

SUMMARY
Deacetylation of nucleosomes by the Rpd3S histone deacetylase along the path of transcribing RNA polymerase II regulates access to DNA, contributing to faithful gene transcription. The association of Rpd3S with chromatin requires its Eaf3 subunit, which binds histone H3 methylated at lysine 36 (H3K36). Eaf3 is also part of NuA4 acetyltransferase that recognizes methylated H3K4. Here we show that Eaf3 in Saccharomyces cerevisiae contains a chromo barrel-related domain that binds methylated peptides, including H3K36 and H3K4, with low specificity and millimolar-range affinity. Nuclear magnetic resonance structure determination of Eaf3 bound to methylated H3K36 was accomplished by engineering a linked Eaf3-H3K36 molecule with a chemically incorporated methyllysine analog. Our study uncovers the molecular details of Eaf3-methylated H3K36 complex formation, and suggests that, in the cell, Eaf3 can only function within a framework of combinatorial interactions. This work also provides a general method for structure determination of low-affinity protein complexes implicated in methyllysine recognition.
INTRODUCTION
It is now widely known that nucleosomes are actively involved in the regulation of many important cellular processes, including DNA transcription, replication, repair, and cell cycle progression. These functions are mediated in part by posttranslational modifications, such as acetylation, methylation, phosphorylation, ADP ribosylation, ubiquitination, glycosylation, and sumoylation of the core histones (Kouzarides, 2007). Histone acetylation is one of the most frequent modifications. It involves the transfer of an acetyl moiety from acetyl coenzyme A onto the ζ-amino group of a lysine. The reaction is catalyzed by a family of enzymes called histone acetyltransferases (HATs). Acetylation is chemically stable, and can be reversed by another family of enzymes called histone deacetylases (HDACs).
Reduced potassium dependency-3 (Rpd3S) is one of the HDACs in Saccharomyces cerevisiae. It is a 0.6 MDa multisubunit protein assembly that deacetylates histones at 3′ coding sequences in response to histone methylation by the SET domain-containing enzyme Set2 (Carrozza et al., 2005; Joshi and Struhl, 2005; Keogh et al., 2005; Reid et al., 2004). Set2 associates with the phosphorylated C-terminal domain of elongating RNA polymerase II and dimethylates histone H3 at lysine 36 (H3K36) on transcribed genes (Kizer et al., 2005; Krogan et al., 2003; Li et al., 2003; Schaft et al., 2003; Xiao et al., 2003). Rpd3S, through its Esa1-associated factor 3 (Eaf3) subunit, recognizes and binds directly to methylated H3K36 (Carrozza et al., 2005; Joshi and Struhl, 2005; Keogh et al., 2005). The deacetylation by Rpd3S ultimately suppresses spurious transcription by RNA polymerase II.
It was shown that the N-terminal region of Eaf3 (Eaf3-NT) binds histone H3 mono-, di-, and trimethylated at lysine 36 (designated as H3K36me, H3K36me2, and H3K36me3, respectively), as well as histone H3 peptides trimethylated at lysine 4 (H3K4me3) immobilized on streptavidin sepharose or agarose beads (Carrozza et al., 2005; Joshi and Struhl, 2005). In these studies, Eaf3-NT was referred to as a chromodomain, although other reports have suggested that it is a chromo barrel domain based on sequence homology with chromo barrel-containing proteins, such as the fly males absent on the first (MOF) HAT and the human MRG15 HDAC (Nielsen et al., 2005; Zhang et al., 2006).
In the context of nucleosomes, it was demonstrated that both Eaf3-NT and the plant homeodomain (PHD) of the transcriptional regulatory protein Rco1 (Rco1-PHD), also a subunit of Rpd3S, are required for the recognition of methylated H3K36 (Li et al., 2007a). In the Rpd3S multiprotein complex, it was shown that Eaf3-NT provides the specificity for H3K36-methylated nucleosomes, while the PHD module of Rco1 enhances the overall affinity for these nucleosomes (Li et al., 2007a). Deletion of Eaf3-NT substantially reduces the affinity of Rpd3S for methylated nucleosomes and impairs the ability of Rpd3S to discriminate between methylated and unmethylated nucleosomes, while deletion of the PHD domain completely abrogates the binding of Rpd3S to nucleosomes. The deletion of either Eaf3-NT or Rco1-PHD domain results in global acetylation and generation of aberrant, internally initiated transcripts, strongly supporting the idea that the coupled actions of Eaf3-NT and the Rco1-PHD domain are essential in regulating acetylation/deacetylation in histones and suppression of spurious transcripts (Li et al., 2007a). It is thought that binding of Rco1-PHD to nucleosomes helps anchor Rpd3S in a configuration that allows Eaf3-NT to recognize methylated H3K36 (Li et al., 2007a). The molecular mechanisms of these interactions remain unknown.
Eaf3 is also a component of the budding yeast HAT-nucleosomal H2A/H4 (NuA4) complex (Eisen et al., 2001). NuA4 is responsible for histone H4 acetylation at lysines 5, 8, and 12 (Allard et al., 1999; Clarke et al., 1999; Loewith et al., 2000; Reid et al., 2000), and may be recruited to nucleosomes by interaction with methylated H3K4 (Ruthenburg et al., 2007). In the context of the NuA4 enzyme, where Eaf3-NT is paired with the PHD domain of Yng2 (Yng2-PHD), Eaf3 does not support stable binding of NuA4 to nucleosomes with histone H3 methylated at Lys36 (Li et al., 2007a). However, substitution of Yng2-PHD with Rco1-PHD increases the affinity of NuA4 for H3K36-methylated nucleosomes, suggesting that the specific combination of Eaf3-NT and Rco1-PHD, in the context of either Rpd3S or NuA4, directs robust binding to nucleosomes methylated at histone H3K36 (Li et al., 2007a).
In this study, we investigate the molecular mechanism of Eaf3 interaction with methylated histone H3K36 by determining the three-dimensional (3D) solution structures of the N-terminal fragment of Eaf3, free and in complex with a dimethylated histone H3K36 analog. Through a combination of molecular biology techniques and chemical modification, we created the complex by integrating the sequences of Eaf3 (1–115 aa), a 4 aa linker, and the 28–42 aa fragment of histone H3 with a dimethylated lysine analog (KC) at position 36 into a single protein molecule that we refer to as Eaf3-H3KC36me2. Inside the cell, Eaf3 interacts tightly with methylated H3K36 in the presence of other subunits of Rpd3S. In isolation, we found that Eaf3 (1–113 aa) binds a methylated H3K36 peptide (31–42 aa), as well as a methylated H3K4 peptide (1–10 aa), but with millimolar-range affinity, making it virtually impossible to carry out structural studies on these complexes. With the fused Eaf3-H3KC36me2 protein, we were able to bring forth a tight interaction between Eaf3 and H3KC36me2, allowing us to perform a structural characterization of the fused complex.
Through the use of nuclear magnetic resonance (NMR) spectroscopy, we show that Eaf3, free and bound to H3KC36me2, adopts a chromo barrel-related fold. The methyllysine binding site is a cage formed by four aromatic residues of Eaf3. Mutating any of these residues either destabilizes Eaf3 or abolishes its interaction with H3KC36me2. Only 4 amino acids from the linked H3KC36me2 sequence (KC36me2, H3V35, H3K37, and H3P38) contact Eaf3, as indicated from identified nuclear Overhauser effect signals (NOEs), likely explaining the low affinity of Eaf3 for a methylated histone H3K36 peptide in vitro and the requirement of Rco1-PHD domain in enhancing the avidity of Eaf3 for methylated nucleosomes in the context of the Rpd3S complex. We show that the isolated chromo barrel domain of Eaf3 has low specificity, supporting the idea that it is its combined action with other domains in multiprotein complexes that determines not only tight binding, but also the specificity of interaction.
RESULTS
Structure of Eaf3 in the Free State
As a step toward understanding the interaction of Eaf3 with methylated histone targets, we first determined the solution structure of the N-terminal 1–113 aa of budding yeast Eaf3 by multidimensional NMR spectroscopy. The final 20 solution structures with the lowest energy of free Eaf3 are shown in Figure 1A. The conformers have an average pairwise root-mean-square deviation (rmsd) of 0.54 Å for the well-defined backbone regions, and do not have distance violations greater than 0.5 Å or angle violations greater than 5°. Table 1 summarizes the statistical analysis.
Figure 1. Solution NMR Structures of Eaf3 in the Free State and Linked to Histone H3KC36me2.

(A) Left: Stereo view of the 20 lowest-energy structures of Eaf3 chromo barrel domain (1–113 aa) showing only backbone N, Cα, and C′ after superposition of residues 8–41 and 55–111. The rmsd is 0.54 and 1.16 Å for the backbone atoms N, Cα, and C′ and for all heavy atoms of residues 8–41 and 55–111, respectively. Right: Ribbon representation of the lowest-energy structure of Eaf3 chromo barrel domain. The helices (α) and β strands (β) are colored green and blue, respectively. N and C termini are also indicated.
(B) Left: Stereo view of the 20 lowest-energy structures of Eaf3-H3KC36me2 (1–134 aa) showing only backbone N, Cα, and C′ after superposition of residues 8–41, 55–111, and 127–130. The Eaf3 chromo barrel domain (1–115 aa) is colored black, while the linker (116–119 aa) and H3KC36me2 (120–134 aa) are shaded blue and orange, respectively. The rmsd is 0.61 and 1.13 Å for the backbone atoms N, Cα, and C′ and for all heavy atoms of residues 8–41, 55–111, and 127–130, respectively. Right: Cartoon representation of the lowest-energy structure of Eaf3-H3KC36me2; ribbon for Eaf3 and stick for a portion of H3KC36me2. The helices (α) and β strands (β) are indicated. Color coding is the same as in (A).
Table 1.
NMR and Structure Refinement Statistics for Eaf3 and Eaf3-H3KC36me2
| Eaf3 | Eaf3-H3KC36me2 | |
|---|---|---|
| NMR distance and dihedral constraints |
||
| Distance constraints | ||
| Total NOEs | 2062 | 2295 |
| Intraresidue | 353 | 355 |
| Interresidue | 1709 | 1940 |
| Sequential (|i − j| = 1) | 568 | 620 |
| Medium range (1 < |i − j| ≤ 4) | 362 | 412 |
| Long range (|i − j| > 5) | 779 | 908 |
| Hydrogen bonds | 27 | 27 |
| Dihedral angle restraints | ||
| ϕ | 58 | 58 |
| ψ | 58 | 58 |
| NOEs between protein and linked peptide | NA | 87 |
| Structure statistics | ||
| Violations (mean ± SD) | ||
| Number of distance constraints (>0.3 Å) | 1.2 ± 0.6 | 1.4 ± 0.4 |
| Number of dihedral angle constraints (>3°) | 1.6 ± 0.6 | 1.8 ± 0.7 |
| Max. dihedral angle violation (°) | 2.8 ± 0.8 | 3.2 ± 1.0 |
| Max. distance constraint violation (Å) | 0.36 ± 0.03 | 0.38 ± 0.06 |
| Deviations from idealized geometry | ||
| Bond (Å) | 0.0079 | 0.0078 |
| Angle (°) | 2.18 | 1.95 |
| Impropers (°) | 2.5 | 2.2 |
| Average pairwise rmsda (Å) | ||
| Heavy | 1.16 ± 0.13 | 1.13 ± 0.12 |
| Backbone | 0.54 ± 0.10 | 0.61 ± 0.13 |
| Ramachandran space (%) | ||
| Most favored regions | 78.7 ± 2.8 | 80.7 ± 2.5 |
| Additionally allowed | 19.2 ± 2.2 | 17.4 ± 2.4 |
| Generously allowed | 1.0 ± 1.5 | 1.3 ± 1.2 |
| Disallowed | 1.1 ± 1.0 | 0.6 ± 0.5 |
Abbreviations: Eaf3, Esa1-associated factor 3; H3KC36, H3 with a dimethylated lysine analog (KC) at position 36; NA, not applicable; NOE, nuclear Overhauser effect; NMR, nuclear magnetic resonance; rmsd = root-mean-square deviation. The mean AMBER energy of the 20 best-refined structures was −6073 ± 18 kcal mol−1 for free Eaf3 and −6846 ± 25 kcal mol−1 for the Eaf3-H3KC36me2-linked complex.
Pairwise rmsd was calculated from 20 structures aligned on well-defined secondary structure elements: residues 8–41 and 55–111 for Eaf3, and residues 8–41, 55–111, and 127–130 for Eaf3-H3KC36me2.
The NMR structures reveal a chromo barrel-like fold with a 38 residue insertion. Eaf3 consists of seven antiparallel β strands: β1 (14–17 aa), β2 (22–31 aa), β3 (38–40 aa), β4 (57–59 aa), β5 (77–81 aa), β6 (89–92 aa), and β7 (97–99 aa); and 2 α helices: α1 (69–72 aa) and α2 (102–112 aa), as shown in Figure 1A. Strands β1, β2, and β5–β7 form the barrel. The long helix at the C terminus (α2) folds along one face (β1, β2, β7) of the barrel. The short α helix (α1) makes contacts with Trp31 in strand β2 and nearby residue Pro33. Helix α1, strands β3 and β4, and a long loop (L3) connecting these strands are part of the 38 aa insertion. L3 seems to contribute to the tertiary fold of the protein. Based on the observed NOEs, residues Gln51 and Thr53–Ile56 of L3 and β4 interact with several residues from β2 (Leu28 and Lys29) and β3 (Tyr38–Ser40) strands, as well as Ile41 and Pro42 in the vicinity of these strands. Nevertheless, L3 is poorly defined in the ensemble of NMR structures due to an insufficient number of constraints. NMR relaxation measurements demonstrate flexibility of L3 in the form of fast motions of backbone 15N-1H vectors on the picosecond to nanosecond time scale (data not shown).
The fragment 64–68, within the inserted segment, adopts an extended conformation stabilized by hydrophobic contacts among Ile67, Trp31, and Trp91. The methyl resonances of Ile67 are markedly upfield shifted with chemical shift values of −1.84 ppm for HD1* and −1.73 ppm for HG2*, likely due to the ring current influence of Trp31 and Trp91.
Several networks of hydrophobic interactions exist in the Eaf3 chromo barrel structure. One network involves Trp31, Leu61, Ile67, Ile71, and Ile72 residues (see Figure S1 available online). The significant upfield shifts of Ile67 HD1* and Ile72 HD1*, likely due to the ring current from the side chains of Trp31 and Trp91, are evidence for the tight packing interactions. A hydrogen bond between Trp31 HE1 and the carbonyl group of Ile72 can be identified from the ensemble of structures (Figure S1), which may strengthen the hydrophobic packing and stabilize helix α1. Another network is formed by the C-terminal helix α2 and the N-terminal β strands 1 and 2. More specifically, Phe17, Leu21, Met22, and Leu111 residues are involved in hydrophobic interactions while conserved residues Glu24 and Lys108 contribute a long-range salt bridge.
Based on 1H/2H exchange experiments, we found that, while most of the slowly exchanging amide protons are from residues in the regular secondary structure elements β1, β2, β3, β6, β7, and α2, none comes from α1, β4, and β5. This is consistent with the structure of Eaf3, where α1 is exposed to the solvent (Figure 1A), and with NMR relaxation measurements, where β4 exhibits greater flexibility on the picosecond to nanosecond time scale than the other β strands (data not shown). The less rigid structure of β5, located near the methylated histone binding site, may be needed for methylated peptide recognition.
A search in the Protein Data Bank (PDB) for structural analogs of Eaf3 with the program Dali (Holm and Sander, 1996) returned the human protein MRG15 (PDB accession code 2F5K) (Zhang et al., 2006) as the closest match, with an rmsd of 2.3 Å for 71 superimposed backbone residues out of 113 residues in Eaf3. MRG15 is thought to be the human homolog of Eaf3, and has the canonical chromo barrel domain (Figure 2A) originally identified in the fly protein MOF (Nielsen et al., 2005). Despite close similarities among Eaf3, MRG15, and MOF, we also note major differences, including more secondary structure elements, such as β3, β4, and α1, and the presence of the long loop, L3, between β3 and β4 in Eaf3 (Figure 2B). The helix α2 in Eaf3 is also shorter than the corresponding helix α1 of MRG15 (Figure 2B). We found that extending Eaf3 by 13 residues—Eaf3 (1–126 aa)—led to its apparent homodimerization, as suggested by a change in gel filtration chromatography retention time (data not shown). Noticeably, residues 108–123 have a high probability to form a coiled-coil motif (Lupas et al., 1991), and so a longer helix α2 could be the basis for the possible dimerization of Eaf3.
Figure 2. Comparison of Eaf3 and MRG15 Chromo Barrel Domains.
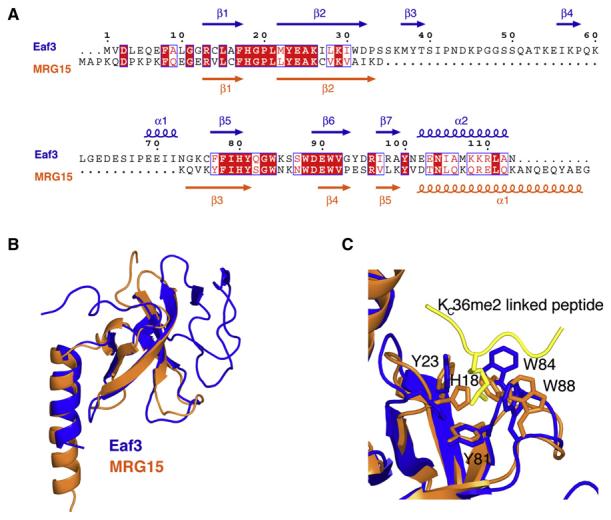
(A) Alignment of Eaf3 and MRG15 amino acids based on their 3D structures. The secondary structure elements are indicated. The alignment was created with ESPript (http://espript.ibcp.fr/ESPript/ESPript/).
(B) Superposition of the structures of Eaf3 and MRG15 (PDB accession code 2F5K) chromo barrel domains.
(C) Expanded view of the overlaid aromatic methyllysine binding cages of Eaf3-H3KC36me2 and MRG15 (PDB accession code 2F5K). H3KC36me2 from the Eaf3-H3KC36me2-linked complex is shown in yellow.
Eaf3 Interactions with H3K4me3 and H3K36me3 Peptides Probed by NMR Chemical Shift Perturbations
Previous functional studies have demonstrated that Eaf3 binds methylated H3K36 and H3K4 peptides, with apparent highest affinities for the trimethylated forms. To investigate the interactions of Eaf3 with both H3K36me3 (ATGGVK36me3KPHRYR) and H3K4me3 (ARTK4me3QTARKS) peptides, we first performed a titration experiment in which we added increasing amounts of nonlabeled H3K4me3 peptide to an 15N-labeled sample of Eaf3, and collected 15N-1H heteronuclear single quantum coherence (HSQC) NMR spectra of Eaf3 before and after each addition of H3K4me3. From Figure 3A, several peaks in the HSQC spectra of Eaf3 shifted, indicating that the exchange between free and H3K4me3-bound Eaf3 is fast on the NMR chemical shifts time scale. Only a limited number of signals were affected, consistent with a small or localized binding interface between Eaf3 and the trimethylated H3K4 peptide. The peaks that shifted most strongly correspond to the protein backbone amide hydrogen and nitrogen atoms of Ala16, Phe17, His18, Leu21, Tyr23, Ala25, Tyr81, Gln82, and Trp88, and side-chain amide atoms (HE1 and NE1) of Trp84 and Trp88. Through the use of a subset of these peaks in a curve-fitting algorithm, we estimated the dissociation constants (KD) of the Eaf3-H3K4me3 interaction to be 1–3 mM (Figure 3D). Considering all other perturbed peaks, mapping is fully consistent with the structure of Eaf3-H3KC36me2 complex (presented below), wherein the methylated lysine analog inserts into an aromatic cage in Eaf3.
Figure 3. Interactions of Methylated Histone H3 with Eaf3 Chromo Barrel Domain.

(A) 15N-1H HSQC titration spectra of 15N-labeled Eaf3 (1–113 aa), free (black) and upon addition of increasing amounts of nonlabeled H3K4me3 peptide (1–10 aa). In this and other panels, the side-chain amide atom signals of Trp84 and Trp88 are labeled W84sc and W88sc, respectively.
(B) 15N-1H HSQC titration spectra of 15N-labeled Eaf3 (1–113 aa), free (black) and upon addition of increasing amounts of nonlabeled H3K36me3 peptide (31–42 aa).
(C) Overlaid 15N-1H HSQC spectra of 15N-labeled Eaf3-H3C36 (1–134 aa) before (black) and after chemical modification Cys128 or H3C36 into a dimethyllysine analog (red). Representative residues of Eaf3, which exhibited strongest perturbations (indicated by arrows) upon addition of the peptides or after dimethylation reaction are labeled in (A), (B), and (C) spectra.
(D) Estimates of KD of Eaf3 for H3K4me3 peptide from the change in chemical shifts of selected 1H-15N Eaf3 resonances upon addition of nonlabeled H3K4me3 (1–10 aa).
(E) Estimates of KD of Eaf3 for H3K36me3 peptide from the change in chemical shifts of selected 1H-15N Eaf3 resonances upon addition of nonlabeled H3K36me3 (31–42 aa).
(F) Portion of the overlaid 15N-1H HSQC spectra of 15N-labeled Eaf3-H3C36 (1–134 aa) before (black) and after alkylation of Cys128 or H3C36 into a dimethyllysine analog (red). Ten resonance signals corresponding to residues of the linked H3KC36me2 sequence are labeled.
Next, a titration experiment was performed with a nonlabeled H3K36me3 peptide and 15N-labeled Eaf3. As shown in Figure 3B, Eaf3 peaks perturbed by the H3K36me3 peptide were the same as those affected by the H3K4me3 peptide, with the additional perturbed residues, Met22, Trp84, and Gly93. By using the backbone amide atoms of Ala25 and Tyr81, and side-chain amide atoms of Trp84 and Trp88, in the same curve-fitting algorithm for KD determination, we obtained a KD range of 1.8–3.4 mM (Figure 3E), comparable to that determined for the Eaf3-H3K4me3 complex. We also titrated 15N-labeled Eaf3 with a version of the H3K36me3 peptide in which trimethyllysine 36 was replaced by a trimethyllysine analog (H3KC36me3: ATGGVKC36me3KPHRYR), produced by cysteine alkylation (Simon et al., 2007) of a chemically synthesized H3C36 peptide. With a KD of 5–7 mM (Figure S2), the affinity of H3KC36me3 for Eaf3 is only slightly lower than that of the H3K36me3 peptide. This result is consistent with the study by Simon et al. (2007), whose antibody analysis suggested that, in some cases, the recognition of chemically alkylated peptides might be slightly weaker than that of peptides with a natural methyllysine.
With such low-affinity interaction of Eaf3 with methylated H3K4 and H3K36 peptides, further structural characterization of these complexes was virtually impossible. This prompted us to work with fused versions of the complexes, taking advantage of the availability of cysteine alkylation procedures for the incorporation of methyllysine analogs into proteins (Simon et al., 2007).
Formation and Structure of Eaf3-H3KC36me2 Complex
We engineered a fused complex of Eaf3 with methylated H3K36 by covalently tethering the Eaf3 protein and H3 peptide sequences via a short linker and chemically incorporating a methyllysine analog at the site of interest. We were not able to trimethylate H3K36 in the linked Eaf3-H3 protein construct, as the reaction entailed an incubation temperature of at least 42°C, which led to precipitation of the protein. Since Eaf3 also binds dimethylated H3K36, thought to be the in vivo target of Eaf3 (Carrozza et al., 2005; Joshi and Struhl, 2005; Keogh et al., 2005), and since the conditions for dimethylation are milder and nondestructive to the protein (Simon et al., 2007), we prepared a fused version of the Eaf3-H3K36me2 complex.
A complex of Eaf3 chromo barrel domain and histone H3 dimethylated at lysine 36 was created by a combination of steps. These include extending the C-terminal end of Eaf3 with a short linker, followed by an H3K36 sequence; incorporating the appropriate mutations (i.e., C14V and C76S in Eaf3, and K36C in H3K36), and carrying out the alkylation of cysteine 36 of the Eaf3-linked K36C-mutated H3 into a dimethyllysine analog (Figure 4A). Previously, it was shown that methyllysine analogs (mono-, di-, and trimethylated) are functionally similar to their natural methylated lysine counterparts (Simon et al., 2007). In order to proceed with the reaction, the cysteines of Eaf3 had to be mutated first. The buried Cys14 was changed to a valine, mimicking the equivalent position in MRG15. Cys76 was replaced by a serine. 15N-1H HSQC spectra were recorded for the wild-type Eaf3 and double-mutant Eaf3-C14V/C76S proteins, as well as for Eaf3-H3K36 and Eaf3-H3C36 fused proteins (data not shown). The positions of the peaks did not change appreciably, except those of the mutated residues (Cys14 and Cys76 of Eaf3) and a few residues nearby the mutations, indicating that the overall structure of Eaf3 chromo barrel domain was practically unaffected by the mutations. 15N-1H HSQC spectra were also acquired before and after the dimethylation reaction (i.e., on Eaf3-H3C36 and Eaf3-H3KC36me2 samples, respectively) (Figure 3C). Only a few peaks of Eaf3 shifted, but strongly, indicating a tight interaction brought about by methylation and a limited binding interface between Eaf3 and linked H3KC36me2. The perturbed Eaf3 residues include Ala16, Phe17, His18, Gly19, Leu21, Tyr23, Ala25, Tyr81, Trp84, Trp88, and Glu90, which, as described below, constitute or surround the methyllysine binding cage of Eaf3. While several 1H-15N backbone resonances of the linked peptide could not be detected before methylation, most of them appeared after methylation of the fused complex, as a result of the Eaf3 and linked H3KC36me2 interaction (Figure 3F).
Figure 4. Details of Eaf3 Chromo Barrel Domain Interaction with the Linked Histone H3KC36me2 Sequence.
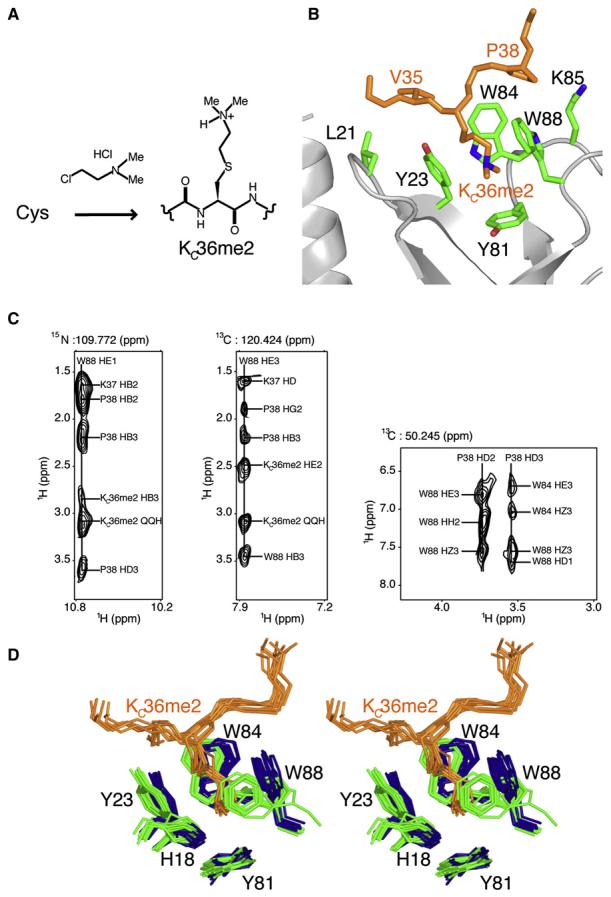
(A) Chemical conversion of a cysteine residue to a dimethyllysine analog (KCme2).
(B) Close-up view of the main interaction site within the Eaf3-H3KC36me2 protein. Tyr23, Tyr81, Trp84, and Trp88 of the chromo barrel domain of Eaf3 form an aromatic cage that accommodates the linked dimethylated lysine analog of H3K36. Other residues (Leu21 and Lys85 of Eaf3; and V35 and Pro38 of linked H3KC36me2) involved in the interaction are also labeled.
(C) Planes from the 3D 15N nuclear Overhauser effect spectroscopy (NOESY) experiment showing NOE correlations of W88HE1 of Eaf3 to KC36me2, Lys37, and Pro38 of linked H3KC36me2 (left); and 13C-edited NOESY experiments showing NOE correlations of W88HE3 of Eaf3 to KC36me2, Lys37, and Pro38 of linked H3KC36me2 (middle), and NOE correlations of the HD protons of P38 of linked H3KC36me2 to the aromatic protons of Trp84 and Trp88 of Eaf3 (right).
(D) Stereo view of the superposition of 10 NMR structures each of free Eaf3 (blue) and Eaf3-H3KC36me2 complex (Eaf3 in green and H3KC36me2 in orange) showing a close-up representation of the aromatic pocket binding site.
To assess if the linker region had any effect on the Eaf3-H3KC36me2 interaction, we also acquired a series of 15N-1H HSQC spectra on an isolated 15N-labeled Eaf3 chromo barrel domain as it was being titrated with nonlabeled H3K36me3 and H3KC36me3 peptides, as described above. Superposition of the titration spectra in Figure 3B and Figure S2 shows that addition of H3K36me3 and H3KC36me3 peptides perturbed similar Eaf3 residues as those affected by dimethylating Eaf3-H3C36 (Figure 3C). Importantly, the directions of the chemical shift changes induced by chemical methylation and titration were similar, suggesting that the linker does not interfere with the interaction of Eaf3 and H3KC36me2, and that the histone sequence is positioned in the correct orientation relative to Eaf3 in the fused Eaf3-H3KC36me2 complex. We noted, however, that the magnitudes of perturbations with the fused Eaf3-H3KC36me2 were much stronger than when Eaf3 protein and excess H3K36me3 or H3KC36me3 peptides were combined (Figure 3C versus Figure 3B; Figure S2), demonstrating a much tighter interaction in the former. Thus, with the linkage, we were able to form a high-affinity 1:1 Eaf3:H3KC36me2 complex, highly amenable to structural studies and that may mimic the Eaf3-H3K36me2 interaction in the Rpd3S complex, despite the absence of other Rpd3S components.
We determined the 3D solution structure of the fused Eaf3-H3KC36me2 complex (Figure 1B) and found that the overall fold of bound Eaf3 is similar to that of Eaf3 in the free state (Figure 1A). In Eaf3-H3KC36me2, the linker is highly disordered and the fused H3KC36me2 sequence has an extended conformation and exhibits backbone flexibility on the picosecond to nanosecond time scale, as determined by NMR relaxation measurements (data not shown). H3KC36me2 is accommodated into an aromatic pocket of Eaf3 formed by the aromatic residues Tyr23, Tyr81, Trp84, and Trp88 (Figure 4B). The planes containing the rings of Tyr23, Tyr81, Trp84, and Trp88 are oriented perpendicular to each other, roughly forming four sides of a cube. In Eaf3-H3KC36me2, the side chains of Trp84 and Trp88 occupy markedly different orientations compared with Eaf3 in the free state, as shown in Figure 4D. In the free protein, the aromatic ring current from Tyr23 causes the HE1 resonance of Trp84 (9.2 ppm) to shift upfield. In Eaf3-H3KC36me2, Trp84 HE1 is shifted to 10.4 ppm, which is consistent with the change in orientation observed in the structure of the complex. Similar observation applies to the Trp88 HE1 resonance that shifts from 9.8 ppm in Eaf3 to 10.7 ppm in Eaf3-H3KC36me2. The large change in chemical shift observed for Gly19 upon complex formation suggests a change in conformation or dynamics of Gly19 and/or adjacent His18, both in the close vicinity of the binding cage (Figure 4D).
The dimethylated lysine analog interacts with all four aromatic residues of Eaf3 (Figure 4B). The side chain of KC36me2 has five NOEs with the aromatic ring of Tyr23, five NOEs with the aromatic ring of Tyr81, 13 NOEs with the aromatic ring of Trp84, and 11 NOEs with the aromatic ring of Trp88. Some of these NOEs are shown in Figure 4C. Other residues nearby the dimethylated lysine analog contribute to the binding affinity of Eaf3-H3KC36me2. The networks of hydrophobic interactions, one encompassing Val127 (or H3V35), and Leu21, Tyr23, and Trp84 of Eaf3; another, Lys129 (or H3K37), and Trp84 and Trp88 of Eaf3; and another, Pro130 (or H3P38), and Trp84, Lys85, and Trp88 of Eaf3, can be seen to help in stabilizing the complex. There is one unambiguous NOE between the side-chain protons of Val127 (or H3V35) and Leu21; five between the side-chain protons of Val127 (or H3V35) and aromatic ring of Tyr23; four between the side-chain protons of Val127 (or H3V35) and the aromatic ring of Trp84; six between the side-chain protons of Lys129 (or H3K37) and the aromatic ring of Trp84; two between the side-chain protons of Pro130 (or H3P38) and Lys85; one between the side-chain protons of Lys129 (or H3K37) and the aromatic ring of Trp88; three between the side-chain protons of Pro130 (or H3P38) and the aromatic ring of Trp84; and 10 between the side-chain protons of Pro130 (or H3P38) and the aromatic ring of Trp88. Some of these NOEs are depicted in Figure 4C.
Mutation of the Aromatic Cage Residues of Eaf3
In order to evaluate the individual contribution of the Eaf3 residues constituting the aromatic cage for interaction with dimethylated histone H3KC36, we made the following single-point mutations in Eaf3-H3C36: Y23A, Y81A, W84A, and W88A. The Y23A and Y81A mutants expressed very poorly in Escherichia coli cells. The purified proteins were unstable and highly prone to precipitation. Nonetheless, 15N-1H HSQC spectra were collected on very dilute samples of these two mutants. When the spectra of these mutants were overlaid with the 15N-1H HSQC spectrum of the Eaf3-H3C36 protein, we observed little correlation, as most of the peaks had changed positions and intensities. The mismatch means that the structures of the Y23A and Y81A mutants were altered with respect to that of Eaf3-H3C36, indicating that the aromatic residues at positions 23 and 81 are not only important for the binding of methylated H3K36, but also for the integrity of the structure. Due to protein instability, dimethylation was not performed on Y23A and Y81A mutant proteins.
The W84A and W88A mutants were well expressed in bacteria. However, the W84A mutant was prone to precipitation, indicating some contribution of Trp84 to protein stability. 15N-1H HSQC spectra were collected on the W84A and W88A mutants before and after dimethylation of fused H3C36. Spectral overlays are shown in Figure S3. Very little spectral change was observed upon methylation, indicating that the strong Eaf3 and H3KC36me2 interaction achieved with the fused Eaf3-H3KC36me2 molecule was lost when Trp84 and Trp88 were mutated. These experiments demonstrate that Trp84 and Trp88 are essential for Eaf3 binding to linked H3KC36me2.
DISCUSSION
Inside the cell, the association of Eaf3 with histone H3 methylated at Lys36 occurs in the presence of multiple proteins or other components of Rpd3S deacetylase. In order to decipher the mechanism of Eaf3 and methylated H3K36 histone interaction, we first simplified the multiprotein system by focusing only on the binding of the Eaf3-NT and an H3K36me3 peptide. We found this interaction to be of very low affinity (KD in the millimolar range), which is consistent with the observation that robust recruitment of Rpd3S to nucleosomes requires multiple subunits of Rpd3S (Li et al., 2007a). By linking the Eaf3 N-terminal region and an analog of the H3K36me2 fragment (H3KC36me2) into a single protein molecule and incorporating a number of mutations, we succeeded in forming a much tighter complex, more amenable to structural studies, allowing us to elucidate the molecular details of Eaf3 and methylated H3K36 interaction. We have confirmed that the linker and the modifications made to produce Eaf3-H3KC36me2 do not interfere with the Eaf3 and H3KC36me2 interaction, but rather help bring the two regions in poised positions for a more favorable methyllysine-driven interaction.
We have determined the solution structures of Eaf3, free and linked to H3KC36me2. Eaf3 folds into a chromo barrel-like domain markedly different from the canonical chromo barrel motif first identified in the fly MOF acetyltransferase (Nielsen et al., 2005), with Eaf3 having more secondary structural elements. An aromatic cage is present in Eaf3 and is formed by four residues, namely: Tyr23, Tyr81, Trp84, and Trp88. Eaf3 uses this pocket to bind H3K36me3 and H3K4me3 peptides. The same cage accommodates the dimethyllysine analog in the Eaf3-H3KC36me2 complex.
From our mutagenesis studies, we demonstrated that Tyr23 and Tyr81 are not only important for methyllysine recognition, but also for the overall stability of the chromo barrel motif. Trp84 and Trp88, on the other hand, are essentially involved in directly binding the dimethylated lysine analog, and do not seem to contribute appreciably to protein stability. Formation of the complex requires reorientation of the planes of these two aromatic residues. Our results are in agreement with previous studies showing that mutations to an alanine, single or combined, at positions 81 and 84 in Eaf3, totally abrogated the ability of Rpd3S to bind H3K36-methylated nucleosomes (Keogh et al., 2005).
In addition to Tyr23, Tyr81, Trp84, and Trp88 that directly contact the dimethyllysine analog (KC36me2) of histone H3, there are residues outside the aromatic cage of Eaf3 and other residues of H3 that are important for the interaction of Eaf3 with H3KC36me2. Val127 (or H3V35), Lys129 (or H3K37), and Pro130 (or H3P38), residues at positions (i − 1), (i + 1), and (i + 2) with respect to H3KC36me2 at position i, also contribute to the formation and stability of the complex within Eaf3-H3KC36me2. From observable NOEs, it is clear that hydrophobic interactions exist between Val127 (or H3V35) and Leu21, Tyr23, and Trp84; between Lys129 (or H3K37) and Trp84 and Trp88; and between Pro130 (or H3P38) and Lys84, Lys85, and Trp88. From the Eaf3-H3KC36me2 structure, the aromatic ring of Trp88 is sandwiched between the dimethylated lysine analog and Pro130 (or H3P38).
Based on these structural studies, we have acquired new understanding in order to explain why the putative human homolog of Eaf3, MRG15, can bind methylated H3K36, despite significant differences in the structures of Eaf3 and MRG15 (Figure 2B). Although MRG15 is missing a 38 residue segment that corresponds to a region containing two β strands (β3 and β4) and a long loop (L3) in Eaf3 (Figure 2A), MRG15 has conserved Tyr26, Tyr46, Trp49, and Trp53, corresponding to the aromatic cage residues (Tyr23, Tyr81, Trp84, and Trp88, respectively) of Eaf3 (Figure 2C) (Zhang et al., 2006). Additionally, His18 and Leu21 in Eaf3, which are in the vicinity of methylated H3K36, are also conserved in MRG15. We note that the side chain of MRG15 His21 (corresponding to His18 in Eaf3) partially occludes the aromatic cage in the crystal structure of MRG15 chromo barrel domain that was determined in the absence of bound peptide (Figure 2C). Therefore, for the methyllysine to be accommodated into the binding cage of MRG15, His21 has to move significantly. Such apparent conformational flexibility of the histidine was also noted in free Eaf3.
The methylated peptide binding mode of Eaf3 chromo barrel domain is reminiscent of that of chromo (Jacobs et al., 2001; Nielsen et al., 2002), double chromo (Flanagan et al., 2005), tandem tudor (Botuyan et al., 2006), hybrid tudor (Huang et al., 2006; Lee et al., 2008), malignant brain tumor (Li et al., 2007b; Min et al., 2007), PHD finger (Li et al., 2006; Pena et al., 2006), and ankyrin repeat domains (Collins et al., 2008). In all cases, a two to four aromatic residue binding cage establishes van der Waals and cation-π interactions with the methylated ammonium group. In a first category of interactions, a selectivity filter, in the form of a carboxylate side chain, discriminates against the trimethylated state by establishing a hydrogen bond and ion pair interactions with the methylammonium protons of mono- and dimethyllysine. In another category, a more open binding cage, with no possibility of methylammonium hydrogen-bonded carboxylate group, leads to a range of affinities that increases from a monomethyllysine- to a trimethyllysine-containing target. The Eaf3 chromo barrel domain does not have a carboxylate selectivity filter, and belongs to the second category of methylated histone binding domains.
In contrast to the different proteins mentioned above, which all have KDs in the 0.5–50 mM range with their targets, Eaf3 chromo barrel domain has an extremely low affinity for methylated histone H3K4 and H3K36 peptides (millimolar range), which also suggests a low selectivity of the isolated domain. Indeed, the H3K4 and H3K36 peptides do not share any significant amino acid sequence similarity with the exception of the methyllysine. Furthermore, NMR titration of 15N-labeled Eaf3 chromo barrel domain with a trimethylated histone H4K20 peptide (H4K20me3: KRHRK20me3VLRDN), for which there is no report in the literature of a functional interaction, gave a KD of 3–4 mM, similar to the KDs measured with H3K4me3 and H3K36me3 peptides (Figures 5A and 5C). The chemical shift perturbations by the three peptides are also similar (Figures 3A, 3B, and 5A). We show that the Eaf3 chromo barrel domain interacts with trimethylated lysine, albeit with an affinity significantly lower than that measured with trimethylated peptides under similar conditions. The KD for the Eaf3 and trimethyllysine interaction is 8.6–19.2 mM (Figures 5B and 5D). The Eaf3 chromo barrel domain does not have any significant affinity for symmetrically and asymmetrically dimethylated arginine (Figures 5E and 5F).
Figure 5. Interactions of Eaf3 Chromo Barrel Domain with Trimethylated Histone H4K20, Trimethylated Lysine and Dimethylated Arginine.

(A) 15N-1H HSQC titration spectra of 15N-labeled Eaf3 (1–113 aa), free (black), and upon addition of increasing amounts of nonlabeled H4K20me3 peptide (16–25 aa). In this and other panels, the side chain amide atom signals of Trp84 and Trp88 are labeled W84sc and W88sc, respectively.
(B) 15N-1H HSQC titration spectra of 15N-labeled Eaf3 (1–113 aa), free (black), and upon addition of increasing amounts of trimethylated lysine.
(C) Estimates of KD of Eaf3 for H4K20me3 peptide from the change in chemical shifts of selected 1H-15N Eaf3 resonances upon addition of nonlabeled H4K20me3 (16–25 aa).
(D) Estimates of KD of Eaf3 for trimethylated lysine from the change in chemical shifts of selected 1H-15N Eaf3 resonances upon addition of nonlabeled trimethylated lysine.
(E) 15N-1H HSQC titration spectra of 15N-labeled Eaf3 (1–113 aa), free (black), and upon addition of increasing amounts of symmetrically dimethylated arginine up to ∼7-fold excess.
(F) 15N-1H HSQC titration spectra of 15N-labeled Eaf3 (1–113 aa), free (black), and upon addition of increasing amounts of asymmetrically dimethylated arginine up to ∼7-fold excess.
The low affinity and permissiveness of the Eaf3 chromo barrel domain for different methyllysine-containing peptide sequences suggests that tight and specific interaction of Eaf3 with methylated chromatin can only occur in the context of the combined action of several domains in multiprotein complexes. In the case of the HDAC Rpd3S, it was convincingly shown that specific recognition of nucleosomes methylated at H3K36 not only requires the chromo barrel domain of Eaf3, but also the PHD domain of the Rco1 subunit (Li et al., 2007a).
In S. cerevisiae, Eaf3 is also a component of the HAT NuA4 complex that acetylates multiple sites of histone H4. NuA4 binds nucleosomes methylated at histone H3K4, but the subunit of NuA4 involved in the recognition of methylated H3K4 has not been unambiguously identified. Our study suggests that, in a manner similar to Rpd3S, NuA4 may be recruited to nucleosomes through direct binding of Eaf3 chromo barrel domain to methylated H3K4. Again, the low specificity of Eaf3 for methylated peptide targets and its low affinity for the H3K4me3 peptide suggest that a specific and tight interaction of NuA4 with nucleosomes methylated at histone H3K4 could only take place within a framework of combinatorial interactions, possibly involving the PHD domain of the Yng2 subunit of NuA4.
A better understanding of the binding mechanism of Eaf3 and other low-affinity methylated histone binding proteins will require the structural characterization of multidomain protein complexes bound to the nucleosome core particle (Luger et al., 1997) methylated at selected lysine residues.
The work presented here was made possible by the recent development of a method for the selective installation of methyllysine analogs into proteins, as demonstrated by the site-specific modification of the nucleosome core particle (Simon et al., 2007). Here, we have adapted this procedure to the 3D structure determination of a methyllysine-dependent protein complex. The low affinity of the Eaf3-H3K36me3 interaction in vitro, precluding structure determination, was overcome by linking Eaf3 chromo barrel domain to its histone H3 target. To our knowledge, the 3D structure of Eaf3-H3KC36me2 is the first of a chromo barrel domain in complex with a methylated target. It is also, to our knowledge, the first structure of a protein complex involving a methylated lysine analog obtained by cysteine alkylation. Only after chemical methylation of H3 at position 36 does the linked histone H3 peptide contact the chromo barrel structure. This work illustrates the remarkable influence of a single methylated lysine residue in bringing two proteins together.
EXPERIMENTAL PROCEDURES
Protein and Peptide Preparation
A detailed description of the preparation of the different Eaf3 constructs, including the fused Eaf3-H3KC36me2 complex, as well as the procedure for selective incorporation of a methyllysine analog, are presented in the Supplemental Data.
NMR Resonance Assignments
All NMR experiments were performed at 300K with Bruker Avance 600 MHz and Avance 700 MHz spectrometers equipped with cryogenic probes. 15N- and 15N/13C-labeled samples of Eaf3, Eaf3-C14V/C76S, Eaf3-H3K36, as well as wild-type and mutant Eaf3-H3C36 and Eaf3-H3KC36me2, with concentrations ranging from 0.3 to 1.5 mM, were prepared in 20 mM NaPi, pH 6.7, 50 mM NaCl, 1 mM EDTA, 2 mM DTT, and either 93% H2O/7% D2O or 100% D2O. 15N-1H HSQC spectra were recorded for all protein samples for various purposes (e.g., routine sample check, titration, to test the effects of various mutations). Free Eaf3 has a tendency to aggregate, producing broad NMR signals. To improve signal line widths, 800 mM urea (final concentration) was added to the protein sample. Addition of this amount of urea did not compromise the native structure of Eaf3, as the 15N-1H HSQC spectra collected on the protein in the absence and presence of 800 mM urea matched exactly in terms of peak positions (data not shown). No urea was added to Eaf3-H3KC36me2, as binding of the methylated target greatly improved the quality of the spectra.
For Eaf3 and Eaf3-H3KC36me2, 3D CBCA(CO)NH, CBCANH, HBHA(CO)NH, HCCH-TOCSY, HNCO and HN(CA)CO, CCH-COSY, CCH-TOCSY, C(CO)NHTOCSY, H(CCO)NH-TOCSY, and 2D HBCGCDCEHE, and HBCGCDHD experiments were acquired for backbone and side-chain assignments following established procedures (Ferentz and Wagner, 2000). 15N- and 13C-edited NOE spectroscopy data, each with a mixing time of 120 ms, were also collected to generate distance restraints. All data were processed and analyzed with the software NMRPipe (Delaglio et al., 1995), NMRView (Johnson and Blevins, 1994), and Sparky (Goddard and Kneller, 2004). A couple of free Eaf3 residues (Gly83 and Ser86) were not assigned, while the side chains of Lys45, Lys54, Lys57, Gln59, and Lys108 were incompletely assigned. For Eaf3-H3KC36me2, assignments were missing for Gly83, Ser86, Tyr133 (or H3Y41), and Arg134 (or H3R42). The resonances of Lys45, Lys54, Gln59, Lys108, Ser117, Thr118, and Gly125 (or H3G33) were only partially assigned.
1H/2H Exchange Experiments
Freeze-dried 15N-labeled samples of free Eaf3 and Eaf3-H3KC36me2 were reconstituted in D2O, and time-course 15N-1H HSQC spectra were collected to locate slowly exchanging amide protons and identify possible hydrogen bonds. The hydrogen bonds were confirmed from NOEs.
NMR Titration Experiments
To map in Eaf3 chromo barrel domain the binding site for various methylated molecules and estimate the corresponding KDs, 15N-1H HSQC spectra were collected with 15N-labeled Eaf3 samples, free and with additions of increasing amounts of nonlabeled H3K36me3 (31–42 aa), H3KC36me3 (31–42 aa), H3K4me3 (1–10 aa), H4k20me3 (16–25 aa) peptides, trimethylated lysine, symmetrically dimethylated arginine, and asymmetrically dimethylated arginine. The trimethylated lysine, symmetrically dimethylated arginine, and asymmetrically dimethylated arginine were purchased from Sigma-Aldrich. Weighted average chemical shift variations (Δ ppm) were calculated according to the formula: Δ ppm = ([δHN]2 + [δN]2)½, where δHN and δN are the changes in HN and N chemical shifts, respectively. From the Δ ppm, the KDs were estimated with the amide peaks of two to four selected amino acids, as shown in Figures 3 and 5, and in Figure S2.
Structure Calculations
The 3D solution structures of free Eaf3 and fused Eaf3-H3KC36me2 were calculated with the program CYANA2.1 (Güntert et al., 1997), with manual assignments as well as semiautomated NOE assignments by SANE (Duggan et al., 2001), and angle restraints derived by CSI (Wishart and Sykes, 1994) and TALOS (Cornilescu et al., 1999). Hydrogen bond restraints were identified from 1H/2H exchange experiments and NOE patterns, and were also included in the calculations.
The structures of free Eaf3 were refined with AMBER 8 (Case et al., 2004) following a previously published calculation protocol (Mer et al., 2000) with the generalized Born model to mimic solvent (Bashford and Case, 2000). A similar procedure was followed to calculate Eaf3-H3KC36me2 structures, but with additional restraints involving the H3KC36me2 fragment. The different restraints used in the calculations are summarized in Table 1.
From 200 initial structures, the final 20 lowest energy conformers of Eaf3, by itself and linked to H3KC36me2, were selected to represent the final ensemble of structures for each protein. The families of structures were of high quality, as evaluated by the program PROCHECK (Laskowski et al., 1996) (Table 1). Molecular representations were generated with PyMol (http://pymol.sourceforge.net/) and MOLMOL (Koradi et al., 1996).
Supplementary Material
ACKNOWLEDGMENTS
We thank Jerry Workman and Bing Li for helpful discussions, and Slobodan Macura, Nenad Juranic, and Prasanna Mishra at the Mayo Clinic NMR Core Facility for assistance. We are very grateful to Emeric Wasielewski for help with the figures. This work was supported by NIH grants CA109449 and CA132878 to G.M.
Footnotes
ACCESSION NUMBERS
The atomic coordinates and NMR constraints of Eaf3 chromo barrel domain in the free state and Eaf3-H3KC36me2-linked complex have been deposited at the Protein Data Bank under accession codes 2K3X and 2K3Y, respectively.
SUPPLEMENTAL DATA
Supplemental Data include Supplemental Experimental Procedures and three figures and can be found with this article online at http://www.structure.org/cgi/content/full/16/11/■■■/DC1/.
REFERENCES
- Allard S, Utley RT, Savard J, Clarke A, Grant P, Brandl CJ, Pillus L, Workman JL, Côté J. NuA4, an essential transcription adaptor/histone H4 acetyltransferase complex containing Esa1p and the ATM-related cofactor Tra1p. EMBO J. 1999;18:5108–5119. doi: 10.1093/emboj/18.18.5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashford D, Case DA. Generalized Born models of macromolecular solvation effects. Annu. Rev. Phys. Chem. 2000;51:129–152. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4–K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, Lee KK, Shia WJ, Anderson S, Yates J, Washburn MP, et al. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell. 2005;123:581–592. doi: 10.1016/j.cell.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Case DA, Darde TA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Wang B, Pearlman DA, et al. AMBER 8 (computer program) University of California; San Francisco: 2004. [Google Scholar]
- Clarke AS, Lowell JE, Jacobson SJ, Pillus L. Esa1p is an essential histone acetyltransferase required for cell cycle progression. Mol. Cell. Biol. 1999;19:2515–2526. doi: 10.1128/mcb.19.4.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins RE, Northrop JP, Horton JR, Lee DY, Zhang X, Stallcup MR, Cheng X. The ankyrin repeats of G9a and GLP histone methyltransferases are mono- and dimethyllysine binding modules. Nat. Struct. Mol. Biol. 2008;15:245–250. doi: 10.1038/nsmb.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Duggan BM, Legge GB, Dyson HJ, Wright PE. SANE (structure assisted NOE evaluation): an automated model-based approach for NOE assignment. J. Biomol. NMR. 2001;19:321–329. doi: 10.1023/a:1011227824104. [DOI] [PubMed] [Google Scholar]
- Eisen A, Utley RT, Nourani A, Allard S, Schmidt P, Lane WS, Lucchesi JC, Côté J. The yeast NuA4 and Drosophila MSL complexes contain homologous subunits important for transcription regulation. J. Biol. Chem. 2001;276:3484–3491. doi: 10.1074/jbc.M008159200. [DOI] [PubMed] [Google Scholar]
- Ferentz AE, Wagner G. NMR spectroscopy: a multifaceted approach to macromolecular structure. Q. Rev. Biophys. 2000;33:29–65. doi: 10.1017/s0033583500003589. [DOI] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY 3 (computer program) University of California; San Francisco: 2004. [Google Scholar]
- Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Mapping the protein universe. Science. 1996;273:595–603. doi: 10.1126/science.273.5275.595. [DOI] [PubMed] [Google Scholar]
- Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–751. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC, Allis CD, Khorasanizadeh S. Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J. 2001;20:5232–5241. doi: 10.1093/emboj/20.18.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BA, Blevins RA. NMRView: a computer program for visualization and analysis of NMR data. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- Joshi AA, Struhl K. Eaf3 chromodomain interaction with methylated H3-K36 links histone deacetylation to Pol II elongation. Mol. Cell. 2005;20:971–978. doi: 10.1016/j.molcel.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, Collins SR, Schuldiner M, Chin K, Punna T, Thompson NJ, et al. Cotranscriptional Set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell. 2005;123:593–605. doi: 10.1016/j.cell.2005.10.025. [DOI] [PubMed] [Google Scholar]
- Kizer KO, Phatnani HP, Shibata Y, Hall H, Greenleaf AL, Strahl BD. A novel domain in Set2 mediates RNA polymerase II interaction and couples histone H3 K36 methylation with transcript elongation. Mol. Cell. Biol. 2005;25:3305–3316. doi: 10.1128/MCB.25.8.3305-3316.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wüthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. 29–32. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol. Cell. Biol. 2003;23:4207–4218. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- Lee J, Thompson JR, Botuyan MV, Mer G. Distinct binding modes specify the recognition of methylated histones H3K4 and H4K20 by JMJD2A-tudor. Nat. Struct. Mol. Biol. 2008;15:109–111. doi: 10.1038/nsmb1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Howe L, Anderson S, Yates JR, 3rd, Workman JL. The Set2 histone methyltransferase functions through the phosphorylated carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 2003;278:8897–8903. doi: 10.1074/jbc.M212134200. [DOI] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Gogol M, Carey M, Lee D, Seidel C, Workman JL. Combined action of PHD and chromo domains directs the Rpd3S HDAC to transcribed chromatin. Science. 2007a;316:1050–1054. doi: 10.1126/science.1139004. [DOI] [PubMed] [Google Scholar]
- Li H, Fischle W, Wang W, Duncan EM, Liang L, Murakami-Ishibe S, Allis CD, Patel DJ. Structural basis for lower lysine methylation state-specific readout by MBT repeats of L3MBTL1 and an engineered PHD finger. Mol. Cell. 2007b;28:677–691. doi: 10.1016/j.molcel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Meijer M, Lees-Miller SP, Riabowol K, Young D. Three yeast proteins related to the human candidate tumor suppressor p33 (ING1) are associated with histone acetyltransferase activities. Mol. Cell. Biol. 2000;20:3807–3816. doi: 10.1128/mcb.20.11.3807-3816.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252:1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, Edwards AM, Chazin WJ. Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell. 2000;103:449–456. doi: 10.1016/s0092-8674(00)00136-7. [DOI] [PubMed] [Google Scholar]
- Min J, Allali-Hassani A, Nady N, Qi C, Ouyang H, Liu Y, MacKenzie F, Vedadi M, Arrowsmith CH. L3MBTL1 recognition of mono- and dimethylated histones. Nat. Struct. Mol. Biol. 2007;14:1229–1230. doi: 10.1038/nsmb1340. [DOI] [PubMed] [Google Scholar]
- Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED. Structure of the HP1 chromodomain bound to histone H3 methylated at lysine 9. Nature. 2002;416:103–107. doi: 10.1038/nature722. [DOI] [PubMed] [Google Scholar]
- Nielsen PR, Nietlispach D, Buscaino A, Warner RJ, Akhtar A, Murzin AG, Murzina NV, Laue ED. Structure of the chromo barrel domain from the MOF acetyltransferase. J. Biol. Chem. 2005;280:32326–32331. doi: 10.1074/jbc.M501347200. [DOI] [PubMed] [Google Scholar]
- Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, Kutateladze TG. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–103. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid JL, Iyer VR, Brown PO, Struhl K. Coordinate regulation of yeast ribosomal protein genes is associated with targeted recruitment of Esa1 histone acetylase. Mol. Cell. 2000;6:1297–1307. doi: 10.1016/s1097-2765(00)00128-3. [DOI] [PubMed] [Google Scholar]
- Reid JL, Moqtaderi Z, Struhl K. Eaf3 regulates the global pattern of histone acetylation in Saccharomyces cerevisiae. Mol. Cell. Biol. 2004;24:757–764. doi: 10.1128/MCB.24.2.757-764.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol. Cell. 2007;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Schaft D, Roguev A, Kotovic KM, Shevchenko A, Sarov M, Neugebauer KM, Stewart AF. The histone 3 lysine 36 methyltransferase, SET2, is involved in transcriptional elongation. Nucleic Acids Res. 2003;31:2475–2482. doi: 10.1093/nar/gkg372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Sykes BD. The 13C chemical-shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J. Biomol. NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- Xiao T, Hall H, Kizer KO, Shibata Y, Hall MC, Borchers CH, Strahl BD. Phosphorylation of RNA polymerase II CTD regulates H3 methylation in yeast. Genes Dev. 2003;17:654–663. doi: 10.1101/gad.1055503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Du J, Sun B, Dong X, Xu G, Zhou J, Huang Q, Liu Q, Hao Q, Ding J. Structure of human MRG15 chromo domain and its binding to Lys36-methylated histone H3. Nucleic Acids Res. 2006;34:6621–6628. doi: 10.1093/nar/gkl989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.