Hardy E, Farahani M, Hall IP. Regulation of histamine H1 receptor coupling by dexamethasone in human cultured airway smooth muscle. Br J Pharmacol. 1996;118(4):1079–84. doi: 10.1111/j.1476-5381.1996.tb15509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nabishah BM, et al. Effect of steroid hormones on muscarinic receptors of bronchial smooth muscle. Gen Pharmacol. 1991;22(2):389–92. doi: 10.1016/0306-3623(91)90469-m. [DOI] [PubMed] [Google Scholar]
- 36.Michel MC, Knapp J, Ratjen H. Sensitization by dexamethasone of lymphocyte cyclic AMP formation: evidence for increased function of the adenyl cyclase catalyst. Br J Pharmacol. 1994;113(1):240–6. doi: 10.1111/j.1476-5381.1994.tb16200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mak JC, et al. Protective effects of a glucocorticoid on downregulation of pulmonary beta 2-adrenergic receptors in vivo. J Clin Invest. 1995;96(1):99–106. doi: 10.1172/JCI118084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mak JC, Nishikawa M, Barnes PJ. Glucocorticosteroids increase beta 2-adrenergic receptor transcription in human lung. Am J Physiol. 1995;268(1 Pt 1):L41–6. doi: 10.1152/ajplung.1995.268.1.L41. [DOI] [PubMed] [Google Scholar]
- 39.Young PG, Skinner SJ, Black PN. Effects of glucocorticoids and beta-adrenoceptor agonists on the proliferation of airway smooth muscle. Eur J Pharmacol. 1995;273(1–2):137–43. doi: 10.1016/0014-2999(94)00679-2. [DOI] [PubMed] [Google Scholar]
- 40.Burgess JK, et al. Dual ERK and phosphatidylinositol 3-kinase pathways control airway smooth muscle proliferation: differences in asthma. J Cell Physiol. 2008;216(3):673–9. doi: 10.1002/jcp.21450. [DOI] [PubMed] [Google Scholar]
- 41.Roth M, et al. Dysfunctional interaction of C/EBPalpha and the glucocorticoid receptor in asthmatic bronchial smooth-muscle cells. N Engl J Med. 2004;351(6):560–74. doi: 10.1056/NEJMoa021660. [DOI] [PubMed] [Google Scholar]
- 42.Larsen JK, et al. Phosphorylation of the 27-kDa heat shock protein via p38 MAP kinase and MAPKAP kinase in smooth muscle. Am J Physiol. 1997;273(5 Pt 1):L930–40. doi: 10.1152/ajplung.1997.273.5.L930. [DOI] [PubMed] [Google Scholar]
- 43.Wang P, Bitar KN. Rho A regulates sustained smooth muscle contraction through cytoskeletal reorganization of HSP27. Am J Physiol. 1998;275(6 Pt 1):G1454–62. doi: 10.1152/ajpgi.1998.275.6.G1454. [DOI] [PubMed] [Google Scholar]
- 44.Ibitayo AI, et al. HSP27 in signal transduction and association with contractile proteins in smooth muscle cells. Am J Physiol. 1999;277(2 Pt 1):G445–54. doi: 10.1152/ajpgi.1999.277.2.G445. [DOI] [PubMed] [Google Scholar]
- 45.Yamada H, et al. Activation of MAP kinase and translocation with HSP27 in bombesin-induced contraction of rectosigmoid smooth muscle. Am J Physiol. 1995;269(5 Pt 1):G683–91. doi: 10.1152/ajpgi.1995.269.5.G683. [DOI] [PubMed] [Google Scholar]
- 46.Fernandes DJ, et al. Do inflammatory mediators influence the contribution of airway smooth muscle contraction to airway hyperresponsiveness in asthma? J Appl Physiol. 2003;95(2):844–53. doi: 10.1152/japplphysiol.00192.2003. [DOI] [PubMed] [Google Scholar]