


Abstract
MicroRNAs (miRNAs) negatively regulate the expression of target genes at the post-transcriptional level. Little is known about the crosstalk between miRNAs and transcription factors (TFs). Here we provide data suggesting that the interaction patterns between TFs and miRNAs can influence the biological functions of miRNAs. From this global survey, we find that a regulated feedback loop, in which two TFs regulate each other and one miRNA regulates both of the factors, is the most significantly overrepresented network motif. Mathematical modeling shows that the miRNA in this motif stabilizes the feedback loop to resist environmental perturbation, providing one mechanism to explain the robustness of developmental programs that is contributed by miRNAs. Furthermore, on the basis of a network motif profile analysis, we demonstrate the existence of two classes of miRNAs with distinct network topological properties. The first class of miRNAs is regulated by a large number of TFs, whereas the second is regulated by only a few TFs. The differential expression level of the two classes of miRNAs in embryonic developmental stages versus adult tissues suggests that the two classes may have fundamentally different biological functions. Our results demonstrate that the TFs and miRNAs extensively interact with each other and the biological functions of miRNAs may be wired in the regulatory network topology.
INTRODUCTION
Regulation of gene expression plays a critical role in development and cellular homeostasis. One class of regulators that contribute to this control is transcription factors (TFs). Previous studies have investigated the regulatory networks controlled by TFs (1). Over the past several years, microRNAs (miRNAs) have emerged as another important class of regulatory factors, and they are distinct from TFs in that they modulate gene expression at the post-transcriptional level (2,3). There is increasing evidence that these two classes of trans-acting factors, TFs and miRNAs, can work cooperatively. A number of groups have provided new insights into their biology by identifying and characterizing for individual miRNAs their regulatory interactions with specific TFs. For example, reciprocal regulation, feedback and feed-forward loop (FFL) motifs involving both TFs and miRNAs have been described (4–7). These intriguing studies raise a number of interesting questions about the generality of miRNA/TF interactions, and about the overall structure of possible miRNA/TF interaction networks. How prevalent are these interactions, what relationships predominate and do the interaction modes have any implication related to the biological functions of miRNAs?
Gene regulatory networks consist of basic structural units. Some units occur more often than would be expected from a random distribution. Such units are termed network motifs (8). Understanding the functionality of these network motifs can help elucidate the design principle and provide insight into the behavior of regulatory networks. Previous work in transcriptional regulatory networks of several species has discovered some network motifs such as feed-forward, feedback loops (8–11). These network motifs can build up useful features, examples of which include the acceleration of response time and filtering out external environmental noise (10,12).
Several bioinformatics studies have further explored the relationships between TFs and miRNAs. Tsang et al. (13) investigated the expression relation between miRNAs and their target genes and suggested that individual miRNAs and their targets can share common regulator(s). Shalgi et al. (14) examined the network motifs through which TFs and miRNAs co-regulate their target genes. Based on our interest in network interactions and gene regulation, we have attempted to expand on these studies, with particular focus on recurring interaction patterns between TFs and miRNAs. To reveal the design principles of the networks involving both transcriptional and post-transcriptional regulation, we investigated the basic interaction patterns between the two types of regulators on a systems level. Our work has two novelties compared to the previous studies. First, our study explored a broader scope of network motifs. We studied not only the network motifs in which both TF and miRNA as regulators, but also other types of network motifs where they could also be the regulatory targets. In total, we examined 46 network motifs (compared to five network motifs studied in Shalgi's work). Second, previous studies placed less emphasis on examining the functionality of these network motifs. We tried not only to identify network motifs, but also attempted to understand the biological roles played by the network motifs. We have utilized a mathematical model to help elucidate the potential functions of the regulated feedback loop in development and have classified network motif patterns related to different stages of development.
MATERIALS AND METHODS
Genomic locations of genes
We used the RefSeq gene set in hg18 version from UCSC genome browser (http://genome.ucsc.edu). Cis-regulatory module (CRM) locations of PReMod (15) were originally based on hg17 and were lifted to hg18 using http://genome.ucsc.edu/cgi-bin/hgLiftOver. The miRNA gene set (pri-miRNA) was downloaded from miRBase (http://microrna.sanger.ac.uk).
Construction of integrated regulatory networks
Similar to previous studies (14,16), we constructed human gene regulatory networks based on computational results due to the scarcity of experimental data. The transcription factor binding sites (TFBSs) were mapped on the human genome sequence based on PReMod, in which CRMs were identified if regions contained phylogenetically conserved binding sites for several TFs (15). If a CRM is located within 5-kb upstream relative to the transcription start site or 5′-UTR, the corresponding TFs of the CRM are predicted as the regulators of the gene. For cross-validation of TF regulation, another TFBS set ‘cTFBS’ is similarly derived from the ‘TFBS Conserved’ track (hg18 version) of UCSC genome browser (http://genome.ucsc.edu).
How miRNAs genes are regulated is less clear. We simply assume TFs adopt the same mechanism to regulate miRNA expression that they use to regulate other protein-coding genes. That is, if we found a TFBS within the upstream 5 kb of a miRNA gene (pri-miRNA), we predicted the TF regulates the miRNA. For the miRNAs located within other protein-coding genes, we used the promoters of the host genes as the promoter of the miRNAs. For miRNA polycistrons, such as mir-17-92, mir-106a-92 and mir-106b-25, we used the promoter of the 5′-most miRNA for all the members in the polycistron. Similarly, the target genes of miRNAs were predicted on the basis of the miRNA binding sites in the 3′-UTRs in the target genes.
Degree-preserving random networks and motif Z-scores
To evaluate the statistical significance, we compare the occurrence of each subgraph in real networks and random networks. In the degree-preserving random networks, we keep the same incoming degree (number of regulators on the node) and outgoing degree (number of targets of the node) for each node as in the IRNs. Furthermore, we also keep the compositions of incoming (i.e. number of TFs or miRNAs regulating the node) and outgoing degree (i.e. number of miRNAs or other genes regulated by the node) for each node the same as in the IRNs. To generate such a random network, we performed ‘permutation’ in the IRNs. For a given edge (say, from a TF t1 to a gene g1) in the IRNs, we first found another random edge with the same type of connected nodes (from a TF t2 to a gene g2). We then swapped the two connections (i.e. connect t1 to g2, connect t2 to g1 and delete the initial connections from t1 to g1, and from t2 to g2). We repeated the same procedure for each edge and obtained a randomized network. The Z-score of a motif is calculated as the difference of its observed occurrence in the IRN and its averaged occurrence in several hundreds random networks, normalized with the standard deviation. The P-value is defined as the fraction of the random networks that have higher occurrences than the observed.
Mathematical modeling for regulated feedback loops
To understand the biological function of regulated feedback loops, we performed a mathematical modeling on it. The rates of two TF concentrations can be approximately described as follows:
where A and B are the concentrations of two TFs (a and b); A0 and B0 are the concentration threshold of a and b to start acting, respectively; θ(x) is the step function (1 for x ≥ 0 and 0 for x < 0); α and β are the decaying coefficients; Kxy is the rate constant that x regulates y; M represents miRNA. There are three terms determining the rate of one TF concentration: (i) regulation from the other TF; (ii) regulation from the miRNA; and (iii) degradation. The above equation group is equivalent to
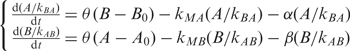 |
Taking transformations 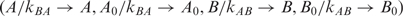 and θ(cx) = θ(x), where positive constant c can be kBA or kAB, we have
and θ(cx) = θ(x), where positive constant c can be kBA or kAB, we have
If the repressions are so strong that kMAA0 + αA0 > 1 or kMBB0 + βB0 > 1, any initial input (Ainitial, Binitial) will lead to the off state (0,0) because one rate is always negative and its concentration will be below the threshold and cause the other rate to be negative. Otherwise, we set the two rates zero and derive a nonzero stable solution (on state) (1/(kMA + α),1/(kMB + β)) because the two stable concentrations should be larger than A0 and B0, respectively. The condition of initial concentrations leading to the on state is
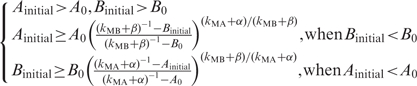 |
These equations are equivalent to a critical curve in phase transition between on and off states. In the first situation (Ainitial > A0 and Bintial > B0), if the initial concentration is less than the on state, its rate is positive; otherwise, its rate is negative. Thus, both lead to the on state. In the region of Ainitial < A0 and Bintial < B0, both initial rates are negative, and the system will go to the off state. For the region (Ainitial > A0 and Bintial < B0), the initial rate of B is positive and that of A is negative. It is required that B has risen to B0 when A decreases to A0. Thus we can obtain the second equation, and similarly, the third one.
Network motif profiles
For any miRNA, we first counted the occurrences of the different subgraphs containing this miRNA in the IRNs and thus obtained its subgraph occurrence profile (M1, M2, …, Mn), where n is the total number of subgraphs we studied. Therefore, the total number of all subgraphs containing this miRNA is 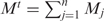 . We also obtained the total occurrence of each of the subgraphs (N1, N2, …, Nn) in the network for all miRNAs, and thus a probability vector (p1, p2, …, pn), where
. We also obtained the total occurrence of each of the subgraphs (N1, N2, …, Nn) in the network for all miRNAs, and thus a probability vector (p1, p2, …, pn), where 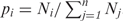 . As the null hypothesis, we assume that any miRNA has a chance of pi to appear in subgraph i. Consequently, the expected occurrence in each subgraph is (p1Mt, p2Mt, …, pnMt). By comparing the expected with the actual occurrence profile, we calculated the z-score (z1, z2, …, zn) as a measure of the deviation from the random expectation, where
. As the null hypothesis, we assume that any miRNA has a chance of pi to appear in subgraph i. Consequently, the expected occurrence in each subgraph is (p1Mt, p2Mt, …, pnMt). By comparing the expected with the actual occurrence profile, we calculated the z-score (z1, z2, …, zn) as a measure of the deviation from the random expectation, where 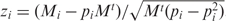 . We then clustered the miRNAs on the basis of Z-scores using program cluster with the option of ‘hierarchical’, ‘correlation (uncentered)’ and ‘complete lineage’ (17).
. We then clustered the miRNAs on the basis of Z-scores using program cluster with the option of ‘hierarchical’, ‘correlation (uncentered)’ and ‘complete lineage’ (17).
Gene expression comparison for two groups
We compared the rank difference of gene expression between the two groups of miRNAs in each condition (18). There are two advantages of using rank instead of their actual expression values. First, the expression levels from different data sets are on different scales. The expression becomes comparable by using their ranking. Second, the comparison is not dominated by single or several large expressed genes. All genes contribute to the comparison equally. To evaluate the statistical significance of the observed ranking difference between two groups, we permutated the miRNA genes in the list and obtained the expected ranking difference. We repeated the permutations for 1000 times and a P-value was defined as the fraction of the permutations that have larger differences than the observed ranking difference of these two subgroups, adjusted by Benjamini and Hochberg False Discovery Rate multiple testing correction (19).
Functional analysis of upstream TFs
Since miRNAs negatively regulate their target genes, the functional categories of their target genes are often not correlated with the miRNA functions. However, we expected the upstream TFs that are regulating the miRNAs are more likely to share the similar functional categories with the miRNAs. At first, we classified the TF regulators into two classes that preferentially regulate one of the two classes of miRNAs. We found 943 gene ontology (GO) occurrences associated with class I TFs, and 225 with class II. For a GO term with nI occurrences in class I and nII occurrences in class II, we calculated Z-scores based on the null hypothesis that this GO item has the same occurrence percentages in the two classes, i.e.
Denoting p as 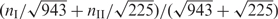 (to keep the sum of Z-scores zero, see below), we derived Z-scores as
(to keep the sum of Z-scores zero, see below), we derived Z-scores as
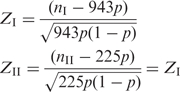 |
If the value of a Z-score is positive, the studied GO item is overrepresented in the class of genes; otherwise, the GO item is under-represented.
RESULTS
To study the regulatory relationships between miRNAs and TFs, we first assembled integrated regulatory networks (IRNs) by superimposing the networks controlled by TFs and miRNAs (Figure 1a). As a first approximation, we used available in vitro and predicted binding activity for TFs and miRNAs for developing the networks. For the transcriptional component we determined 96 371 regulatory relationships between 405 TFs and 24 582 genes (including miRNA genes), by detecting the presence of the TF binding sites in the promoters of the genes based on PReMod data set (15). Similarly, the post-transcriptional regulatory relationships were obtained based on miRNA recognition sites in 3′UTRs of the genes. For cross-validation of our findings, we used two separate sources of miRNA target predictions [miRanda (20) and PicTar (21)] to prepare two sets of IRNs. miRanda predicted 39 801 miRNA-target relationships for 157 miRNAs, and PicTar predicted 75 968 relationships for 178 miRNAs (for the details of the IRNs construction, see Materials and methods section and Supplementary Table S1).
Figure 1.

Network motifs involving both TFs and miRNAs. (a) Integrated regulatory network (IRN). (b) The 46 possible three-node subgraphs involving at least one TF and one miRNA. The numbers under the subgraphs are the Z-scores of their occurrences as compared with random networks. The number pairs formatted as x|y represent the statistics from miRanda|PicTar-derived IRNs. The correlation coefficient between the Z-scores from two data sets is 0.73 (P < 10−9). The only subgraph whose Z-scores are not consistent between two data sets is marked with star, which occurs only a few times in the IRNs. (c) Top network motifs. Rows with m are for miRanda and p for PicTar.
TFs and miRNAs tend to regulate each other and co-regulate genes
Our preliminary inspection of the two types of regulators in the IRNs showed that TFs and miRNAs tend to regulate each other. Genes encoding TFs have more miRNA binding sites in their 3′-UTRs than do other protein-coding genes (3.4 versus 1.5 miRNA sites per gene, P ∼ 0−968). Conversely, miRNA genes have more TF binding sites in their promoters than do protein-coding genes (5.4 versus 3.9 TF sites per gene, P ∼ 10−46). This cross-regulation relationship has been previously shown in isolated instances, such as the reciprocal negative regulatory feedback between TF Yan and miR-7 in the Drosophila eye that promotes photoreceptor differentiation (6).
Identification of overrepresented network motifs and cross-validation of different data sets
Motivated by the preferential cross-regulation and cooperation between TFs and miRNAs, we further investigated their interaction patterns by calculating the occurrence of different subgraphs in the IRNs. For three-node subgraphs, there are 46 possible regulatory configurations (Figure 1b) involving at least one TF and one miRNA. We exhaustively enumerated the occurrences of these subgraphs in IRNs. The actual occurrence was compared with that expected from degree-preserving random networks, which randomize the edges while keeping the same numbers of incoming and outgoing edges for each node as in actual IRNs (Supplementary Table S2). A Z-score is calculated based on this comparison (see Materials and methods section). The subgraphs that occur significantly more than expected are defined as network motifs (8–11).
Following previous work, we chose both z-score and cluster number of the each type of subgraph (all nodes involved in one type of subgraph form a number of distinct clusters within the IRNs) as the criteria for definition of significant motifs. We used Z-score of larger than 3.5 and cluster number of larger than 5 as cutoffs for significant motifs (the Z-score cutoff corresponds to P-value of 0.01, after multiple testing corrections). Interestingly, the network motifs identified from the two data sets are almost identical (Supplementary Table S2). In total, we discovered 17 significant motifs from both data sets, with only one being inconsistent (P = 2.8 × 10−10). Furthermore, the same procedure was applied to random networks and none of these motifs were recovered. This suggested that the identified network motifs are likely to be true motifs even though the input networks contain a certain amount of noise.
Figure 1c illustrated some of the network motifs (for more network motifs see Supplementary Table S2D). In one motif, TFs and miRNAs tend to co-regulate their target genes. A more detailed analysis reveals that 5082 pairs of TFs and miRNAs are likely to cooperate to co-regulate target genes (Supplementary Figure S1). Also, miRNA-associated FFLs are abundant in the IRNs. The functionality of FFLs consisting of only TFs and target genes has been extensively studied (8,10,12). The miRNA-associated FFLs might have the similar functionality.
Regulated feedback loop is the most significantly overrepresented network motif
The regulated feedback loop, where two TFs regulate each other and one miRNA regulates both of them, is the most significant network motif according to the results from both data sets (Figure 1c). It occurs 10 times more than expected from random networks. Feedback loops have been extensively studied in biological systems, such as synaptic connections between neurons and the developmental transcription networks of the fruit fly and sea urchin (9,22,23). One elegant feature of this type of feedback loop is that it can convert a transient stimulus into a stable and irreversible response. This feature is particularly useful in developmental transcription networks in that such irreversible decisions can lock a cell into a particular fate. The observed interaction between miRNAs and feedback loops coincides with the important roles miRNAs are known to play during development.
Mathematical modeling of the feedback loop
To understand the functionality of miRNA-associated feedback loops, we performed a mathematical analysis of the dynamics behavior of the motifs (see Materials and methods section). We examined the steady state of the system under different conditions such as initial TF concentration and the strength of miRNA control over the TFs. In simple feedback loops without contribution from the miRNA (Figure 2a), activating one TF is often sufficient to cause the system to be in the on state. With higher effects of miRNA in the regulated feedback loops, higher starting concentrations of TFs are required (Figure. 2b), and in the extreme case, both TFs must be simultaneously activated to turn on the system (Figure 2c). Therefore, one major benefit from a regulated feedback loop as compared with a simple feedback loop is that it can prevent inadvertent activation and thus provide the stability and robustness needed for the developmental process. Recently, miRNAs have been suggested to contribute to the canalization of genetic programs, the tendency to maintain the phenotypic reproducibility of development (24–27) For example, several studies have shown that miRNAs are not required for a normal development, but can affect the developmental process in stressful conditions, suggesting that miRNAs play a role in stabilizing development. Our analysis offers one possible mechanism for the molecular basis of the canalization of development by miRNA. As an additional property, regulated feedback loops have lower expression levels for the on state (see Materials and methods section). Depending on the activity of the miRNA, the feedback loop could have a low or high on state, providing more options for finer regulatory control (28). The mathematical modeling of the regulated feedback loop suggests that the basic repressive function of miRNAs when combined with other regulatory factors can build up more complex and higher-order functions such as canalization and fine-tuning of development.
Figure 2.

Mathematical modeling of regulated feedback loop. For simplicity, we let 1/(kMA + α) = 1/(kMB + β) and A0 = B0 = 1. The x-axis and y-axis are the initial concentrations of the two TFs at time 0. Different initial concentrations of TFs will lead to either on or off states. (a–c) represent the scenario with different levels of miRNA repression efficiency.
Two classes of miRNAs with distinct preference on network motifs emerge
While regulated feedback loops represent the most significantly overrepresented global network motif in the IRNs, specific miRNAs show distinct preferences for occurring in different interaction subgraphs. For each miRNA, this preference for different subgraphs can be combined as a network motif profile. When miRNAs are clustered according to the similarity of network motif profiles using an unsupervised hierarchical clustering method, a surprising pattern emerges (Figure 3). The clustering on both data sets (i.e. PicTar and miRanda) shows there exist two classes of miRNAs with distinct preference for different network subgraphs (Supplementary Figure S2). Class I miRNAs are enriched for network subgraphs in which miRNAs are regulated by TFs, whereas class II miRNAs are enriched for subgraphs in which miRNAs regulate TFs. There is general agreement about which miRNAs fall into each class between the two data sets (P-values for overlap between two data sets are 10−3.4, and 10−6.6 for classes I and II, respectively). For further analysis, we used the 26 class I miRNAs and 57 class II miRNAs that were identified from both data sets (Figure 3; Supplementary Table S4).
Figure 3.

Two classes of miRNAs with distinct interaction patterns with TFs. The miRNAs were clustered based on their relative occurrence in different subgraphs. The values in the map are the Z-scores for the enrichment of the occurrence in a subgraph as compared to a random expectation. Class I miRNAs are regulated by large numbers of TFs whereas class II miRNAs are regulated by a few TFs.
We characterized the two classes of miRNAs by examining their regulatory relationship with TFs. In both classes, there was a similar number of putative target TF genes for each miRNA (i.e. 10.8 and 12.3 TFs for classes I and II, respectively), suggesting that the two classes have similar regulatory influences on their downstream TFs. However, there was a significant difference in how many TFs were regulating each of the miRNAs. On average, each miRNA in class I was regulated by 23 TFs. In contrast, each miRNA in class II was targeted by only 0.8 TF, indicating that the class I miRNAs are under more complex combinatorial regulation by TFs than are class II miRNAs (Figure 3). We also performed a random simulation and found that we were not able to recover the two classes that have a perfect division between inbound and outbound miRNAs.
To confirm the regulatory differences in the upstream regulation of the two classes of miRNAs, we analyzed experimental data sets on whole genome in vivo binding of human TFs (data on 16 TFs available) (29–36). On average, 1.31 and 0.56 TF binding sites were found in each miRNA promoter from classes I and II, respectively (P = 5× 10−4 from comparison with the random expected). Note that the difference between the two classes is not as dramatic as with IRNs. One possible reason is that multiple TFBSs in the promoters of class I miRNAs are functional in different physiological conditions, while a ChIP–chip experiment captures only one snapshot at a particular time under specific conditions.
Gene expression patterns indicate the two classes have different biological functions
To investigate how the different topological properties shape the biological functions of the miRNAs, we compared the expression levels of the two classes during development as approximated by embryonic developmental stages and adult tissues. Strikingly, in ~90% (19/21) of embryonic developmental conditions, class I miRNAs were more highly expressed than class II miRNAs (Figure 4). Among these, in 52.4% (11/21) of the embryonic developmental conditions the average expression level of class I miRNAs was statistically significantly higher than that of class II miRNAs (P < 0.05, for detailed P-values for each condition, see Supplementary Table S6). On the other hand, in most of the normal adult tissues (30/32) class II miRNAs were more highly expressed than class I miRNAs. In 12.5% (4/32) of the tissues, the expression difference between two classes is statistically significant. This trend is preserved even in tumor tissues (21), where in a remarkable 99% (67/68) of tumor samples class II miRNAs were expressed higher than class I (Supplementary Figure S3). In summary, during embryonic developmental conditions, the average expression levels of class I miRNAs are higher than that of class II, whereas in adult tissues, class II was more highly expressed than class I. Among total 54 conditions, only five of them violate the general trend (P < 10−10).
Figure 4.

miRNA expression levels at embryonic developmental conditions and adult tissues. For each condition, we ranked the miRNAs based on their expression level and then compared the average ranks between two classes. We compared the observed rank difference with that expected from two random groups. P-values were calculated to evaluate the significance for the rank difference. The miRNA expression data sets are obtained from (38) for zebra fish (39,40) for mouse and (41) for human.
The observed correlation between network motif type and miRNA gene expression pattern is likely to be true because the miRNA expression patterns are conserved across species as the expression data we used in the analysis included human, mouse and zebra fish. In addition, the platforms for gene expression measurement in these studies are diverse (from microarray to sequencing), suggesting the observation is not due to technical systematic error arising from the method of gene expression measurement.
Besides the gene expression of the miRNAs, we also investigated the gene functions of the upstream TFs that control the two classes of miRNAs. TFs regulating class I miRNAs are enriched for the functional categories of ‘organogenesis’ and ‘development’, while TFs regulating class II miRNAs are enriched for diverse functional categories such as ‘immune response’ and ‘lipid metabolism’ (Supplementary Table S7). Limited by the numbers of two classes of TFs, the total occurrence number of a typical GO item in two classes is <10, resulting in nonsignificant Z-scores.
Tolerance to false regulatory relations and other factors
Our results are supported by the consistency between the two different data sets and the independent gene expression studies. For a more stringent evaluation, we performed additional tests on our observations. First, we added a conserved TFBS (cTFBS) set (conservation z-score > 2.33) as TF–target relationship for further cross-validation. Now we have totally four combinations for TF (PReMod and cTFBS) and miRNA (PicTar and miRanda) predictions. Although the predictions do not have large overlap (for example, among all the predicted miRNA-target relationships, only 8629 relationships are shared by PicTar and miRanda), the identified network motifs are consistent between these data sets. A total of 11 consistent significant motifs were obtained (Figure 1c and Supplementary Table S2D) from the four data sets: PReMod:miRanda (17), PReMod:PicTar (17), cTFBS:PicTar (19) and cTFBS:miRanda (19) (P-value ∼10−22). In contrast, for random data sets, any subgraph has a chance of 0.0233 [(17/46)2 × (19/46)2] to be a common motif of the four data sets, thus only 1.1 common motifs would be expected by chance. This suggests that the network motifs are robust and tolerant to the noise input.
As an additional test, we simulated false inputs to both miRanda and PicTar data sets by shuffling the IRN following the same method that was used to construct random networks (14,16). The results show that our conclusions are robust to noise additions up to 50% for both sets, meaning that even if 50% of the regulatory relationships are noise, we can still detect the miRNA-regulated TF feedback loop as a statistically significant network motif (see Supplementary Table S3). We also changed the noise level by changing the conservation cutoff for the cTFBS-based data sets. Our conclusion stands even for the available minimal conservation cutoff of 1.64 (see Supplementary Table S4). These results provide additional evidence that the network motifs we identified are significant in describing the relationships of miRNAs and TFs.
CONCLUSION AND DISCUSSION
Together, our results demonstrate that there are two classes of miRNAs with distinct local network topological properties and that these network properties may contribute to the biological functions of the miRNAs. Class I miRNAs, which are regulated by more TFs, are likely functional components of complex developmental programs orchestrated by TF-mediated combinatorial regulation. In contrast, class II miRNAs may be involved in processes that maintain tissue identity in the adult by repressing inappropriate expression of TFs characteristic of earlier states of differentiation or other tissue types. The development process is dynamic and goes through various statuses, while the adult state represents a relatively stable condition. Therefore, class I miRNAs might be regulated by different sets of TFs during development. Since the integrated regulated networks represent a composite picture for regulatory relationships at various conditions, we observed a large number of TFs regulating class I miRNAs.
Our study provides a global picture of regulatory networks involving both transcriptional and post-transcriptional mechanisms. Although our analysis is based on predicted regulatory networks as other studies (14,16), several independent lines of evidence strongly support the validity of our findings. First, we prepared two IRNs based on two sets of predictions and compared the results side by side. Although the two sets of IRNs have different individual predicted relationships, the topological properties are robust to large perturbation and our findings obtained from the two IRNs are largely consistent. Second, our study revealed two different classes of miRNA, distinguished by their participation in different types of network subgraphs. Strikingly, the two classes of miRNAs show high correlation between network topological properties and gene expression patterns, a correlation that is unlikely to be seen from random networks. Third, in vivo TF binding results support our observations on the differential regulation of the two classes of miRNAs.
Previous computational approaches to predicting miRNA functions have frequently been based on the known roles of the putative target genes (3). Due to the negative regulatory nature of miRNAs, there often lacks correlation between the functions of the miRNAs and their target genes. While a few studies on relationship between miRNA and other regulatory factors have revealed several network motifs (13,14,16), there has been less emphasis on examining the functionality of these network motifs. Our results provide a new perspective on analyzing the possible functions of miRNAs, as well as deeper understanding of regulatory networks. As ontological classification and biological understanding of miRNAs is being developed, these findings suggest that one important aspect of miRNA functionality is their network relationship with other regulatory factors. Our results indicate that the biological functions of miRNAs are encoded not only at the post-transcriptional regulatory level, but also at the transcriptional level.
Several factors can contribute to the classification of miRNAs in our study. Since evolutionarily conserved promoters tend to harbor more functional TFBSs, one possibility is that these two classes of miRNAs have distinct conservation levels. We calculated the conservation scores for the promoter of each miRNA and the average conservation scores for the classes I and II are 0.36 and 0.22, respectively. Furthermore, we examined the correlation between average conservation score and the number of TFBSs in promoter of each miRNA. If the difference in number of TFBSs is solely determined by the conservation level, the expected numbers of TFs for two classes of miRNAs would be 11.0 and 6.1, respectively. This difference is much smaller than we observed in the IRNs (23 versus 0.8). Therefore, although conservation contributes to the classification of miRNAs, the classification is not solely determined by conservation. One further complication caused by conservation is that there might be correlation of conservation levels between different genomic regions. For example, a gene with a conserved promoter might tend to have a conserved 3′-UTR. This correlation could cause the over-representation of TF-miRNA co-regulation motif (the second motif in Figure 1c). To exclude the possibility that the network motif is due to the correlation between conservation levels in promoter and 3′-UTR regions, we examined whether we can still recover the network motif if the conservation constraint is removed. We used TRANSFAC matrices to search the binding sites in the promoter sequences with the same match cutoffs we obtained in our previous work (37). We also searched the miRNA binding sites in 3′-UTR sequence of each gene. Even without any conservation constraint, we observed the significant TF-miRNA co-regulation motif (the second motif in Figure 1c). The actual occurrence of the motif is 2.420 × 107, while the expectation from random simulations is (1.927 ± 0.01) × 107. The z-score is 469.
Another possible contribution to the classification of miRNAs is that the two classes of miRNAs could be regulated by different types of TF subfamilies. Since the TF members in the same subfamily tend to have similar binding sites and the sizes of TF subfamilies vary greatly, the miRNAs regulated by TFs from large subfamilies might have more TFBSs in their promoters. However, even if we count only once for the overlapped TFBSs, the numbers of upstream TFBSs targeting classes I and II are 12.2 and 0.7, respectively. This difference is still significant enough to yield two classes of miRNAs. Therefore, the sizes of TF-subfamilies do not contribute significantly to the classification of miRNAs.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
The National Institutes of Health (to J.Q., D.J.Z. and J.T.M.); Guerrieri Center for Genetic Engineering and Molecular Ophthalmology; a gift from Mr and Mrs Robert and Clarice Smith. Funding for open access charges: NIH.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENT
We are grateful to Dr Uri Alon for suggestions.
REFERENCES
- 1.Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–355. doi: 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- 3.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 4.Johnston RJ, Jr, Chang S, Etchberger JF, Ortiz CO, Hobert O. MicroRNAs acting in a double-negative feedback loop to control a neuronal cell fate decision. Proc. Natl Acad. Sci. USA. 2005;102:12449–12454. doi: 10.1073/pnas.0505530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston RJ, Hobert O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature. 2003;426:845–849. doi: 10.1038/nature02255. [DOI] [PubMed] [Google Scholar]
- 6.Li X, Carthew RW. A microRNA mediates EGF receptor signaling and promotes photoreceptor differentiation in the Drosophila eye. Cell. 2005;123:1267–1277. doi: 10.1016/j.cell.2005.10.040. [DOI] [PubMed] [Google Scholar]
- 7.O’Donnell KA, Wentzel EA, Zeller KI, Dang CV, Mendell JT. c-Myc-regulated microRNAs modulate E2F1 expression. Nature. 2005;435:839–843. doi: 10.1038/nature03677. [DOI] [PubMed] [Google Scholar]
- 8.Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science. 2002;298:824–827. doi: 10.1126/science.298.5594.824. [DOI] [PubMed] [Google Scholar]
- 9.Milo R, Itzkovitz S, Kashtan N, Levitt R, Shen-Orr S, Ayzenshtat I, Sheffer M, Alon U. Superfamilies of evolved and designed networks. Science. 2004;303:1538–1542. doi: 10.1126/science.1089167. [DOI] [PubMed] [Google Scholar]
- 10.Shen-Orr SS, Milo R, Mangan S, Alon U. Network motifs in the transcriptional regulation network of Escherichia coli. Nat. Genet. 2002;31:64–68. doi: 10.1038/ng881. [DOI] [PubMed] [Google Scholar]
- 11.Yeger-Lotem E, Sattath S, Kashtan N, Itzkovitz S, Milo R, Pinter RY, Alon U, Margalit H. Network motifs in integrated cellular networks of transcription-regulation and protein-protein interaction. Proc. Natl Acad. Sci. USA. 2004;101:5934–5939. doi: 10.1073/pnas.0306752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mangan S, Itzkovitz S, Zaslaver A, Alon U. The incoherent feed-forward loop accelerates the response-time of the gal system of Escherichia coli. J. Mol. Biol. 2006;356:1073–1081. doi: 10.1016/j.jmb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 13.Tsang J, Zhu J, van Oudenaarden A. MicroRNA-mediated feedback and feedforward loops are recurrent network motifs in mammals. Mol. Cell. 2007;26:753–767. doi: 10.1016/j.molcel.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shalgi R, Lieber D, Oren M, Pilpel Y. Global and local architecture of the mammalian microRNA-transcription factor regulatory network. PLoS Comput. Biol. 2007;3:e131. doi: 10.1371/journal.pcbi.0030131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blanchette M, Bataille AR, Chen X, Poitras C, Laganiere J, Lefebvre C, Deblois G, Giguere V, Ferretti V, Bergeron D, et al. Genome-wide computational prediction of transcriptional regulatory modules reveals new insights into human gene expression. Genome Res. 2006;16:656–668. doi: 10.1101/gr.4866006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cui Q, Yu Z, Purisima EO, Wang E. Principles of microRNA regulation of a human cellular signaling network. Mol. Syst. Biol. 2006;2:46. doi: 10.1038/msb4100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl Acad. Sci. USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Breitling R, Armengaud P, Amtmann A, Herzyk P. Rank products: a simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Lett. 2004;573:83–92. doi: 10.1016/j.febslet.2004.07.055. [DOI] [PubMed] [Google Scholar]
- 19.Benjamini Y, Hochberg Y. Controlling the false discovery Rate: a practical and powerful approach to multiple testing. J.R. Stat. Soc. Ser. B. 1995;57:289–300. [Google Scholar]
- 20.John B, Enright AJ, Aravin A, Tuschl T, Sander C, Marks DS. Human MicroRNA targets. PLoS Biol. 2004;2:e363. doi: 10.1371/journal.pbio.0020363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, et al. Combinatorial microRNA target predictions. Nat. Genet. 2005;37:495–500. doi: 10.1038/ng1536. [DOI] [PubMed] [Google Scholar]
- 22.Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, et al. A genomic regulatory network for development. Science. 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- 23.Stathopoulos A, Levine M. Genomic regulatory networks and animal development. Dev. Cell. 2005;9:449–462. doi: 10.1016/j.devcel.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Cohen SM, Brennecke J, Stark A. Denoising feedback loops by thresholding–a new role for microRNAs. Genes Dev. 2006;20:2769–2772. doi: 10.1101/gad.1484606. [DOI] [PubMed] [Google Scholar]
- 25.Hornstein E, Shomron N. Canalization of development by microRNAs. Nat. Genet. 2006;38(Suppl):S20–S24. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
- 26.Ronshaugen M, Biemar F, Piel J, Levine M, Lai EC. The Drosophila microRNA iab-4 causes a dominant homeotic transformation of halteres to wings. Genes Dev. 2005;19:2947–2952. doi: 10.1101/gad.1372505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sokol NS, Ambros V. Mesodermally expressed Drosophila microRNA-1 is regulated by Twist and is required in muscles during larval growth. Genes Dev. 2005;19:2343–2354. doi: 10.1101/gad.1356105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartel DP, Chen CZ. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 2004;5:396–400. doi: 10.1038/nrg1328. [DOI] [PubMed] [Google Scholar]
- 29.Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. doi: 10.1016/s0092-8674(04)00127-8. [DOI] [PubMed] [Google Scholar]
- 31.Conaco C, Otto S, Han JJ, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc. Natl Acad. Sci. USA. 2006;103:2422–2427. doi: 10.1073/pnas.0511041103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Euskirchen GM, Rozowsky JS, Wei CL, Lee WH, Zhang ZD, Hartman S, Emanuelsson O, Stolc V, Weissman S, Gerstein MB, et al. Mapping of transcription factor binding regions in mammalian cells by ChIP: Comparison of array- and sequencing-based technologies. Genome Res. 2007;17:898–909. doi: 10.1101/gr.5583007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee TI, Jenner RG, Boyer LA, Guenther MG, Levine SS, Kumar RM, Chevalier B, Johnstone SE, Cole MF, Isono K, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125:301–313. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc. Natl Acad. Sci. USA. 2003;100:12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Odom DT, Dowell RD, Jacobsen ES, Nekludova L, Rolfe PA, Danford TW, Gifford DK, Fraenkel E, Bell GI, Young RA. Core transcriptional regulatory circuitry in human hepatocytes. Mol. Syst. Biol. 2006;2:2006 0017. doi: 10.1038/msb4100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl Acad. Sci. USA. 2005;102:16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu X, Lin J, Zack DJ, Qian J. Computational analysis of tissue-specific combinatorial gene regulation: predicting interaction between transcription factors in human tissues. Nucleic Acids Res. 2006;34:4925–4936. doi: 10.1093/nar/gkl595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wienholds E, Kloosterman WP, Miska E, Alvarez-Saavedra E, Berezikov E, de Bruijn E, Horvitz HR, Kauppinen S, Plasterk RH. MicroRNA expression in zebrafish embryonic development. Science. 2005;309:310–311. doi: 10.1126/science.1114519. [DOI] [PubMed] [Google Scholar]
- 39.Mineno J, Okamoto S, Ando T, Sato M, Chono H, Izu H, Takayama M, Asada K, Mirochnitchenko O, Inouye M, et al. The expression profile of microRNAs in mouse embryos. Nucleic Acids Res. 2006;34:1765–1771. doi: 10.1093/nar/gkl096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomson JM, Parker J, Perou CM, Hammond SM. A custom microarray platform for analysis of microRNA gene expression. Nat. Methods. 2004;1:47–53. doi: 10.1038/nmeth704. [DOI] [PubMed] [Google Scholar]
- 41.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. RNA. 2005;11:241–247. doi: 10.1261/rna.7240905. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.