

Abstract
Matrix metalloproteinase-9 (MMP-9) and NADPH oxidase contribute to blood brain barrier (BBB) disruption after ischemic stroke. We have previously shown that normobaric hyperoxia (NBO) treatment reduces MMP-9 and oxygen free radical generation in ischemic brain. In this study, we tested the hypothesis that NBO protects the BBB through inhibiting NADPH oxidase-mediated MMP-9 induction in transient focal cerebral ischemia. Male Sprague-Dawley rats (n = 69) were given NBO (95% O2) or normoxia (21% O2) during 90-min filament occlusion of the middle cerebral artery (MCAO). Cerebral microvessels were isolated for analyzing MMP-9 and NADPH oxidase. BBB damage was non-invasively quantified with MRI. In normoxic rats, both NADPH oxidase catalytic subunit gp91phox and MMP-9 expression were upregulated in ischemic hemispheric microvessels after 90-min MCAO with 22.5 hrs reperfusion. Inhibition of NADPH oxidase with apocynin reduced the MMP-9 increase, indicating a causal link between NADPH oxidase-derived superoxide and MMP-9 induction. NBO treatment inhibited gp91phox expression, NADPH oxidase activity, and MMP-9 induction, which led to significantly less BBB damage and brain edema in the ischemic brain. These results suggest that gp91phox containing NADPH oxidase plays an important role in MMP-9 induction in ischemic BBB microvasculature, and that NBO treatment may attenuate MMP-9 induction and brain edema through inhibiting NADPH oxidase after transient cerebral ischemia.
Keywords: blood-brain barrier, matrix metalloproteinase, oxygen, stroke, NADPH oxidase
Introduction
Improving tissue oxygenation with oxygen therapy has long been considered a logical and important therapeutic strategy for acute ischemic stroke, however, the potential possibility of increased ischemic brain damage due to the detrimental effects of oxygen-derived free radicals has caused considerable concern and uncertainty for oxygen use (Chan 2001; Liu and Rosenberg 2005). Under certain conditions, oxygen therapy could enhance free radical generation that may exacerbate ischemic damage to the blood brain barrier (BBB), leading to serious vasogenic brain edema and hemorrhage (Liu and Rosenberg 2005; Kelly et al. 2006), two major causes of death in acute stroke patients. In rodent experiments, we and others have shown that normobaric hyperoxia (NBO) treatment markedly reduces infarction volumes and improves neurological outcome following transient cerebral ischemia (Flynn and Auer 2002; Singhal et al. 2002a; Singhal et al. 2002b; Liu et al. 2004; Kim et al. 2005; Liu et al. 2006; Henninger et al. 2007; Shin et al. 2007). More importantly, several of these studies showed that NBO treatment salvaged ischemic brain tissue without increasing the risk of oxidative damage or worsening BBB damage (Singhal et al. 2002b; Kim et al. 2005; Liu et al. 2006). In fact, NBO treatment decreased oxygen free radical generation as well as matrix metalloproteinase-9 (MMP-9) expression (Kim et al. 2005; Liu et al. 2006), two major contributors to BBB disruption in ischemic stroke (Liu and Rosenberg 2005). These findings suggest that NBO may protect the BBB against ischemic damage. Indeed, our very recent experiments showed that NBO treatment during ischemia attenuated early BBB disruption in a rat model of focal cerebral ischemia (manuscript submitted). However, the mechanisms of BBB protection by NBO remain largely unknown.
Oxygen free radicals are important mediators for BBB disruption in the ischemic brain (Chan 2001; Gasche et al. 2001; Liu and Rosenberg 2005). NADPH oxidase is an important source for superoxide (O2•−) generation in many cells. It is composed of membrane bound subunits (p22phox, gp91phox), cytoplasmic subunits (p40phox, p47phox, and p67phox), and the small G protein Rac-1 (Babior 2004). Substantial experimental evidence suggests that NADPH oxidase is upregulated in the ischemic brain, critically contributing to neuronal death after ischemic stroke (Walder et al. 1997; Infanger et al. 2006; Wang et al. 2006). Using genetic knockout mice models, several recent studies have demonstrated that the catalytic subunit gp91phox is critically implicated in several neurological disorders, including ischemic stroke (Park et al. 2005; Ostrowski et al. 2006; Abramov et al. 2007; Kahles et al. 2007). Besides the neural tissue, NADPH oxidase is also an important source of oxygen free radicals within the cerebral vasculature (Miller et al. 2005). Excessive generation of O2•−and its derivatives by NADPH oxidase from the BBB microvasculature may cause lipid peroxidation and membrane disruption, thus damaging BBB integrity. An important role of NADPH oxidase in BBB disruption was demonstrated in a recent experimental stroke study in which gp91phox knockout mice showed significantly less BBB damage than wild-type mice (Kahles et al. 2007). Oxygen at hyperbaric pressure was previously shown to suppress gp91phox containing NADPH oxidase in experimental subarachnoid hemorrhage (Ostrowski et al. 2006). However, it is unknown whether NBO protects against ischemia-induced BBB damage through inhibiting NADPH oxidase at the BBB microvascular level.
In addition to direct oxidative damage to the BBB, oxygen free radicals are also important triggers for MMP-9 in cerebral ischemia (Asahi et al. 2000; Gasche et al. 2001; Liu and Rosenberg 2005; Kelly et al. 2008), thus enhancing MMP-induced proteolytic degradation of the BBB structural components. NADPH oxidase-mediated O2•−generation has been shown to participate in MMP-2 and 9 activation in the vasculature outside of the CNS (Inoue et al. 2001; Deem and Cook-Mills 2004; Lai et al. 2006; Pagano and Haurani 2006). However, to date, no data are available on its ability to regulate MMP-9 in the BBB microvasculature in ischemic stroke.
Therefore, our present study was performed to answer the following questions: 1) whether gp91phox-containing NADPH oxidase is upregulated in the ischemic BBB microvasculature; 2) whether there is a causal link between NADPH oxidase and MMP-9 induction in the ischemic BBB; 3) whether NBO protects against ischemia-induced BBB damage through inhibiting NADPH oxidase-mediated MMP-9 induction in the ischemic BBB microvasculature.
Materials and Methods
Rat model of focal cerebral ischemia and reperfusion
The Laboratory Animal Care and Use Committee of the University of New Mexico approved all experimental protocols.0 Sprague–Dawley rats (Charles River Laboratories, Wilmington, MA, USA) weighing 290 to 320 g were anesthetized with isoflurane (4% for surgical induction, 1.75% for maintenance) in NO2:O2 (70%:30%) during surgical procedures. Body temperature was monitored continuously with a rectal probe and maintained at 37.5°C ± 0.5°C using a heating pad. The middle cerebral artery occlusion (MCAO) model was established by proximal occlusion of the right middle cerebral artery with the use of a 4-0 silicone-coated nylon monofilament (5 mm-length of the coated end with a diameter of 0.35 mm), as we previously described (Liu et al. 2006). After 90-min occlusion, reperfusion was accomplished by careful withdrawal of the monofilament and the rats were returned to their cages. The reperfusion time duration (3 or 22.5 hrs) depended on experimental design. For a total number of 69 rats included in this study, successful MCAO was confirmed by 2,3,5-triphenyltetrazolium chloride (TTC) staining of the 1-mm thick brain coronal section 6 mm away from the tip of the front lobe. TTC staining was perform exactly as we described previously (Liu et al. 2006).
The normoxic and NBO rats were ventilated (3 L/min) with medical air (21% O2) or a gas mixture of 95% O2 + 5% CO2, respectively, during the 90-min ischemic period. In our earlier study, we found that rats breathing 95% O2 + 5% CO2 were able to maintain the ischemic penumbral pO2 close to the preischemic level, and showed a relatively normal blood pH and breathing rhythm compared to rats breathing 95% O2 + 5% N2 or 100% O2 (Liu et al. 2006). Therefore, in this study, we chose 95% O2 + 5% CO2 as the NBO treatment. For the NADPH oxidase inhibitor studies, apocynin (Sigma, St. Louis, MO, USA) was delivered at a dose of 30 mg/kg body weight by intraperitoneal injection 1 hr before the onset of MCAO.
Postischemic tissue processing
Rats were sacrificed by decapitation at the end of reperfusion. Brains were quickly removed and chilled in ice-cold PBS for 5 min. Four 2-mm thick and one 1-mm thick coronary slices were sectioned from an 9-mm thick region 3 mm away from the tip of the frontal lobe, which contained the main infarction area according to our earlier studies with TTC staining (Liu et al. 2004; Liu et al. 2006). After digitally photographing the 2-mm thick brain slices, they were carefully cleaned of meninges, and then a longitudinal cut was made 2 mm away from the midline between two hemispheres to exclude tissue primarily supplied by the anterior cerebral artery. Nonischemic and ischemic hemispheric tissue was then collected from each brain slice, and freshly used for cerebral microvessel isolation. The 1-mm thick brain section 6 mm away from the tip of the frontal lobe was used for TTC staining to confirm successful MACO.
Measurement of brain edema
Brain edema was assessed 24 hrs after MCAO by measuring the hemispheric areas of each 2-mm thick brain slices using ImageJ software (NIH, Bethesda, MA, USA). Edema formation was calculated as hemispheric enlargement and was expressed as relative increase of the brain area in the ischemic hemisphere versus the nonischemic hemisphere as described previously (Manley et al. 2000).
Isolation of cerebral microvessels
Isolation of cerebral microvessels was performed as described previously (Kago et al. 2006). The hemispheric brain tissue was minced and homogenized in 4 ml ice-cold PBS using a Dounce homogenizer. The homogenate was filtered through a 41-μm nylon mesh (Spectrum, Irving, TX, USA), and the nylon mesh was washed three times with 5 ml PBS. Microvessels retained on the mesh were then washed off with PBS and pelleted by centrifugation at 4000 g for 10 min at 4 °C. The pellets were resuspended in 15% dextran T-500 and then added onto 20% dextran T-500, followed by centrifugation at 25,000 g for 10 min at 4 °C. The pellets were collected as the cerebral microvessels and were stored at −80 °C until further analysis. The purity and property of microvessel preparation were confirmed by microscopic observation after immuno-staining for tight junction protein claudin-5. Representative fluorescent micrograph of isolated microvessels was shown in Figure 1 after incubation with primary antibody against claudin-5 (Invitrogen, Carlsbad, CA, USA) and FITC-conjugated goat anti-mouse secondary antibody (Chemicon, Temecula, CA, USA) as we previously described (Yang et al. 2007).
Figure 1.

Representative fluorescent micrograph of cerebral microvessl preparation after immunostaining of tight junction protein claudin-5.
Gelatin zymography analysis for MMP-9
Isolated cerebral microvessels were homogenized on ice in 20 μl lysis buffer (Tris-Cl: 50 mM, pH 7.6, NaCl: 150 mM, CaCl2: 5 mM, Brij-35: 0.05%, NaN3: 0.02%, Triton X-100: 1%) containing protease inhibitor cocktail (Sigma, St. Louis, MO, USA). The protein concentration was determined using Bradford reagent (Bio-Rad, Hercules, CA, USA). MMP-9 was analyzed by gelatin zymography as previously described with modifications (Yang et al. 2007). Equal amounts of samples (50 μg protein) were separated on 10% SDS-polyacrylamide gels copolymerized with 1 mg/ml gelatin (Sigma, St. Louis, MO, USA) under nonreducing conditions. Gels were washed in 2.5% Triton X-100 and then incubated for 72 hrs with a developing buffer containing 50 mM Tris, pH 7.6, 5 mM CaCl2, 0.2 mM NaCl, and 0.02% (w/v) Brij-35 at 37 °C before they were stained with 0.125% Coomassie blue R-250 for 30 min in 10% (v/v) acetic acid and 50% methanol. Gels were destained with a solution containing 10% acetic acid and 10% methanol until clear bands of gelatinolysis appeared on a dark blue background. To confirm that detected activities were zinc-dependent gelatinases, some zymogram gels were incubated with developing solution in the presence of 10 mM EDTA. Disappearance of lytic bands in these gels confirmed the metal dependence of gelatinolytic activity characteristic of MMPs. The intensities of gelatinolytic bands were analyzed using Kodak 4000 image station (Carestream Molecular Imaging, New Haven, CT, USA). A mixture of human MMP-2/9 (Chemicon, Temecula, CA, USA) was used as gelatinase standards.
Western blot analysis for gp91phox
Isolated cerebral microvessels were homogenized on ice in 20 μl RIPA buffer. Homogenates (50 μg of total protein) were boiled for 5 min and then electrophoresed in 10% SDS-PAGE acrylamide gels, transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA, USA), and incubated for 1 hr in TBS-T (Tris-buffered saline and 0.1% Tween 20) containing 5% nonfat milk at room temperature. Membranes were then incubated overnight at 4 °C with primary antibody against gp91phox (BD Transduction Laboratories, Lexington, KY, USA) at a 1:1000 dilution, washed in TBS-T, and incubated for 1 hr at room temperature with HRP-conjugated anti-mouse antibody (1:1000; Santa Cruz Biotech, CA, USA). Subsequently, the membranes were developed with the SuperSignal West Pico HRP substrate kit (Pierce, Rockford, IL, USA) and photographed on a Kodak 4000 image station (Carestream Molecular Imaging, New Haven, CT, USA). To control sample loading and protein transfer, the membranes were stripped and reprobed with β-actin antibody (1:4000, Sigma, St. Louis, MO, USA).
Measurement of NADPH oxidase activity
NADPH oxidase enzymatic activity was quantified by lucigenin-enhanced chemiluminescence as described previously (Jacobson et al. 2003) with modifications. Cerebral microvessels freshly isolated from ischemic hemispheric brain tissue were resuspended in modified Krebs-HEPES buffer containing (in mM): 119 NaCl, 20 HEPES, 4.6 KCl, 1.0 MgSO4, 0.15 Na2HPO4, 0.4 KH2PO4, 5 NaHCO3, 1.2 CaCl2, and 5.5 glucose (pH 7.4). NADPH oxidase activity was measured in the presence of 5 μM lucigenin and 100 μM substrate NADPH (Sigma, St. Louis, MO, USA). No enzymatic activity could be detected in the absence of NADPH. After addition of NADPH, microvessels in small plastic tubes were placed in a luminometer (Model TD-20/20; Turner Designs, Sunnyvale, CA, USA). Luminescence measurements were integrated for 30-s periods, and the cycle was repeated 9 times; the 10 values were then averaged. After 10 cycles, the cell membrane-permeable O2•−scavenger 10 mM Tiron (Sigma, St. Louis, MO, USA) was added and incubated for 4 min. Thereafter, three more cycles were read and the values were averaged. After measurement, microvessels were lysed in RIPA buffer and the protein concentration was determined using Bradford reagent (Bio-Rad, Hercules, CA, USA). NADPH oxidase activity was calculated as luminescence difference before and after Tiron treatment, and expressed as relative luminescence units per minute per mg protein (RLU/min/mg protein).
Magnetic resonance imaging measurements for BBB permeability
MRI measurements for BBB permeability were performed exactly as described in our previous studies (Sood et al. 2007; Sood et al. 2008) using a 4.7T Biospec dedicated research MR scanner (Bruker-Biospin, Billerica), equipped with 500 mT/m (rise time 80-120 μs) gradient set (for performing small animal imaging) and a small bore linear RF coil (ID 72 mm). In brief, after 21 hrs of reperfusion, NBO-treated or normoxic rats were catheterized with PE-50 polyethylene tubing through left femoral vein for injection of gadolinium-diethylenetriamine pentaacetic acid (Gd-DTPA). During MRI measurements, anesthesia was maintained using a gas mixture of nitrous oxide (70%), oxygen (30%), and isoflurane (1.5 to 2.0%). Rectal temperature was kept at 37°C ± 1.0°C using a feedback controlled water bath.
A tripilot imaging sequence was used for reproducible positioning of the animal in the magnet at each MRI session. T2 weighted and diffusion weighted imaging (DWI), and the barrier permeability coefficient (or blood-to-brain transfer constant) Ki of gadolinium-diethylenetriamine pentaacetic acid (Gd-DTPA) measurement were performed. These sequences required approximately 1.5 hrs of scan time.
To measure the barrier permeability coefficient Ki, the Patlak plot method was used to acquire T1 maps. After 1 baseline T1 map was acquired, a bolus of 0.1 mmol/kg Gd-DTPA was manually injected into femoral vein via the place catheter. Fourteen T1 maps were then continuously acquired. The acquired data were transferred to a dedicated computer workstation for post processing. Post processing of the raw data involved generating Apparent Diffusion Coefficient (ADC) maps from DWI images, T1 maps from the raw data, reconstruction of permeability coefficient maps and construction of the permeability plots.
Statistical analysis
The Data are presented as means ±SEM. Statistical analysis was carried out using student t-test or ANOVA. Significant effects were probed using Newman–Keuls post hoc comparisons. A value of P ≤0.05 was considered statistically significant.
Results
Verification of successful MCAO
TTC staining of the 1-mm thick brain slice showed that 90 min MCAO and 22.5 hrs of reperfusion induced a large infarction in the ischemic hemispheres, while NBO-treated rats showed a smaller infarct area compared to normoxic rats (Figure 2A). When the reperfusion time was shortened to 3 hrs, all normoxic rats still showed infarction on TTC staining, with an infarct area mainly restricted to the striatum (Figure 2B). However, no obvious infarction was observed on the TTC-stained brain slices from NBO-treated rats, though these animals still displayed neurological deficits typical of MCAO, circling to the left (nonischemic side). Interestingly, in the NBO-treated rats, brain slices showed noticeable weaker TTC staining (less red color) in the region that encompassed the same infarct region observed in the normoxic rats, which may represent a mixture of surviving and dead neuronal tissue or dying neuronal tissue (Figure 2B). As such, TTC staining verified successful MCAO in all ischemic rats included in this study. Moreover, these TTC results also confirmed the neuroprotective effect of NBO as demonstrated in previous studies (Singhal et al. 2002a; Kim et al. 2005; Liu et al. 2006; Henninger et al. 2007; Shin et al. 2007).
Figure 2.
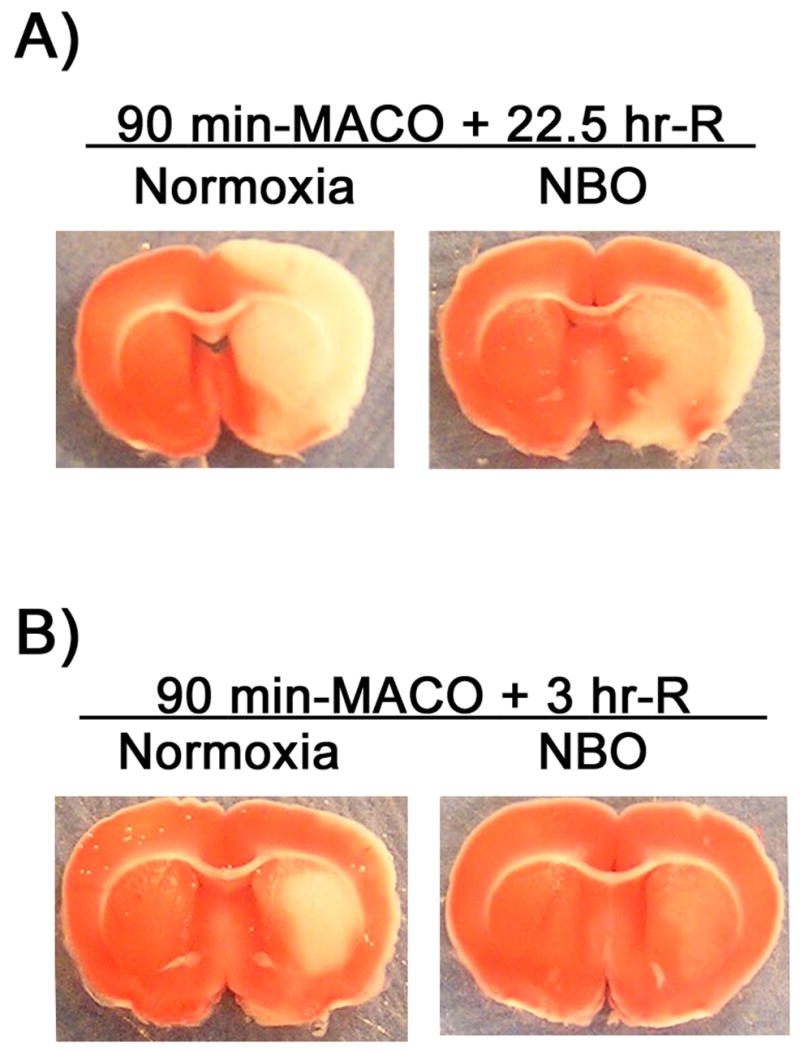
TTC staining verified successful MCAO in normoxic and NBO-treated rats. Typical TTC staining of the 1-mm thick brain slice 6 mm away from the tip of frontal lobe after 90-min MCAO with 22.5 (A) or 3 (B) hrs of reperfusion. NBO was delivered during 90-min ischemia.
Gp91phox is upregulated in ischemic cerebral microvessels
BBB disruption after transient cerebral ischemia exhibits a biphasic pattern with an early opening occurring within several hours after ischemia onset, and a late opening at 24 hr or later (Kuroiwa et al. 1985). To determine the role of NADPH oxidase in BBB disruption, we first examined the change of gp91phox protein, the catalytic subunit of NADPH oxidase, in the cerebral microvessels following 90-min MCAO with 3 or 22.5 hrs of reperfusion, which represent time points of early and late BBB opening. As shown in Figure 3, after 90 min of ischemia and 22.5 hrs of reperfusion, the level of gp91phox protein was dramatically increased (about 2.5-fold) in the microvessels from the ischemic hemisphere versus nonischemic hemisphere (P < 0.05). When the reperfusion time was shortened to 3 hrs, the level of gp91phox protein was only slightly increased in the ischemic hemispheric microvessels (1.30 ±0.28 versus 1.06 ±0.04, P > 0.05). These results suggest that gp91phox upregulation may be closely associated with late BBB disruption. Therefore, we selected 22.5 hr as the reperfusion time duration for the rest of this study.
Figure 3.

Gp91phox protein expression in cerebral microvessels after 90-min MCAO with 3- or 22.5-hr reperfusion. Cerebral microvessel lysates (50 μg) were analyzed for gp91phox protein by western blot. As a loading control, the blots were stripped and re-blotted with β-actin antibody. A) Representative blots of gp91phox and corresponding β-actin are shown. Non-I: nonischemic hemispheric microvessels; I: ischemic hemispheric microvessels. B) The relative quantity of protein was calculated after normalization to β-actin. The gp91phox protein expression was significantly increased in the ischemic hemispheric microvessels in those rats reperfused for 22.5 hrs (n = 6, *p < 0.05 versus nonischemic hemispheric microvessels). Only a slight increase of gp91phox was observed for those rats reperfused for 3 hrs (n =5, P > 0.05). Data are expressed as mean ± SEM.
NADPH oxidase is involved in MMP-9 induction in ischemic microvessels
Using NADPH oxidase inhibitor apocynin, we attempted to determine the involvement of this enzyme in MMP-9 induction in the ischemic hemispheric microvessels. MMP-9 expression was analyzed by gelatin zymography. As shown in Figure 4, MMP-9 was expressed at low level in the nonischemic hemispheric microvessels of the vehicle treated rats (0.15 ml ethanol/kg body weight), while its expression was drastically increased in the ischemic hemispheric microvessels (P < 0.05). Compared to MMP-9, MMP-2 was constitutively expressed at much lower levels in both hemispheric microvessels, indicating that MMP-9 is the main gelatinolytic enzyme produced by cerebral microvascular cells. Pretreatment with apocynin (30 mg/kg body weight) significantly reduced the MMP-9 increase in the ischemic hemispheric microvessels (P < 0.05 versus vehicle-treated rats). Interestingly, apocynin also reduced MMP-9 levels in the nonischemic hemispheric microvessels (P < 0.05 versus vehicle-treated rats). These results indicate that there is a causal link between NADPH oxidase-derived O2•−and MMP-9 induction in ischemic hemispheric microvessels.
Figure 4.

Effect of apocynin on MMP-9 induction in ischemic cerebral microvessels after 90-min MCAO with 22.5-hr reperfusion. Apocynin (30 mg/kg body weight) or vehicle (0.15 ml/kg body weight) was intraperitoneally injected to rats 1 hr before the onset of MCAO. Cerebral microvessel lysates (50 μg) were analyzed for MMP-9 expression with gelatin zymography. A) Representative gelatin zymogram showing MMP-9 expression in the nonischemic (Non-I) and ischemic (I) hemispheric cerebral microvessels. MMP-9 was clearly visualized on the gelatin zymogram, while MMP-2 was barely visible on the gel. STD is a mixture of standard MMP-2 and MMP-9. B) The relative band intensity of MMP-9 was quantified. A significant increase was observed for MMP-9 expression in the ischemic hemispheric microvessels in both vehicle- and apocynin-treated rats (*p < 0.05 versus nonischemic hemispheric microvessels). Apocynin significantly reduced MMP-9 expression in both nonischemic and ischemic hemispheric microvessels (#p < 0.05 versus vehicle-treated rats). Data are expressed as mean ± SEM, n = 3 in the vehicle-treated group, n = 6 in apocynin-treated group.
NBO treatment inhibits gp91phox upregulation in ischemic microvessels
We next examined whether NBO treatment could inhibit gp91phox upregulation in the ischemic cerebral microvessels. As shown in Figure 5A and 5B, gp91phox protein level is significantly higher in microvessels from the ischemic versus nonischemic hemisphere in the normoxic rats (P < 0.05). NBO treatment significantly inhibited the gp91phox protein increase in the ischemic hemispheric microvessels (P < 0.05 versus normoxic group), but did not change its expression in the nonischemic hemispheric microvessels.
Figure 5.

The effect of NBO on gp91phox-containing NADPH oxidase in ischemic hemispheric microvessels after 90-min MCAO with 22.5-hr reperfusion. A) Representative blots of gp91phoxand corresponding β-actin are shown. Cerebral microvessel lysates (50 μg protein) were analyzed for gp91phox protein by western blot. As a loading control, the blots were stripped and reblotted with β-actin antibody. Non-I: nonischemic hemispheric microvessels; I: ischemic hemispheric microvessels. B) The relative quantity of protein was calculated after normalization to β-actin. Ischemia and reperfusion significantly increased gp91phox protein expression in the ischemic hemispheric microvessels (*p < 0.05 versus nonischemic hemispheric microvessels), which was significantly inhibited by NBO treatment (*p < 0.05 versus normoxic rats). Data are expressed as mean ±SEM, n = 6 in each group. C) NADPH oxidase activity in ischemic hemispheric microvessels was assayed using lucigenin-enhanced chemiluminescence. NBO orapocynin treatment significantly reduced NADPH oxidase activity (*p < 0.05). Data are expressed as mean ±SEM, n = 6 in each of normoxic and NBO-treated group, n = 5 in apocynin-treated group.
Since gp91phox is the catalytic subunit of NADPH oxidase, we reasoned that gp91phox inhibition by NBO could result in reduction of NADPH oxidase activity. Because NBO did not change the low level of gp91phox expression in the nonischemic hemispheric microvessels (Figure 5A and 5B), we measured NADPH oxidase activity in the ischemic hemispheric microvessels. As shown in Figure 5C, microvascular NADPH oxidase activity was significantly lower in NBO-treated rats than that of normoxic rats (P < 0.05). As expected, pretreatment with apocynin markedly attenuated NADPH oxidase activity in the ischemic hemispheric microvessels (P < 0.05). These results demonstrate that NBO treatment inhibits gp91phox upregulation and NADPH oxidase activity in the ischemic cerebral microvessels.
NBO treatment reduces MMP-9 induction in ischemic microvessels
Since NADPH oxidase is shown to mediate MMP-9 expression following cerebral ischemia and reperfusion (Figure 4), we next asked whether inhibition of NADPH oxidase by NBO could result in reduction of MMP-9 expression in the ischemic hemispheric microvessels. As shown in Figure 6, NBO treatment significantly inhibited the MMP-9 increase in the ischemic hemispheric microvessels (P < 0.05 versus normoxic group), but did not change its low level of expression in the nonischemic hemispheric microvessels (Figure 6).
Figure 6.

Effect of NBO on MMP-9 induction in ischemic cerebral microvessels after 90-min MCAO with 22.5-hr reperfusion. Cerebral microvessel lysates (50 μg) were analyzed for MMP-9 expression with gelatin zymography. A) Representative gelatin zymogram showing MMP-9 expression in the nonischemic (Non-I) and ischemic (I) hemispheric cerebral microvessels. STD is a mixture of standard MMP-2 and MMP-9. B) The relative band intensity of MMP-9 was quantified. A significant increase was observed for MMP-9 expression in the ischemic hemispheric microvessels in both normoxic and NBO-treated rats (*P < 0.05 versus nonischemic hemispheric microvessels). NBO significantly reduced MMP-9 expression in the ischemic hemispheric microvessels (#P < 0.05 versus vehicle-treated rats). Data are expressed as mean ±SEM, n = 6 in each group.
NBO treatment attenuates BBB disruption and brain edema
MMP-9 and gp91phox-containing NADPH oxidase are important mediators of BBB disruption and brain edema in ischemic stroke (Rosenberg et al. 1998; Kahles et al. 2007). Therefore, using brain edema as an assessment of the final outcome, we determined whether inhibition of MMP-9 and NADPH oxidase by NBO or apocynin could lead to the attenuation of brain edema formation. Brain edema was evaluated as hemispheric enlargement. As shown in Figure 7A and 7B, 90-min MCAO and 22.5 hrs reperfusion under normoxic condition led to a significant enlargement of the right hemisphere (ischemic side). Importantly, both NBO and NADPH oxidase inhibitor (apocynin) treatment significantly reduced the hemispheric enlargement, while no significant difference was observed for hemispheric enlargement between normoxic and vehicle-treated rats. These results suggest that NBO treatment can attenuate brain edema, likely through inhibition of NADPH oxidase.
Figure 7.

Effect of NBO on brain edema formation and BBB permeability after 90-min MCAO with 22.5-hr reperfusion. A) Representative brain sections of the normoxic, NBO-, vehicle- and apocynin-treated rats. Significant right (ischemic) hemispheric enlargement is seen in rats of all groups. B) Quantitative image analysis showed a significant reduction of hemispheric enlargement in the NBO- or apocynin-treated rats when compared to normoxic rats. Data are expressed as mean ±SEM, n = 4 in the vehicle-treated group, n = 7 in each of other three groups. *P < 0.05 versus normoxic rats. C) Effect of NBO on BBB permeability coefficient assessed by MRI based technique. A significant increase was observed for BBB permeability coefficient in the ischemic hemisphere of both normoxic and NBO-treated rats (*P < 0.05 versus nonischemic hemisphere). NBO significantly reduced BBB permeability in the ischemic hemisphere (#P < 0.05 versus normoxic rats). Data are expressed as mean ± SEM, n = 6 in each group.
We also carried out a study to investigate whether NBO treatment could attenuate ischemia-mediated BBB disruption, using an MRI based method to directly estimate the BBB permeability coefficient Ki in ischemic rats treated with normoxia or NBO. Figure 7C shows the mean permeability coefficient values in these animal groups. In the normoxic rats, the Ki values in the ischemic and nonischemic hemispheres were 5.1 ± 1.68 × 10−3 ml/g/min and 0.55 ± 0.18 × 10−3 ml/g/min, respectively, suggesting that ischemia resulted in a dramatic increase of BBB permeability in the ischemic tissue. Importantly, NBO treatment significantly reduced the permeability coefficient value to 3.27 ± 1.99 × 10−3 ml/g/min in the ischemic hemisphere, while the nonischemic hemisphere value was maintained at 0.51 ± 0.16 × 10−3 ml/g/min. These results provide the direct in vivo evidence that NBO treatment reduces BBB disruption in the ischemic tissue.
Discussion
Despite great promising results from both experimental and clinical studies, oxygen therapy in stroke is still under debate. One major safety concern with oxygen therapy is the possibility of increased generation of toxic free radicals, which in the setting of ischemic stroke, could exacerbate brain edema, pos-ischemic hemorrhage and tissue necrosis (Chan 2001; Liu and Rosenberg 2005). Recent reports indicate that NBO treatment salvages ischemic brain tissue without increasing the risk of oxidative damage (Singhal et al. 2002b; Kim et al. 2005; Liu et al. 2006), and in fact, the generation of oxygen free radicals is reduced in the ischemic brain (Liu et al. 2006). These findings have allayed the concern over exacerbated oxidative damage to neuronal tissue due to hyperoxia exposure. Besides neuronal tissue, the BBB microvasculature is another vulnerable target for oxidative damage due to relatively low antioxidant capacity, high membrane polyunsaturated fatty acid content, and ready accessibility to pro-oxidant iron (Shukla et al. 1993; Yang and Betz 1994). The potential of BBB damage exacerbation by oxygen free radicals due to hyperoxia exposure is of particular concern (Singhal 2007) because it can result in serious brain edema, hemorrhage, and high mortality in acute ischemic stroke patients (Liu and Rosenberg 2005; Kelly et al. 2006). Our present study reveals that NBO treatment during ischemia protects against brain swelling through inhibiting gp91phox containing NADPH oxidase-mediated MMP-9 induction at the BBB microvascular level. Specifically, we found that NADPH oxidase catalytic subunit gp91phox protein level is significantly increased in the ischemic BBB microvasculature after 90 min-MCAO with 22.5 hrs of reperfusion. Moreover, the upregulation of this enzyme was closely associated with MMP-9 induction in the ischemic BBB. Most importantly, we found that NBO treatment inhibited gp91phox upregulation as well as its activity in the ischemic BBB microvasculature, leading to reductions in the MMP-9 level, brain swelling, and BBB disruption.
Oxygen free radicals are long-recognized contributors to BBB disruption in cerebral ischemia and reperfusion (Chan 1996, 2001; Liu and Rosenberg 2005). Recently, NADPH oxidase, particularly its catalytic subunit gp91phox, has attracted considerable attention as a contributor to ischemic damage to the brain tissue after stroke (Walder et al. 1997; Infanger et al. 2006; Ostrowski et al. 2006; Wang et al. 2006). The contribution of this enzyme to BBB disruption was demonstrated by the finding that gp91phox knockout mice showed significantly less BBB damage than wild-type mice after stroke (Kahles et al. 2007). However, the cellular origin of gp91phox in the ischemic brain and the mechanism of its role in BBB disruption remain to be defined. It is conceivable that oxygen free radicals involved in BBB disruption are most likely generated at the BBB microvascular level because they are highly reactive chemical species that do not travel long distances (Stamler 1996). Therefore, in the present study, we analyzed gp91phox-containing NADPH oxidase in the cerebral microvessels after cerebral ischemia and reperfusion. Our results demonstrate that gp91phox protein expression is significantly increased in the ischemic cerebral microvessels after 90-min ischemia with 22.5 hrs of reperfusion, indicating that gp91phox containing NADPH oxidase may be an important source of oxygen free radical generation in the ischemic BBB. A previous study showed that 120-min ischemia with 2 hrs of reperfusion led to an approximate 0.5-fold increase of gp91phox protein in the ischemic brain tissue (Hong et al. 2006). In this study, we only observed a slight increase (but not significant) of gp91phox protein level in the ischemic cerebral microvessels at 3 hrs after reperfusion. One possible explanation for this difference may be that there might be a different temporal profile of gp91phox expression between whole brain tissue and cerebral microvessels. Our results suggest that gp91phox upregulation may be an important mechanism responsible for NADPH oxidase-derived oxygen free radical generation at the ischemic BBB microvasculature after prolonged reperfusion.
A body of experimental evidence has demonstrated that oxygen free radicals participate in MMP-9 induction in the ischemic brain (Asahi et al. 2000; Gasche et al. 2001; Liu and Rosenberg 2005). Among these free radical species, O2•−overproduction is demonstrated to be an important cause of MMP-9 induction and BBB disruption in a recent ischemic stroke study wherein SOD1 transgenic rats (SOD1 overexpression) showed less MMP-9 induction and Evan’s blue leakage in the ischemic brain (Kamada et al. 2007). However, the source of O2•−which is involved in MMP-9 regulation is not clear in the BBB. In non-neural systems, results from several studies using apocynin, a specific intracellular inhibitor of NADPH oxidase assembly, suggest that NADPH oxidase-derived O2•−mediates MMP-9 induction in systemic cardiovascular cells such as cardiomyocytes (Rude et al. 2005), endothelial cells (Deem and Cook-Mills 2004) and smooth muscle cells (Inoue et al. 2001; Lai et al. 2006; Pagano and Haurani 2006). However, no data are available on its ability to regulate MMP-9 in the BBB microvasculature. In the present study, our finding that apocynin inhibits NADPH oxidase activity and MMP-9 induction in the ischemic cerebral microvessels, demonstrates for the first time that NADPH oxidase may be an important source of O2•−that regulates MMP-9 expression in the BBB microvasculature. It is important to point out that apocynin also decreased the basal MMP-9 expression in the nonischemic microvessels, suggesting that the low level of NADPH oxidase activity in the BBB microvasculature may be important in regulating the normal turnover of neurovascular matrix through maintaining basal MMP-9 expression
The potential possibility of enhanced oxygen free radical generation is the major concern with oxygen therapy in acute ischemic stroke. Several of recent studies showed that NBO treatment salvaged ischemic brain tissue without increasing the risk of oxidative damage (Singhal et al. 2002b; Kim et al. 2005; Liu et al. 2006). In fact, NBO treatment decreased oxygen free radical generation in the ischemic brain tissue (Liu et al. 2006). In this context, our finding that NBO treatment inhibits gp91phox upregulation and NADPH oxidase activity in the ischemic cerebral microvessels, demonstrates that NBO also reduces oxygen free radical generation in the ischemic BBB microvasculature. As shown in the present study, NADPH oxidase is a major contributing factor to the induction of MMP-9 in the ischemic BBB microvasculature (Figure 4). It is thus likely that inhibition of NADPH oxidase by NBO would result in reductions in MMP-9 expression. In support of this possibility, we found that NBO treatment reduces MMP-9 induction in the ischemic cerebral microvessels (Figure 6). We and others have previously shown that NBO treatment reduces MMP-9 expression in the ischemic brain tissue (Kim et al. 2005; Liu et al. 2006). In this context, our results provide important evidence that NADPH oxidase inhibition may represent one of the missing mechanistic links between NBO treatment and its suppressive action on MMP-9 in the ischemic brain. However, it is worth pointing out that apocynin inhibited NADPH oxidase activity to a much greater extent than NBO (Figure 5C), while both treatments reduced MMP-9 expression in the ischemic cerebral microvessels to the similar level, suggesting that the inhibition of NADPH oxidase is an important, but not the only mechanism accounting for NBO-mediated MMP-9 suppression in the ischemic microvasculature.
NADPH oxidase-derived O2•−and its derivatives can enhance lipid peroxidase and membrane disruption, leading to direct oxidative BBB damage (Mickel et al. 1987; Shin et al. 2007). In addition, these oxygen free radicals can also stimulate MMP-9 expression and thus enhance MMP-9-mediated proteolytic degradation of the BBB structural components (Gasche et al. 2001; Liu and Rosenberg 2005). Therefore, it is likely that inhibition of NADPH oxidase by NBO or apocynin could result in reduction of BBB damage in ischemic stroke. Indeed, we observed a significant reduction in hemispheric enlargement in NBO- or apocynin-treated rats, indicating their protective effect on BBB integrity. To unambiguously demonstrate a protective effect of NBO on the ischemic BBB, we directly compared BBB permeability in the ischemic hemispheres of the normoxic and NBO-treated rats using a well established MRI based technique. Our results show clearly a reduction in ischemic damage to the BBB in NBO-treated rats. These results suggest that inhibition of NADPH oxidase-MMP-9 pathway may represent an important mechanism underlying NBO-mediated neuroprotection.
In summary, the findings of this study indicate that gp91phox-containing NADPH oxidase contributes to MMP-9 induction in the ischemic BBB microvasculature. More importantly, our results demonstrate that NBO treatment may attenuate MMP-9 induction, BBB disruption and brain edema formation through inhibiting NADPH oxidase after experimental stroke.
Acknowledgments
The work was supported in part by grants from National Institutes of Health (P20 RR15636 and R01 AG031725), and American Heart Association (0555669Z and 0765461Z).
Abbreviations used
- MMP
Matrix metalloproteinase
- NBO
normobaric hyperoxia
- BBB
blood brain barrier
- MCAO
middle cerebral artery occlusion
- TTC
2,3,5-triphenyltetrazolium chloride
- MRI
magnetic resonance imaging
References
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahi M, Asahi K, Wang X, Lo EH. Reduction of tissue plasminogen activator-induced hemorrhage and brain injury by free radical spin trapping after embolic focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2000;20:452–457. doi: 10.1097/00004647-200003000-00002. [DOI] [PubMed] [Google Scholar]
- Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- Chan PH. Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab. 2001;21:2–14. doi: 10.1097/00004647-200101000-00002. [DOI] [PubMed] [Google Scholar]
- Deem TL, Cook-Mills JM. Vascular cell adhesion molecule 1 (VCAM-1) activation of endothelial cell matrix metalloproteinases: role of reactive oxygen species. Blood. 2004;104:2385–2393. doi: 10.1182/blood-2004-02-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn EP, Auer RN. Eubaric hyperoxemia and experimental cerebral infarction. Ann Neurol. 2002;52:566–572. doi: 10.1002/ana.10322. [DOI] [PubMed] [Google Scholar]
- Gasche Y, Copin JC, Sugawara T, Fujimura M, Chan PH. Matrix metalloproteinase inhibition prevents oxidative stress-associated blood-brain barrier disruption after transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:1393–1400. doi: 10.1097/00004647-200112000-00003. [DOI] [PubMed] [Google Scholar]
- Henninger N, Bouley J, Nelligan JM, Sicard KM, Fisher M. Normobaric hyperoxia delays perfusion/diffusion mismatch evolution, reduces infarct volume, and differentially affects neuronal cell death pathways after suture middle cerebral artery occlusion in rats. J Cereb Blood Flow Metab. 2007;27:1632–1642. doi: 10.1038/sj.jcbfm.9600463. [DOI] [PubMed] [Google Scholar]
- Hong H, Zeng JS, Kreulen DL, Kaufman DI, Chen AF. Atorvastatin protects against cerebral infarction via inhibition of NADPH oxidase-derived superoxide in ischemic stroke. Am J Physiol Heart Circ Physiol. 2006;291:H2210–2215. doi: 10.1152/ajpheart.01270.2005. [DOI] [PubMed] [Google Scholar]
- Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- Inoue N, Takeshita S, Gao D, Ishida T, Kawashima S, Akita H, Tawa R, Sakurai H, Yokoyama M. Lysophosphatidylcholine increases the secretion of matrix metalloproteinase 2 through the activation of NADH/NADPH oxidase in cultured aortic endothelial cells. Atherosclerosis. 2001;155:45–52. doi: 10.1016/s0021-9150(00)00530-x. [DOI] [PubMed] [Google Scholar]
- Jacobson GM, Dourron HM, Liu J, Carretero OA, Reddy DJ, Andrzejewski T, Pagano PJ. Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ Res. 2003;92:637–643. doi: 10.1161/01.RES.0000063423.94645.8A. [DOI] [PubMed] [Google Scholar]
- Kago T, Takagi N, Date I, Takenaga Y, Takagi K, Takeo S. Cerebral ischemia enhances tyrosine phosphorylation of occludin in brain capillaries. Biochem Biophys Res Commun. 2006;339:1197–1203. doi: 10.1016/j.bbrc.2005.11.133. [DOI] [PubMed] [Google Scholar]
- Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. NADPH oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- Kamada H, Yu F, Nito C, Chan PK. Influence of hyperglycemia on oxidative stress and matrix metalloproteinase-9 activation after focal cerebral ischemia/reperfusion in rats: relation to blood-brain barrier dysfunction. Stroke. 2007;38:1044–1049. doi: 10.1161/01.STR.0000258041.75739.cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly MA, Shuaib A, Todd KG. Matrix metalloproteinase activation and blood-brain barrier breakdown following thrombolysis. Exp Neurol. 2006;200:38–49. doi: 10.1016/j.expneurol.2006.01.032. [DOI] [PubMed] [Google Scholar]
- Kelly PJ, Morrow JD, Ning M, Koroshetz W, Lo EH, Terry E, Milne GL, Hubbard J, Lee H, Stevenson E, Lederer M, Furie KL. Oxidative stress and matrix metalloproteinase-9 in acute ischemic stroke: the Biomarker Evaluation for Antioxidant Therapies in Stroke (BEAT-Stroke) study. Stroke. 2008;39:100–104. doi: 10.1161/STROKEAHA.107.488189. [DOI] [PubMed] [Google Scholar]
- Kim HY, Singhal AB, Lo EH. Normobaric hyperoxia extends the reperfusion window in focal cerebral ischemia. Ann Neurol. 2005;57:571–575. doi: 10.1002/ana.20430. [DOI] [PubMed] [Google Scholar]
- Kuroiwa T, Ting P, Martinez H, Klatzo I. The biphasic opening of the blood-brain barrier to proteins following temporary middle cerebral artery occlusion. Acta Neuropathol (Berl) 1985;68:122–129. doi: 10.1007/BF00688633. [DOI] [PubMed] [Google Scholar]
- Lai CF, Seshadri V, Huang K, Shao JS, Cai J, Vattikuti R, Schumacher A, Loewy AP, Denhardt DT, Rittling SR, Towler DA. An osteopontin-NADPH oxidase signaling cascade promotes pro-matrix metalloproteinase 9 activation in aortic mesenchymal cells. Circ Res. 2006;98:1479–1489. doi: 10.1161/01.RES.0000227550.00426.60. [DOI] [PubMed] [Google Scholar]
- Liu JK, Rosenberg GA. Matrix metalloproteinases and free radicals in cerebral ischemia. Free Radic Biol Med. 2005;39:71–80. doi: 10.1016/j.freeradbiomed.2005.03.033. [DOI] [PubMed] [Google Scholar]
- Liu S, Shi H, Liu W, Furuichi T, Timmins GS, Liu KJ. Interstitial pO2 in ischemic penumbra and core are differentially affected following transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:343–349. doi: 10.1097/01.WCB.0000110047.43905.01. [DOI] [PubMed] [Google Scholar]
- Liu S, Liu W, Ding W, Miyake M, Rosenberg GA, Liu KJ. Electron paramagnetic resonance-guided normobaric hyperoxia treatment protects the brain by maintaining penumbral oxygenation in a rat model of transient focal cerebral ischemia. J Cereb Blood Flow Metab. 2006;26:1274–1284. doi: 10.1038/sj.jcbfm.9600277. [DOI] [PubMed] [Google Scholar]
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, Chan P, Verkman AS. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6:159–163. doi: 10.1038/72256. [DOI] [PubMed] [Google Scholar]
- Mickel HS, Vaishnav YN, Kempski O, von Lubitz D, Weiss JF, Feuerstein G. Breathing 100% oxygen after global brain ischemia in Mongolian Gerbils results in increased lipid peroxidation and increased mortality. Stroke. 1987;18:426–430. doi: 10.1161/01.str.18.2.426. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, Schmidt HH, Sobey CG. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ Res. 2005;97:1055–1062. doi: 10.1161/01.RES.0000189301.10217.87. [DOI] [PubMed] [Google Scholar]
- Ostrowski RP, Tang J, Zhang JH. Hyperbaric oxygen suppresses NADPH oxidase in a rat subarachnoid hemorrhage model. Stroke. 2006;37:1314–1318. doi: 10.1161/01.STR.0000217310.88450.c3. [DOI] [PubMed] [Google Scholar]
- Pagano PJ, Haurani MJ. Vascular cell locomotion: osteopontin, NADPH oxidase, and matrix metalloproteinase-9. Circ Res. 2006;98:1453–1455. doi: 10.1161/01.RES.0000231258.23378.a6. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998;29:2189–2195. doi: 10.1161/01.str.29.10.2189. [DOI] [PubMed] [Google Scholar]
- Rude MK, Duhaney TA, Kuster GM, Judge S, Heo J, Colucci WS, Siwik DA, Sam F. Aldosterone stimulates matrix metalloproteinases and reactive oxygen species in adult rat ventricular cardiomyocytes. Hypertension. 2005;46:555–561. doi: 10.1161/01.HYP.0000176236.55322.18. [DOI] [PubMed] [Google Scholar]
- Shin HK, Dunn AK, Jones PB, Boas DA, Lo EH, Moskowitz MA, Ayata C. Normobaric hyperoxia improves cerebral blood flow and oxygenation, and inhibits peri-infarct depolarizations in experimental focal ischaemia. Brain. 2007;130:1631–1642. doi: 10.1093/brain/awm071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla A, Shukla R, Dikshit M, Srimal RC. Alterations in free radical scavenging mechanisms following blood-brain barrier disruption. Free Radic Biol Med. 1993;15:97–100. doi: 10.1016/0891-5849(93)90128-h. [DOI] [PubMed] [Google Scholar]
- Singhal AB. A review of oxygen therapy in ischemic stroke. Neurol Res. 2007;29:173–183. doi: 10.1179/016164107X181815. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Dijkhuizen RM, Rosen BR, Lo EH. Normobaric hyperoxia reduces MRI diffusion abnormalities and infarct size in experimental stroke. Neurology. 2002a;58:945–952. doi: 10.1212/wnl.58.6.945. [DOI] [PubMed] [Google Scholar]
- Singhal AB, Wang X, Sumii T, Mori T, Lo EH. Effects of normobaric hyperoxia in a rat model of focal cerebral ischemiareperfusion. J Cereb Blood Flow Metab. 2002b;22:861–868. doi: 10.1097/00004647-200207000-00011. [DOI] [PubMed] [Google Scholar]
- Sood R, Taheri S, Estrada EY, Rosenberg GA. Quantitative evaluation of the effect of propylene glycol on BBB permeability. J Magn Reson Imaging. 2007;25:39–47. doi: 10.1002/jmri.20802. [DOI] [PubMed] [Google Scholar]
- Sood RR, Taheri S, Candelario-Jalil E, Estrada EY, Rosenberg GA. Early beneficial effect of matrix metalloproteinase inhibition on blood-brain barrier permeability as measured by magnetic resonance imaging countered by impaired long-term recovery after stroke in rat brain. J Cereb Blood Flow Metab. 2008;28:431–438. doi: 10.1038/sj.jcbfm.9600534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS. Alzheimer’s disease. A radical vascular connection. Nature. 1996;380:108–111. [PubMed] [Google Scholar]
- Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- Wang Q, Tompkins KD, Simonyi A, Korthuis RJ, Sun AY, Sun GY. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006;1090:182–189. doi: 10.1016/j.brainres.2006.03.060. [DOI] [PubMed] [Google Scholar]
- Yang GY, Betz AL. Reperfusion-induced injury to the blood-brain barrier after middle cerebral artery occlusion in rats. Stroke. 1994;25:1658–1664. doi: 10.1161/01.str.25.8.1658. discussion 1664-1655. [DOI] [PubMed] [Google Scholar]
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697–709. doi: 10.1038/sj.jcbfm.9600375. [DOI] [PubMed] [Google Scholar]