


Abstract
The function of a protein is intimately tied to its subcellular localization. Although localizations have been measured for many yeast proteins through systematic GFP fusions, similar studies in other branches of life are still forthcoming. In the interim, various machine-learning methods have been proposed to predict localization using physical characteristics of a protein, such as amino acid content, hydrophobicity, side-chain mass and domain composition. However, there has been comparatively little work on predicting localization using protein networks. Here, we predict protein localizations by integrating an extensive set of protein physical characteristics over a protein's extended protein–protein interaction neighborhood, using a classification framework called ‘Divide and Conquer k-Nearest Neighbors’ (DC-kNN). These predictions achieve significantly higher accuracy than two well-known methods for predicting protein localization in yeast. Using new GFP imaging experiments, we show that the network-based approach can extend and revise previous annotations made from high-throughput studies. Finally, we show that our approach remains highly predictive in higher eukaryotes such as fly and human, in which most localizations are unknown and the protein network coverage is less substantial.
INTRODUCTION
For a protein to operate properly, it must reside in the correct compartment of a cell. Knowing the subcellular localization of a protein, therefore, is an important step to understanding its function (1,2). In budding and fission yeast (1–4), systematic protein localization experiments have been carried out through GFP fusions to each open reading frame at the 3′- or 5′-end. Such studies have not yet been performed in higher eukaryotes such as Caenorhabditis elegans, Drosophila melanogaster or mammals, due to the larger proteome sizes and the technical difficulties associated with protein tagging in those species (5–7). In the interim, reliable and efficient computational methods are required to predict the subcellular localization of a newly identified protein.
A considerable number of classification methods have been developed for this purpose (5–24). Typically, these algorithms input a list of features with which to characterize a protein, such as its molecular weight, amino acid content, codon bias, hydrophobicity, side-chain mass and so on. During the training phase, they learn to recognize which features, or patterns of features, are best able to classify a set of gold-standard proteins whose localizations are well known. To date, amino acid content has been a very successful and widely used feature (5,6,8,11–16). Other informative features have been protein sorting signal motifs near the N-terminus (18), as well as protein sequence motifs (7,9–12,16,24) and Gene Ontology terms (5). Classification of these features has relied on a variety of algorithms, including Least Distance Algorithms (20,21), an Artificial Neural Network (10), a Nearest Neighbor approach (5,14), a Markov Model (22), a Bayesian Network approach (9), Support Vector Machines (SVMs) (13,15,16) and Support Vector Data Description (SVDD) (6).
Early methods attempted to classify proteins into a small number of compartments, e.g. intracellular versus extracellular (19). More recently, many compartmental localizations have been defined, including not only membrane-enclosed organelles but also categories such as spindle pole or microtubule association. Current prediction algorithms in yeast cover as many as 22 distinct cellular localizations (5,6). Not surprisingly, approaches which limit their predictions to smaller numbers of localizations have performed better than approaches which attempt to predict many. Moreover, most of these studies have demonstrated their predictions assuming a single localization per protein within a single species such as yeast. Therefore, some open challenges for new methods development are to: (i) increase the classification accuracy when predicting across many cellular compartments; (ii) allow for multiple predictions per protein; and (iii) stabilize performance across many species, some of which may have far fewer data available for training and classification than does yeast.
The recent availability of large protein–protein interaction networks in yeast, fly, worm and human (25–34) provides one means to at least partially address these challenges. To interact physically, two proteins must localize to the same or adjacent cellular compartments, suggesting that interaction may serve as an indicator for co-localization. Integrated analysis of genome-wide protein localization and protein–protein interaction data in Saccharomyces cerevisiae (SC) supports this hypothesis, showing that interactions are strongly enriched between co-localized proteins (1). However, there have been relatively few attempts to use interacting proteins in the prediction of localization (7). Moreover, in recent years the numbers of protein interaction measurements have increased exponentially. This increase has been driven by various proteomics technologies, such as co-immunoprecipitation followed by tandem mass spectrometry, the yeast two-hybrid system and its variants, and large screens for genetic interactions (26,35,36). As a result, there were more than 170 000 protein interactions in the public databases as of this writing (http://www.ebi.ac.uk/intact/); prior to 2002 there were no more than several hundred. Given these developments, protein interactions have become a basic feature available for many proteins. It is therefore of significant interest to ask whether, and to what extent, protein interaction networks can impinge on the prediction of subcellular localization.
Here, we pursue a protein network-based approach for summarizing diverse sequence and functional information of interacting proteins into useful predictors of localization. A variant of the k-Nearest Neighbors classification algorithm (5,14) is developed to exploit the synergy between the physical characteristics of an individual protein and the properties of its interacting neighbors. After generating useful features based on single proteins and their neighbors, the method extracts the best combination of feature sets for each cellular localization. We apply this network-based prediction method to predict the localizations of 5681 SC proteins, in which a protein is not given a single annotation but is characterized by its predicted distribution across 22 subcellular compartments. Through further GFP imaging experiments, we show that the predictions can provide novel leads even when the localization of a protein has already been measured experimentally.
MATERIALS AND METHODS
Overview of protein network-based localization prediction
We integrated three major types of features to predict the localization of a protein, which we term S, N and L (Figure 1). S (single protein) features, nine in total, were used to describe various characteristics of the protein. Seven of the nine S features were extracted from the protein's primary sequence, depicting its amino acid composition and chemical properties. Occurrences of known signaling motifs in the primary protein sequence, downloaded from cross-references in UniProt or FlyBase, was also used as one S feature. The final S feature encoded functional annotations of the protein downloaded from the Gene Ontology database. N and L are network-dependent: N summarizes the S features of the protein's extended network neighborhood, while L represents the distribution of known localizations in the neighborhood. Our modified k-Nearest Neighbor classifier, called DC-kNN, integrates the diverse information of all these features for each localization in a Divide-and-Conquer manner, in which a single kNN classifier is built using each type of feature and the predictions are made through majority voting of the kNN classifiers. A protein can be assigned to multiple localizations if the protein has an estimated probability over a meaningful threshold for each localization.
Figure 1.

Schematic overview of the integrated network-based framework. (a) Generation of single-protein feature vectors (Ss). Nine kinds of Si (AA, diAA, gapAA, three kinds of chemAA, pseuAA, Motif and GO) were generated for each protein Pi based on its sequence, chemical properties, motifs and functions. (b) Calculation of Neighbors’ Significance Matrixes (NSMs). These were calculated based on the number of distinct localizations covered by proteins falling along the path with the highest weight from a target protein to a neighbor protein (see Materials and methods section). (c) Calculation of PLCPs. They were calculated based on a weighted counting with normalization (see Materials and methods section). (d) Generation of network feature vector NiD. Each NiD was generated using up to D-th neighborhood's Ss with neighbors’ significance degrees from NSMs. (e) Generation of network feature vector LiD. Each LiD was generated using Pi's network neighbors up to distance D, weighted by NSMs and PLCPs to reflect each neighbor's significance and the conditional probabilities of interactions between localization pairs, respectively. (f) Model selection for each localization. The best combination of feature sets was selected for each localization based on a forward approach with the DC-kNN classifier. (g) Prediction of unknown localizations. After generating all feature vectors using all known localization and network information, a confidence degree and a decision (on whether an unknown protein has a specific localization or not) were computed for each localization.
To generate the network features (N and L), we pooled protein–protein interactions for SC from the BioGRID (BiG) (37), the Database of Interacting Proteins (DIP) (38), and the Saccharomyces Genome Database (SGD) (39). Known localizations of 3914 proteins from Huh et al. (1) were used for L features (Table 1). Most interactions (>57%) in the protein networks connected known co-localized protein pairs, which implies a high degree of correlation between interaction and localization (Figure 2a and Supplementary Table S5; P << 10−16 compared to 100 random networks of same topology). Among the three databases, BiG has the largest coverage and highest enrichment of co-localized proteins. We also found that proteins in some localizations (e.g. endoplasmic reticulum) tend to interact with proteins in different localizations (e.g. vacuole). To reflect the possibility of interacting pairs being in different localizations, we incorporated such conditional probability into the L features.
Table 1.
Data sources integrated to predict localization information
| Species | Data set | Proteins | Localizations |
|---|---|---|---|
| Localization | |||
| Saccharomyces cerevisiae (22 localizationsa) | Localization-known proteins | 3914 | 5184 |
| Localization-known and having interactions | 3206 | 4284 | |
| Ambiguous | 237 | 189 335 | |
| Localization-unknown | 1530 | 0 | |
| Drosophila melanogaster (12 localizationsb) | Localization-known | 2187 | 2398 |
| Localization-known and having interactions | 1610 | 1778 | |
| Localization-unknown and having interactions | 5656 | 0 | |
| Homo sapiens (13 localizationsc) | Localization-known | 4570 | 5251 |
| Localization-known and having interactions | 2684 | 3093 | |
| Localization-unknown and having interactions | 3767 | 0 | |
| Species | Data set | Proteins | Interactions |
| Interaction | |||
| Saccharomyces cerevisiae | BioGRID | 5184 | 70 700 |
| DIP | 4931 | 17 471 | |
| SGD | 5395 | 56 035 | |
| Drosophila melanogaster | BioGRID | 7545 | 25 463 |
| DIP | 7038 | 20 719 | |
| Homo sapiens | BioGRID | 7378 | 20 968 |
| Feature | Description | ||
| Protein feature | |||
| Sequences | UniProt (for SC and HS) and FlyBase (for DM) | ||
| Chemical property | Hydrophobicity, hydrophilicity and side-chain mass | ||
| Motifs | InterPro | ||
| Functions | InterPro and GO | ||
Here, we only considered the proteins with sequence information.
a22 SC localizations are actin, bud, bud neck, cell periphery, cytoplasm, early Golgi, endosome, ER, ER to Golgi, Golgi, late Golgi, lipid particle, microtubule, mitochondrion, nuclear periphery, nucleolus, nucleus, peroxisome, punctate composite, spindle pole, vacuolar membrane, vacuole.
b12 DM localizations are actin, cell periphery, centrosome, cytosol, ER, golgi, lysosome, mitochondrion, nucleolus, nucleus, peroxisome, vacuole.
c13 HS localizations are actin, cell cortex, centrosome, cytosol, ER, golgi, lysosome, mitochondrion, nucleolus, nucleus, peroxisome, plasma membrane, vacuole. Further details regarding localizations and interactions of SC, DM and HS are in Supplementary Figure S1 and Tables S1–S4.
Figure 2.

Correlation between known localizations and protein interactions of yeast proteins. (a) The number of interactions (inside the circles) and the fraction of interactions whose proteins share localization information (outside the circles) of three interaction databases: BiG, DIP and SGD. (b–d) The PLCPs of BiG, DIP and SGD, respectively. Given a protein at a particular localization (row), each cell corresponds to the conditional probability of the localization of its interacting partners (column). The squares on the diagonal (or off-diagonal) indicate the locations with relatively low (or high) degrees of location-sharing interactions within (or between) locations; the dotted circles on the diagonal indicate different patterns among three interaction databases for proteins in the lipid particle.
Localization and network data
For SC, we downloaded the localization data of Huh et al. (1), who used GFP-tagging experiments to annotate 3914 proteins with up to 22 distinct localizations (Table 1). The 22 localizations are actin (actin cytoskeleton), bud, bud neck, cell periphery, cytoplasm, early Golgi (early Golgi/COPI), endosome, ER (endoplsmic reticulum), ER to Golgi (endoplasmic reticulum to Golgi), Golgi (Golgi apparatus), late Golgi (late Golgi/clathrin), lipid particle, microtubule, mitochondrion, nuclear periphery, nucleolus, nucleus, peroxisome, punctate composite, spindle pole, vacuolar membrane and vacuole (see Supplementary Table S1 for more information). The remaining 1530 SC proteins have no known localization at present and were designated ‘localization-unknown’. For DM and HS, we first downloaded all proteins which had sequence information in FlyBase and UniProt, respectively. We assigned localization information to the 2187 DM and 4570 HS proteins with GO cellular component annotations. To define the corresponding set of localization unknown proteins, we identified 5656 DM and 3767 HS proteins in the BiG protein network with sequences available but that did not have known localizations, i.e. missing GO annotations. For the interaction data, we downloaded the contents of BiG, DIP, and SGD for SC, of BiG and DIP for DM and of BiG for HS.
Generation of single protein feature vectors (S)
Using sequences from UniProt for SC and HS and FlyBase for DM, we generated three kinds of amino acid features for each protein: amino acid composition frequencies (AA), pair-coupled amino acid frequencies (diAA) and pair-coupled amino acid frequencies with a gap (length = 1) (gapAA). AA is a vector of length 20; the diAA and gapAA vectors contain 400 elements enumerating frequencies over all ordered amino acid pairs. For incorporating chemical properties, we generated three kinds of chemical amino acid compositions (chemAA) using normalized hydrophobicity (40) (HPo), hydrophilicity (41) (HPil) or side-chain mass (42) (SCM), respectively (see Supplementary Table S6 for the normalized values of each chemical property). The chemAA compositions were computed by scanning a window of length k along the amino acid sequence (1 ≤ k ≤ 40) and recording the mean squared difference in the chemical property value across all window positions. The k-th element of chemAA using hydrophobicity was defined as:
where HPo(Rl) is the normalized hydrophobicity value of the l-th residue, and n is the length of the protein sequence. The pseudo-amino acid composition (pseuAA) (43) was generated by combining the three chemical properties into one. Formally stated:
where U = (HPo(Rl) - HPo(Rl + k))2, V = (HPil(Rl) - HPil(Rl + k)2 and W = (SCM(Rl) - SCM(Rl + k)2. For the Motif and GO feature vectors, we downloaded InterPro Motifs and GO information from UniProt (SC and HS proteins) and FlyBase (DM proteins). After extracting the motif or GO set using all localization-known proteins for each species, we constructed a binary feature vector (5,6) in which each element was set to ‘1’ if the protein had the corresponding motif (or GO) annotation, otherwise ‘0’. Note that GO terms also include cellular component annotations, which are also used as class labels especially for DM and HS. Thus, to reduce circularity we omitted these annotations while generating the GO feature vectors even though most previous studies used all three branches of GO terms (5,44).
Pair-localizations’ conditional probability
We calculated a pair-localizations’ conditional probability (PLCP) matrix for each protein network (BiG SC, DIP SC, SGD SC, BiG DM, DIP DM and BiG HS) to capture the probability of a protein being in localization lj given that its interaction partner is in localization li:
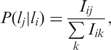 |
Iij is the normalized number of interactions between protein pairs spanning (li and lj). Iij is defined as:
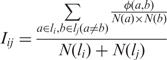 |
where N(li) is the total number of proteins in localization li, N(a) is the number of localizations in protein a, and φ (a, b) is ‘1’ if there is an interaction between proteins a and b; otherwise, zero.
Network-dependent interacting protein-group feature generation
In this study, we generated two kinds of network feature vector: ND and LD. ND of protein Pi is defined as the weighted average of the S feature vectors over proteins up to distance D from Pi in the network, including Pi itself (called the D-th neighborhood of Pi and represented by the variable CiD):
The weightings wki, which make up the Neighbors’ Significance Matrix (Figure 1), represent the significance of neighbor Pk, defined as:
where Ψki is the number of distinct localizations covered by proteins along a path from Pi to Pk, and ρ is a pseudo-counter for handling incompleteness of localization data (in this study ρ = 1 for SC, ρ = 2 for HS and ρ = 3 for DM; different values were used because the portions of known localizations for DM and HS are less than that of SC—see Figure 6b). Note that we assigned max wki among multiple paths from Pi to Pk and assigned less weight on a neighbor protein that interacts with other proteins having many distinct localizations.
Figure 6.

Performance of predicting yeast protein localization as the available interaction (a) or localization (b) data are eroded. In (a), interactions were randomly deleted to reduce the average degree of the yeast PPI network to that specified (x-axis). In (b), known yeast protein localizations were randomly deleted. In either case, AUC was estimated using the leave-one-out approach. To avoid over fitting, the selected feature sets were taken from Supplementary Figure S6 and not re-optimized. Worm, fly, and human were mapped onto these yeast performance curves using the average degree of their available protein networks (a) or the fraction of known localizations for network proteins (b). The blue diamond represents the performance of a conventional approach using all nine single protein features without feature set selection. The red ‘X’ marks denote the performance of the proposed method when applied to recover known protein localizations in fly and human, using LOOCV.
LiD is a vector representing the probability that Pi has each of the 22 localizations, given the D-th neighborhood of protein Pi and considering the probabilities of interaction between proteins in distinct localization pairs:
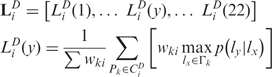 |
where lx is one element of the localization set Γk of Pk, and p(ly |lx) is the conditional probability of label ly given the label lx (from the PLCP matrix). Note that we choose the maximum value among multiple choices for the conditional probability of each localization, owing to the multiple localization property. Moreover, to satisfy the symmetric property, we also include the single protein feature vector of input protein Pi when generating network feature vectors.
Divide-and-Conquer k-Nearest Neighbor Classifier
The DC-kNN has three main steps: dividing, choosing, and synthesizing. In the dividing step, the full feature vector is divided into m meaningful feature subvectors. In this study, each single protein feature set and each network-dependent protein group feature set were treated as meaningful sub-vectors, yielding m = 69 subvectors in total for yeast: the 9 S vectors (AA, diAA, gapAA, three kinds of chemAA, pseuAA, Motif, GO), the 54 N vectors [= 9 S vectors × 2 (up to second neighborhood) × 3 (the number of network databases)], and the 6 L vectors [= 2 (up to second neighborhood) × 3 (the number of network databases)]. In the choosing step, the k-nearest neighbors are chosen for each protein and subvector (in this study, k = 5). Finally, the synthesizing step averages the m sets of k neighbors with a weight on each set, and it generates a confidence for each label by means of a normalization process with m and k. Formally, the confidence mul for label l is defined as:
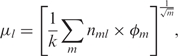 |
where nml is the number of k-nearest neighbors that have label l according to sub-vector m. 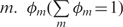 is the weight of the m-th subvector. Instead of using all sub-vectors, DC-kNN finds the best combination of feature subvectors for each label, based on a forward approach. At each iteration, DC-kNN chooses the most predictive feature subvector among those remaining, i.e. the vector that shows the best AUC when added to the previously selected feature subvectors. In the first iteration, feature subvectors are used individually for finding the most predictive one. For the weights φm, DC-kNN uses the AUC obtained using each feature subvector alone. DC-kNN produces a confidence degree (0–1) and a decision on whether a protein has a specific localization or not, using a threshold based on a false positive rate (in this study, <0.01).
is the weight of the m-th subvector. Instead of using all sub-vectors, DC-kNN finds the best combination of feature subvectors for each label, based on a forward approach. At each iteration, DC-kNN chooses the most predictive feature subvector among those remaining, i.e. the vector that shows the best AUC when added to the previously selected feature subvectors. In the first iteration, feature subvectors are used individually for finding the most predictive one. For the weights φm, DC-kNN uses the AUC obtained using each feature subvector alone. DC-kNN produces a confidence degree (0–1) and a decision on whether a protein has a specific localization or not, using a threshold based on a false positive rate (in this study, <0.01).
Microscopic localization analysis
Yeast cells grown to mid-logarithmic phase in SC medium were microscopically analyzed in 96-well glass bottom microplates (Whatman, Florham Park, NJ, USA) pretreated with concanavalin A (Sigma, St. Louis, MO, USA) to ensure cell adhesion. Microscopy was performed on a Zeiss Axiovert 200M inverted microscope with a Plan-NeoFluar 100×/1.3 NA oil immersion objective. Images were recorded on a Zeiss Axiocam MRm with 2 × 2 binning. Fluorescence images for GFP were taken using a standard fluorescein isothiocyanate filter set (excitation band pass filter, 450–490 nm; beam splitter, 510 nm; emission band pass filter, 515–565 nm).
RESULTS AND DISCUSSION
Network information improves localization prediction in yeast
We compared the predictive performance of different features during prediction of localization: S features only, N features only, L features only, all three features together (S + N + L) and random guesses. DC-kNN classification was used in all cases, and performance was evaluated using the technique of leave-one-out cross-validation (LOOCV). In every run of LOOCV, the known localization of one of the 3914 SC proteins in Huh et al. (1) was designated as ‘test’ data and withheld during classifier training.
Three metrics, Top-K, Total and Balanced, were used to summarize the performance of the 3914 runs. The Top-K measure is the fraction of correctly predicted runs, in which the prediction is considered correct if at least one of the known localizations of the test protein is included in the top-K predicted localizations. We used K = 3 assuming most yeast proteins have less than or equal to three localizations (6). The Total measure is the fraction of correctly predicted localizations in the 3914 runs, counting all predictions for all proteins. The Balanced measure calculates the averaged fraction of correctly predicted localizations in distinct localizations (see Supplementary Figure S2 for the metrics used). The Balanced measure is used because predictions based on localization categories with few proteins are usually not as good as predictions based on localization categories with many proteins annotated. For the random guesses, we randomly permuted the assignment of localizations to proteins preserving both the number of localizations per protein and the number of proteins per localization; the measures (Top-3, Total and Balanced) were averaged over 30 runs.
Although all classifiers were clearly better than random (based on the background distribution of proteins in the 22 localizations; Figure 3a), the combination of all three features provided the highest predictive accuracy regardless of the measure. Moreover, according to the Balanced metric, either of the network features N or L achieved higher accuracy than S features. These results suggest that when the number of proteins was not sufficient to learn sequence-level rules for classifying smaller compartments like ‘bud’ or ‘peroxisome’, interaction networks provided one alternative to amplify the weak signals encoded in the individual protein sequences.
Figure 3.

Usefulness of protein interaction networks. (a) The performance of five cases, including (i) random guess of localization, (ii) S features only, (iii) N1 only, (iv) L1 only and (v) all three kinds of features. (b–e) The performance of the ND features for amino acid frequencies (b), chemical amino acid properties (c), and GO terms (d) as well as performance of the feature LD (e). Performance is based on the five interaction networks BiG, DIP, SGD, Combined, and Random (different color curves). The performance of other ND network features is shown in Supplementary Figure S3. The x-axis is the radius of neighborhood D; D = 0 means only the single protein feature vector S was used, which is a conventional approach. For Combined, the three interactome datasets BiG, DIP and SGD were pooled into a single network. For Random, localizations were randomly assigned on the BiG network. The solid lines and the dotted lines represent the Total and Balanced measures, respectively.
In all of the above cases, the network neighborhood was defined as a protein's immediate interactors (N1 or L1, designating network distance = 1). Next, we explored the impact of expanding a protein's network neighborhood to incorporate not only immediate neighbors, but all proteins at network distances up to and including distance D. As seen in Figure 3b–d, incorporating network information up to distance 2 generally improved the accuracy of the amino acid, chemical AA properties and GO features. However, network distances larger than 2 did not have a significant increase in performance, which is understandable given the diameter of the yeast network was six. Similar findings were observed for the L features (Figure 3e). The L features alone (Total accuracies range from 60% to 66% depending on the network used) outperformed any kind of S feature (42–55%), but their accuracies did not increase significantly when more than distance 2 neighbors were included.
Interestingly, a network pooled from all three interaction databases did not improve the performance over any single network alone (Figure 3b–e). It achieved equivalent performance as the SGD network and sometimes worse than the BiG network, indicating that the network quality played a bigger role than the coverage in generating useful N and L features. Overall, the BiG network had the best performance.
The best combination of single-protein features and network features for each localization
Using a subset of features may reduce the possibility of overfitting and therefore lead to a more robust classifier (45,46). To further optimize the predicted localizations, we applied a forward selection which combined feature sets of high predictive power from a pool of S, N and L features from up to distance 2 network neighborhoods. During feature set selection, we used the common measure of Area Under receiver operator characteristic Curve (AUC) (47,48) to rank the predictive power of features and also to evaluate the performance of the resulting classifiers. To reduce overfitting further, we withheld two examples from each training round of cross-validation, and then used one for feature selection and one for performance reporting. Without feature selection, DC-kNN with all single-protein S features achieved 0.65 AUC averaged from the prediction of the 22 compartments. This accuracy increased to 0.79 if feature set selection for each localization was applied during classifier training using all single-protein features (Figure 4a).
Figure 4.

Performance of the network-based approach. (a) The averaged AUC values of three cases: (i) all S features without feature set selection (FSS), (ii) all S features with FSS and (iii) all S, N and L features with FSS for each localization. (b) Performance comparison with two well-known methods. Performance is computing using the Total versus Balanced metrics (top three versus bottom three bars).
Lastly, we explored the effect of selecting the best combination of single-protein features S and network features N and L for each localization separately. We found that selecting different features per localization using single and network features resulted in a dramatic increase in performance, with average AUC of 0.94 for the 22 localizations (see Supplementary Figures S4–S6 for the forward feature set selection, the ROC curves of each approach, and the selected feature sets for each compartment, respectively). This means that the combinatorial effect between single-protein features and network features is indispensable for capturing functional characteristics of proteins.
Another issue in the localization prediction of proteins might be the influence of homologous data in training data. To evaluate the influence of sequence similarity in the developed network-based approach, we checked the performance of DC-kNN with only nonhomologous yeast proteins (see Supplments.doc for more information). We observed similar performance (average AUC value of 0.94) with the previous result with all known yeast proteins. It implies that the network-based DC-kNN is insensitive to the presence of close sequence homologs in a training data set.
Novel localization predictions can revise previous high-throughput experiments
Based on its good performance, we applied this last method to comprehensively predict 5184 localizations for 3914 yeast proteins. Although these yeast predictions were in good agreement with the GFP localization experiments performed by Huh et al. (1) (as expected since the Huh data were used as features), to our surprise we found that for 61 proteins the predicted localizations were novel (Supplementary Tables S7 and S8). For example, Noc4/Ypr144c and Utp21/Ylr409c were localized to the nucleus by Huh et al. (1), whereas our predictions produced the highest signal (5 × 10−4 false positive rate for Noc4 and 1 × 10−3 for Utp21) at the nucleolus. To determine whether a nucleolar localization could be corroborated experimentally, we re-examined the strains containing GFP-tagged Noc4 and Utp21 using fluorescence microscopy (see Materials and methods section). The resulting images show that both proteins do indeed accumulate at the nucleolus with some spread to the nucleoplasm (Figure 5a and b). In some cases, therefore, it appears that network-based predictions can correct or complement the image readouts of high-throughput experiments. This power owes mainly to the fact that our framework synthesizes evidence from multiple interacting partners. For example, Noc4 interacts with many other proteins in the nucleolus, hence the prediction (Figure 5c).
Figure 5.


Validation of novel localizations for yeast proteins. New localization images for two yeast proteins, Noc4/Ypr144c (a) and Utp21/Ylr409c (b), for which the network-based prediction (nucleolus) was different than previously measured (nucleus) (1). The near-complete overlap area between the GFP and RFP images (‘Merge A’), marking the protein and nucleolus, respectively, is consistent with a nucleolar localization (Sik1-RFP was used as a nucleolus marker). Here, DAPI is used for marking the nucleus, and ‘Merge B’ is the overlap among GFP, DAPI and RFP images. (c) Proteins that interact with Noc4/Ypr144c and their localizations. The values in the upper-left box represent the interacting protein pairs’ localization purity (IPLP, or enrichment) among interacting protein pairs for distinct localizations (see the ‘Supplements.doc’ for more information). Panel (c) is drawn using Cytoscape (55).
In Huh et al. (1), 237 SC proteins had ambiguous image readouts for determining their localizations. Among these, 80 proteins were nonetheless annotated with ‘low confidence’ localizations and 157 were never annotated (1). Moreover, an additional 1530 yeast proteins could not be localized by the previous experiments owing to low GFP signals (1). We used the DC-kNN network-based classifier to predict the localization of all of these proteins (Supplementary Figure S7 and Tables S9–S10). For the 80 ‘low confidence’ proteins in Huh et al. (1), our predicted localizations significantly overlapped with their assignments (Supplementary Figure S7c; P < 2.0 × 10−31 based on a hypergeometric distribution). We also found significant overlap between our predictions and the literature-curated annotations recorded in the cellular component branch of the GO database (see Supplementary Figure S7d and Table S11 for the overlap degree and the mapping relationship between 22 localizations and GO terms, respectively; P < 2.6 × 10−71).
Comparison with previous methods
We compared DC-kNN with two popular methods, ISort (5) and PSLT2 (7,17), for the prediction of yeast protein localization. ISort (5) is one of most comprehensive sequence-based methods and also the first of the few machine-learning-based methods to predict more than 15 compartments. PSLT2 (7) is a method that previously incorporated protein interaction networks into localization prediction. In the original PSLT2 paper (7), the authors demonstrated its accuracy in predicting SC proteins in nine general compartments. Therefore, we ran our method and ISort (5) for the same nine compartments with the same data used in the PSLT2 paper (7). Using both sequence and network features, DC-kNN significantly outperformed ISort and PSLT2 based on the Total and Balanced measures [Top-K and AUC measurements are not available in the PSLT2 paper (7)] (Figure 4b). Between ISort and PSLT2, ISort had higher Total accuracy but PSLT2 surpassed ISort in terms of the Balanced measure, which down-weights bigger compartments with more proteins (see Supplementary Table S12 for the performance of each compartment among three methods).
Extrapolation to higher eukaryotes
Given the power of protein network information to predict protein localization, an important question is whether a network-based approach can be extended to other eukaryotes with less network coverage than yeast. To address this question, we ran a series of simulations in which increasing numbers of interactions in the yeast network were successively removed. As expected, the performance of DC-kNN decreased as less network information was available (Figure 6a). However, the rate of decrease was gradual, such that when the average degree of the network was reduced by approximately half (27 versus 13), the associated decrease in AUC was 0.94–0.91. At an average degree of five, the AUC was still ∼0.89. We note that the available protein networks for worm, fly and human are in this range (average degrees from 3 to 7; see Figure 6a). Thus, these results suggest that the protein network-based DC-kNN will achieve high accuracy in predicting protein localization in these species. At average degrees below three, the performance dropped more precipitously to approach 0.79, the AUC achievable without network information (S features only).
Another potential problem is that in eukaryotes other than yeast, few known protein localizations are available for classifier training. Thus, our second simulation was to test the robustness of prediction as the number of proteins with known localization data was decreased. As expected, the AUC decreased when less localization data were available (Figure 6b), but with an even slower rate of degradation than that observed for loss of interaction data (Figure 6a). Dramatically, with only 1% of network proteins having known localizations, the network-based approach still achieved ∼0.83 AUC, which is significantly higher than the ∼0.65 AUC obtained from a conventional sequence-based approach. The improvement results from both the consideration of network features and the feature selection implemented in DC-kNN. These simulations suggest that the proposed network-based method can be applied to predict localization of proteins in higher eukaryotes where only little protein network information is available and only few proteins have previously determined localizations.
To cross-check these simulation results, we applied the proposed framework to predict protein localizations in both fly and human. The currently available fly and human networks, containing 25 463 and 20 968 interactions among 7545 and 7378 proteins, respectively, were downloaded from BiG. Because no high-throughput experimental studies have been conducted to measure the localizations of fly and human proteins, we trained the classifier using literature-curated protein localizations documented in the Cellular Component branch of the GO database. According to GO, 1709 fly and 2684 human proteins in the BiG network have known localizations covering 12 (fly) and 13 (human) cellular compartments in total. Approximately 77% (fly) or 64% (human) proteins had no known localizations, in contrast to only 33% of proteins in yeast (Supplementary Table S13). Nonetheless, consistent with our above simulation results, DC-kNN achieved ∼0.88 (fly) or ∼0.95 (human) AUC in cross validation (red ‘X's in Figure 6b). In terms of network coverage, the performance in human was slightly higher than predicted in simulation (red ‘X's in Figure 6a). (See Supplementary Figure S8 for forward feature set selection of fly and human and Supplementary Figures S9–S10 for selected feature sets for each compartment.) Overall, we predicted 7058 (fly) and 4366 (human) new localizations for proteins with no localizations previously known (see Supplementary Tables S14–S15 for all predicted results and Supplementary Figure S11 for distribution of the results).
In this work, we obtained an average AUC of 0.94 for yeast, 0.88 for fly and 0.95 for human (see the ‘Supplements’ for the discussion of the localization-specific predictions of yeast, fly and human proteins). The high performance of the proposed approach results from both the consideration of network features, in addition to single protein features, and the feature selection implemented in DC-kNN. The performance may be further improved by efforts to specify further details about the type of relationship each interaction represents. For instance, interactions fall into specific biological categories, including physical binding events, genetic interactions such as synthetic lethals or suppressor relationships, and functional associations. Each of these interaction types may have different capacity to predict specific protein localizations. Moreover, protein interactions are dynamic according to external stimuli or environmental conditions (49,50). Where condition-specific expression or interaction data are available, it would be of high interest to predict dynamic changes in protein localization. It is increasingly recognized that such changes are the cornerstone of many cellular regulatory events (51–54), such as the translocation of transcription factors to the nucleus or the trafficking of proteins to the vacuole or cellular membrane.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Korea Research Foundation Grant funded by the Korean Government (MOEHRD) (KRF-2006-352-D00171, partially); NIGMS (GM070743 to T.I.); Korea Science and Engineering Foundation (#2006-04090 to B.L.); 21C Frontier Functional Proteomics Project (FPR08A1-060) funded by the Ministry of Education, Science and Technology, Republic of Korea. Funding for open access charge: NIH/NIGMS (NIGMS is the National Institute of General Medical Sciences); grant no. 1 R01 GM070743.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr Michael Hallett at McGill University and Dr Jenn-Kang Hwang at National Chiao Tung University for sharing their localization data sets, and Dr Kwang Hyung Lee and Dr Doheon Lee at KAIST for valuable discussion on this research. Our special thanks to Hyun-Min Kang at UCLA for advice on statistical analysis of results and Hye-Young Cho at KAIST for preparation of localization data sets.
REFERENCES
- 1.Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 2.Matsuyama A, Arai R, Yashiroda Y, Shirai A, Kamata A, Sekido S, Kobayashi Y, Hashimoto A, Hamamoto M, Hiraoka Y, et al. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat. Biotechnol. 2006;24:841–847. doi: 10.1038/nbt1222. [DOI] [PubMed] [Google Scholar]
- 3.Ross-Macdonald P, Coelho PS, Roemer T, Agarwal S, Kumar A, Jansen R, Cheung KH, Sheehan A, Symoniatis D, Umansky L, et al. Large-scale analysis of the yeast genome by transposon tagging and gene disruption. Nature. 1999;402:413–418. doi: 10.1038/46558. [DOI] [PubMed] [Google Scholar]
- 4.Kumar A, Agarwal S, Heyman JA, Matson S, Heidtman M, Piccirillo S, Umansky L, Drawid A, Jansen R, Liu Y, et al. Subcellular localization of the yeast proteome. Genes Dev. 2002;16:707–719. doi: 10.1101/gad.970902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou KC, Cai YD. Predicting protein localization in budding yeast. Bioinformatics. 2005;21:944–950. doi: 10.1093/bioinformatics/bti104. [DOI] [PubMed] [Google Scholar]
- 6.Lee K, Kim DW, Na D, Lee KH, Lee D. PLPD: reliable protein localization prediction from imbalanced and overlapped datasets. Nucleic Acids Res. 2006;34:4655–4666. doi: 10.1093/nar/gkl638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott MS, Calafell SJ, Thomas DY, Hallett MT. Refining protein subcellular localization. PLoS Comput. Biol. 2005;1:e66. doi: 10.1371/journal.pcbi.0010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhasin M, Raghava GP. ESLpred: SVM-based method for subcellular localization of eukaryotic proteins using dipeptide composition and PSI-BLAST. Nucleic Acids Res. 2004;32:W414–W419. doi: 10.1093/nar/gkh350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drawid A, Gerstein M. A Bayesian system integrating expression data with sequence patterns for localizing proteins: comprehensive application to the yeast genome. J. Mol. Biol. 2000;301:1059–1075. doi: 10.1006/jmbi.2000.3968. [DOI] [PubMed] [Google Scholar]
- 10.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 11.Gardy JL, Spencer C, Wang K, Ester M, Tusnady GE, Simon I, Hua S, deFays K, Lambert C, Nakai K, et al. PSORT-B: Improving protein subcellular localization prediction for Gram-negative bacteria. Nucleic Acids Res. 2003;31:3613–3617. doi: 10.1093/nar/gkg602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35:W585–W587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hua S, Sun Z. Support vector machine approach for protein subcellular localization prediction. Bioinformatics. 2001;17:721–728. doi: 10.1093/bioinformatics/17.8.721. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, Li Y. Prediction of protein subcellular locations using fuzzy k-NN method. Bioinformatics. 2004;20:21–28. doi: 10.1093/bioinformatics/btg366. [DOI] [PubMed] [Google Scholar]
- 15.Park KJ, Kanehisa M. Prediction of protein subcellular locations by support vector machines using compositions of amino acids and amino acid pairs. Bioinformatics. 2003;19:1656–1663. doi: 10.1093/bioinformatics/btg222. [DOI] [PubMed] [Google Scholar]
- 16.Shatkay H, Hoglund A, Brady S, Blum T, Donnes P, Kohlbacher O. SherLoc: high-accuracy prediction of protein subcellular localization by integrating text and protein sequence data. Bioinformatics. 2007;23:1410–1417. doi: 10.1093/bioinformatics/btm115. [DOI] [PubMed] [Google Scholar]
- 17.Scott MS, Thomas DY, Hallett MT. Predicting subcellular localization via protein motif co-occurrence. Genome Res. 2004;14:1957–1966. doi: 10.1101/gr.2650004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakai K, Horton P. PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem. Sci. 1999;24:34–36. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- 19.Nakashima H, Nishikawa K. Discrimination of intracellular and extracellular proteins using amino acid composition and residue-pair frequencies. J. Mol. Biol. 1994;238:54–61. doi: 10.1006/jmbi.1994.1267. [DOI] [PubMed] [Google Scholar]
- 20.Wang M, Yang J, Xu ZJ, Chou KC. SLLE for predicting membrane protein types. J. Theor. Biol. 2005;232:7–15. doi: 10.1016/j.jtbi.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 21.Chou KC, Elrod DW. Protein subcellular location prediction. Protein Eng. 1999;12:107–118. doi: 10.1093/protein/12.2.107. [DOI] [PubMed] [Google Scholar]
- 22.Yuan Z. Prediction of protein subcellular locations using Markov chain models. FEBS Lett. 1999;451:23–26. doi: 10.1016/s0014-5793(99)00506-2. [DOI] [PubMed] [Google Scholar]
- 23.Gardy JL, Brinkman FS. Methods for predicting bacterial protein subcellular localization. Nat. Rev. Microbiol. 2006;4:741–751. doi: 10.1038/nrmicro1494. [DOI] [PubMed] [Google Scholar]
- 24.Mott R, Schultz J, Bork P, Ponting CP. Predicting protein cellular localization using a domain projection method. Genome Res. 2002;12:1168–1174. doi: 10.1101/gr.96802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature. 2000;403:623–627. doi: 10.1038/35001009. [DOI] [PubMed] [Google Scholar]
- 26.Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- 27.Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, et al. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415:180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- 28.Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al. A protein interaction map of Drosophila melanogaster. Science. 2003;302:1727–1736. doi: 10.1126/science.1090289. [DOI] [PubMed] [Google Scholar]
- 29.Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, et al. High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell. 2004;13:225–239. doi: 10.1016/s1097-2765(04)00003-6. [DOI] [PubMed] [Google Scholar]
- 30.Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain PO, Han JD, Chesneau A, Hao T, et al. A map of the interactome network of the metazoan C. elegans. Science. 2004;303:540–543. doi: 10.1126/science.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, Young J, Berriz GF, Brost RL, Chang M, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303:808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- 32.Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437:1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- 33.Mendelsohn AR, Brent R. Protein interaction methods—toward an endgame. Science. 1999;284:1948–1950. doi: 10.1126/science.284.5422.1948. [DOI] [PubMed] [Google Scholar]
- 34.Formstecher E, Aresta S, Collura V, Hamburger A, Meil A, Trehin A, Reverdy C, Betin V, Maire S, Brun C, et al. Protein interaction mapping: a Drosophila case study. Genome Res. 2005;15:376–384. doi: 10.1101/gr.2659105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burckstummer T, Bennett KL, Preradovic A, Schutze G, Hantschel O, Superti-Furga G, Bauch A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods. 2006;3:1013–1019. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
- 36.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440:631–636. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 37.Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xenarios I, Salwinski L, Duan XJ, Higney P, Kim SM, Eisenberg D. DIP, the database of interacting proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002;30:303–305. doi: 10.1093/nar/30.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cherry JM, Adler C, Ball C, Chervitz SA, Dwight SS, Hester ET, Jia Y, Juvik G, Roe T, Schroeder M, et al. SGD: Saccharomyces Genome Database. Nucleic Acids Res. 1998;26:73–79. doi: 10.1093/nar/26.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanford C. Contribution of hydrophobic interactions to the stability of the globular conformation of proteins. J. Am. Chem. Soc. 1962;84:4240–4274. [Google Scholar]
- 41.Hopp TP, Woods KR. Prediction of protein antigenic determinants from amino acid sequences. Proc. Natl Acad. Sci. USA. 1981;78:3824–3828. doi: 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawashima S, Ogata H, Kanehisa M. AAindex: amino acid index database. Nucleic Acids Res. 1999;27:368–369. doi: 10.1093/nar/27.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chou KC, Shen HB. Recent progress in protein subcellular location prediction. Anal. Biochem. 2007;370:1–16. doi: 10.1016/j.ab.2007.07.006. [DOI] [PubMed] [Google Scholar]
- 44.Chou KC, Cai YD. Predicting protein-protein interactions from sequences in a hybridization space. J. Proteome Res. 2006;5:316–322. doi: 10.1021/pr050331g. [DOI] [PubMed] [Google Scholar]
- 45.Lu C, Devos A, Suykens JA, Arus C, Van Huffel S. Bagging linear sparse Bayesian learning models for variable selection in cancer diagnosis. IEEE Trans. Inf. Technol. Biomed. 2007;11:338–347. doi: 10.1109/titb.2006.889702. [DOI] [PubMed] [Google Scholar]
- 46.Mao KZ. Orthogonal forward selection and backward elimination algorithms for feature subset selection. IEEE Trans. Syst. Man Cybern. B Cybern. 2004;34:629–634. doi: 10.1109/tsmcb.2002.804363. [DOI] [PubMed] [Google Scholar]
- 47.Molodianovitch K, Faraggi D, Reiser B. Comparing the areas under two correlated ROC curves: parametric and non-parametric approaches. Biom. J. 2006;48:745–757. doi: 10.1002/bimj.200610223. [DOI] [PubMed] [Google Scholar]
- 48.Streiner DL, Cairney J. What's under the ROC? An introduction to receiver operating characteristics curves. Can. J. Psychiatr. 2007;52:121–128. doi: 10.1177/070674370705200210. [DOI] [PubMed] [Google Scholar]
- 49.Gasch AP, Werner-Washburne M. The genomics of yeast responses to environmental stress and starvation. Funct. Integr. Genomics. 2002;2:181–192. doi: 10.1007/s10142-002-0058-2. [DOI] [PubMed] [Google Scholar]
- 50.Rinner O, Mueller LN, Hubalek M, Muller M, Gstaiger M, Aebersold R. An integrated mass spectrometric and computational framework for the analysis of protein interaction networks. Nat. Biotechnol. 2007;25:345–352. doi: 10.1038/nbt1289. [DOI] [PubMed] [Google Scholar]
- 51.Liu XD, Thiele DJ. Oxidative stress induced heat shock factor phosphorylation and HSF-dependent activation of yeast metallothionein gene transcription. Genes Dev. 1996;10:592–603. doi: 10.1101/gad.10.5.592. [DOI] [PubMed] [Google Scholar]
- 52.Mohri-Shiomi A, Garsin DA. Insulin signaling and the heat shock response modulate protein homeostasis in the Caenorhabditis elegans intestine during infection. J. Biol. Chem. 2008;283:194–201. doi: 10.1074/jbc.M707956200. [DOI] [PubMed] [Google Scholar]
- 53.Conlin LK, Nelson HC. The natural osmolyte trehalose is a positive regulator of the heat-induced activity of yeast heat shock transcription factor. Mol. Cell Biol. 2007;27:1505–1515. doi: 10.1128/MCB.01158-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hahn JS, Hu Z, Thiele DJ, Iyer VR. Genome-wide analysis of the biology of stress responses through heat shock transcription factor. Mol. Cell Biol. 2004;24:5249–5256. doi: 10.1128/MCB.24.12.5249-5256.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.