

Abstract
Many pathogen recognition genes, such as plant R-genes, undergo rapid adaptive evolution, providing evidence that these genes play a critical role in plant-pathogen coevolution. Surprisingly, whether rapid adaptive evolution also occurs in genes encoding other kinds of plant defense proteins is unknown. Unlike recognition proteins, plant chitinases attack pathogens directly, conferring disease resistance by degrading chitin, a component of fungal cell walls. Here, we show that nonsynonymous substitution rates in plant class I chitinase often exceed synonymous rates in the plant genus Arabis (Cruciferae) and in other dicots, indicating a succession of adaptively driven amino acid replacements. We identify individual residues that are likely subject to positive selection by using codon substitution models and determine the location of these residues on the three-dimensional structure of class I chitinase. In contrast to primate lysozymes and plant class III chitinases, structural and functional relatives of class I chitinase, the adaptive replacements of class I chitinase occur disproportionately in the active site cleft. This highly unusual pattern of replacements suggests that fungi directly defend against chitinolytic activity through enzymatic inhibition or other forms of chemical resistance and identifies target residues for manipulating chitinolytic activity. These data also provide empirical evidence that plant defense proteins not involved in pathogen recognition also evolve in a manner consistent with rapid coevolutionary interactions.
Host-pathogen interactions are an important force shaping organismal diversity, yet little is known about the evolution of genes responsible for resistance in the host or virulence in the pathogen. Most genetic studies of plant-pathogen coevolution focus on gene-for-gene interactions that arise from pathogen-detection proteins deployed by the plant host (1–4). These proteins, encoded by R-genes, detect pathogen-produced elicitors, causing induced transcription of pathogenesis response (PR) proteins that can confer local or systemic resistance. R-genes exist in large multigene families that undergo rapid adaptive diversification, apparently in response to intense selection for new resistance specificities (5–7), supporting the hypothesis that these genes are principle targets of selection in the coevolution of hosts and pathogens.
Nevertheless, plants have evolved a complex array of chemical and enzymatic defenses, both constitutive and inducible, that are not involved in pathogen detection but whose effectiveness influences pathogenesis and disease resistance (8–10). The genes underlying these defenses comprise a substantial portion of the host genome. Based on genomic sequencing it is estimated that some 14% of the 21,000 genes in Arabidopsis are directly related to defense (11). Many of these ≈3,000 genes are not involved in pathogen detection but their products do interact directly with pathogen gene or protein products. Among the PR proteins, for example, are chitinases and endoglucanases that attack and degrade the cell walls of pathogens, and which pathogens counterattack with inhibitors (12). Such systems of antagonistically interacting proteins provide the opportunity for molecular coevolution of individual systems of attack and resistance.
Currently, the magnitude of pathogen-imposed selection is not known for any PR gene, nor have the molecular adaptations composing the response to selection been identified for any plant defense gene. Under the neutral theory of molecular evolution, most replacement substitutions are not expected to have adaptive consequences, even in genic regions where neutral evolution can be rejected. For this reason, identifying which replacements are responsible for enzymic adaptation has proven difficult. Recently, the availability of x-ray crystallographic structures has revolutionized the understanding of protein anatomy, and with it, the relationship between structure and function at the molecular level. By providing insight into which residues act in substrate binding and catalysis, crystal structure analysis provides a new tool for identifying adaptive amino acid substitutions by virtue of their structural and functional consequences (13). Here we combine tests of neutrality with crystal structures to analyze selection on a PR protein, class I chitinase.
Members of the chitinase (EC 3.2.1.14) gene family are found in all plants, which express them inducibly as PR proteins and constitutively in tissues vulnerable to pathogen attack (14, 15). Several lines of evidence indicate that chitinases play a direct role in plant defense by attacking chitin, a β-1,4-linked polymer of N-acetyl-d-glucosamine and a major component of fungal cell walls: (i) Purified chitinases can inhibit hyphal growth in vitro (15), (ii) constitutive overexpression of certain chitinases in transgenic plants can confer increased pathogen resistance in vivo (16–18), and (iii) chitinolytic breakdown products induce the production of defense compounds (phytoalexins) and systemic acquired resistance (19). These antifungal properties are greatly enhanced in the presence of β-1,3-endoglucanase (15), another PR protein that attacks the glucan matrix in which chitin is embedded. Nevertheless, chitinases often fail to confer resistance to certain pathogens in both in vivo and in vitro experiments, even in instances where the absence of antifungal effects cannot be attributed to failure to detect and respond to the invading pathogen (19–23). Although the mechanisms protecting fungal cell walls from chitinolytic activity remain obscure, inhibitors of plant chitinases have been isolated from several bacteria (24, 25).
The variable effectiveness of specific chitinases against different pathogens and the existence of microbial chitinase inhibitors suggest the hypothesis that chitinases may coevolve with fungi in response to variation in pathogen defenses against chitinolytic activity. One prediction of this hypothesis is that chitinase genes will be subject to positive selection, and that high rates of adaptive evolution will indicate high intensity or frequency of selective episodes. Moreover, adaptive substitutions should affect enzyme function, an effect that may be detectable by structural analyses. To test these predictions we applied neutral rate tests, capable of distinguishing selectively favored from selectively neutral substitution patterns, to a set of 22 DNA sequences encoding class I chitinase (sometimes called basic chitinase) from 13 species of Arabis (Cruciferae), a group that includes the nearest relatives of the model plant Arabidopsis thaliana (26). We chose class I chitinase because in A. thaliana this class is encoded by a single gene, is expressed in a manner consistent with a defensive role (14), inhibits hyphal growth of certain fungal species but not others (20), and in tobacco degrades chitin more rapidly than do other classes of chitinase (19).
Materials and Methods
Sequence Data.
The coding regions corresponding to 23 mature class I chitinases (Fig. 1) were sequenced in 14 accessions of Arabis spp. as follows. PCR primers based on conserved regions of A. thaliana and Brassica napus class I chitinase (GenBank accession nos. M38240 and M95835) were used to amplify partial genomic sequences of two genes from Arabis fecunda genomic DNA extracts. PCR products were cloned (TA cloning kit, Stratagene) and sequenced. Each gene was amplified with gene-specific primers and direct-sequenced from an additional 60 A. fecunda individuals, demonstrating nonallelism of the two loci in this species. Total RNA was extracted (RNeasy kit, Qiagen, Chatsworth, CA) from A. fecunda, A. drummondii, and A. lemmonii, and 3′ rapid amplification of cDNA ends was used to amplify the carboxyl terminal region of five genes, verifying their transcription in these species. These products were cloned (pGEM-T Vector kit, Promega) and sequenced. Sets of conserved primers within the 5′ and 3′ signal and target peptides were designed to amplify regions corresponding to the mature protein from the remaining species. These products were either gel-purified, cloned, and sequenced or they were direct-sequenced. Primer sequences are available on request. Sequencing was performed on both strands of several independent clones for each gene by using Big Dye Terminators (Applied Biosystems) analyzed on a 377XL DNA sequencer (Applied Biosystems). The number of genes for each species included in the study are: one Arabis alpina; one A. blepharophylla; one A. drummondii; two A. fecunda; one A. glabra; two A. gunnisoniana; one A. holboellii from Colorado; one A. holboellii from Greenland; two A. lemmonii; three A. lignifera; one A. lyallii; three A. microphylla; three A. parishii; and one Halimolobos perplexa [the Arabis clade includes H. perplexa (26)].
Figure 1.

A. parishii class I chitinase amino acid sequence. Residue number 1 corresponds to the start codon, but only residues 25–325 are included in this study. The mature protein is ≈298 residues long, consisting of a cysteine-rich chitin binding domain (5′) and a chitinolytic domain (3′), connected by a hypervariable proline-glycine rich hinge (lowercase). Residues 1–22 and 319–325 form the signal peptide and vacuolar targeting peptide, respectively, and are cleaved from the mature peptide. Positively selected residues are red, catalytic residues Glu-141 and Glu-163 are green, putative substrate-binding residues are shown in blue (40), active site residues (defined as residues within 0.6 nm of bound substrate) are underlined, blocks denote indels, and * denotes importance for enzyme function confirmed by directed mutagenesis (51, 52). Alternative residues found among the 19 sequences are shown above the A. parishii sequence. Positively selected residues are identified as sites having a posterior probability > 0.95 of being in the positive category, for a majority of phylogenies tested.
Sequence Analysis.
Sequences of the single chitinase intron, available for 20 of 23 genes, allowed grouping of genes into three sets distinguished by numerous indels. The first two sets (denoted 1 and 2 in Fig. 2) are from North American Arabis that have n = 7 chromosomes and are paralogous to each other. A third set corresponds to the A. alpina group (denoted 3 in Fig. 2). Coding region sequences corresponding to Fig. 1 were aligned and positions with gaps were excluded from the analyses of nonsynonymous substitution rates (Ka), synonymous substitution rates (Ks), and phylogenetic analyses based on coding region.
Figure 2.

Combined phylogeny of several parsimony phylogenies rooted with B. napus. See Materials and Methods for details of construction. Numbers along each branch show the number of nonsynonymous substitutions/number of synonymous substitutions for that branch, estimated by maximum likelihood for an unrooted tree, and numbers in parentheses show the percent support among 1,000 bootstrap data sets, calculated for each of the component parsimony trees. Branches showing only bootstrap support have estimated branch length = 0, but bootstrap support > 70%, whereas those with no bootstrap values have support < 70%. Numbers shown in bold highlight Ka > Ks (but do not denote significance), and gene names in bold indicate genes involved in significant pairwise comparisons. * denotes phylogenetic position based on coding sequence only. Substitution counts differ from direct pairwise comparisons because the former are estimated by likelihood methods and the latter by approximate methods. Branch lengths are drawn so as to make clades easily discernable.
Chitinase gene phylogenies were estimated by using parsimony and DNA and protein-maximum likelihood methods implemented in phylip (27) and molphy (28), using B. napus as an outgroup (26). Bootstrap analysis indicated that phylogenies based on coding sequence were reliable only at their roots. Further resolution was attained by using intron as well as coding sequences, but it was not possible to align all introns. Therefore, majority-rule consensus parsimony trees were estimated for each of the three intron groups, and resulting clades were rooted by using one gene from each other group. Rooted trees for each of the three sets then were attached at the appropriate location in the consensus phylogeny based on coding sequence. The resulting “combined” phylogeny (Fig. 2) was consistent with phylogenies estimated based on coding sequence alone.
Ka and Ks were estimated for all pairwise comparisons of 22 mature protein sequences by using the program li93 (29, 30). A. glabra was omitted from this and other analyses because part of its active site is deleted. Significance tests for Ka > Ks were evaluated for a small subset of comparisons, using a one-tailed t test with df = infinite (29). Because t is biased toward greater significance when the number of substitutions is small (31), we adopted t > 2.2 (P < 0.014) as an appropriate significance threshold. Simulation models of sequence evolution in lysozyme, which is homologous to class I chitinase, about 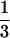 the length of chitinase and has similar substitution rates (32), suggest even when biased this value of t corresponds to P < 0.05 (31). Because codon bias can affect Ks a measure of codon bias, the effective number of codons (33), was estimated for each of the 22 sequences by using the codons program (34).
the length of chitinase and has similar substitution rates (32), suggest even when biased this value of t corresponds to P < 0.05 (31). Because codon bias can affect Ks a measure of codon bias, the effective number of codons (33), was estimated for each of the 22 sequences by using the codons program (34).
Maximum-likelihood models of codon substitution (35) for 19 sequences were implemented in codeml 1.4, a program in the paml package (36). These models address the question of whether Ka > Ks over the entire set of sequences, while taking into account the phylogenetically correlated structure of the data. codeml fits a likelihood model of codon evolution to sequence data, given a phylogeny for the sequences. We compared two scenarios: (i) a null model in which nonsynonymous mutations are assumed to be either neutral or deleterious, and (ii) a positive selection model that includes a third category for advantageous substitutions. The likelihood ratio comparison of the two models tests whether the positive selection model fits the data better than the null hypothesis. We fitted the models by using the set of 99 equally most parsimonious trees produced by dnapars (27), as well as trees estimated by maximum-likelihood methods and the combined tree described above, to avoid any possibility of an outcome dependent on a phylogeny in which some branches receive little statistical support. H. perplexa (uncertain phylogenetic position), A. alpina (C-terminal sequence not available), and A. parishii 2.3 (probably allelic to A. parishii 2.2) were not included in this analysis. Model results were robust to assumptions regarding equilibrium codon frequency. An empirical Bayesian approach implemented in codeml and described in ref. 35 was used to infer which substitution category (neutral, deleterious, or advantageous) each amino acid site in the protein most likely belonged to. Advantageous sites with posterior probability > 0.95 were considered significant. A different model in codeml was used to estimate the number of synonymous and nonsynonymous substitutions for each branch of the unrooted combined Arabis gene phylogeny. That model (described in refs. 37 and 38) specifies a single Ka/Ks ratio across sites, but allows the ratio to vary freely across branches of the phylogeny (36). Both likelihood models average synonymous and nonsynonymous substitutions over all possible ancestral sequences at each interior node, weighted by their likelihoods of occurrence (38).
The number of replacements at each site in the protein was estimated by parsimony (53) applied to phylogenies estimated by using DNA and protein maximum-likelihood methods. Graphics were implemented by mapping the number of replacements onto structural models by using insightii (BioSym Technologies, San Diego). Phylogenies estimated by using different methods yielded similar results. Class I chitinase is modeled by using the crystal structure of barley class II chitinase (39) (Protein Data Bank code 1CNS) with bound hexa-N-acetyl-d-glucosamine (40). The two enzymes are very similar, although class II enzymes lack an N-terminal chitin-binding domain (15). Class I sequences were truncated just 3′ of the proline-rich hinge connecting the chitin-binding domain to the chitinolytic domain (Fig. 1).
For comparative purposes, similar structural analyses were performed on sequence alignments for 12 Solanaceae class I chitinases, 22 primate lysozymes, and 31 plant class III chitinases. Human lysozyme with bound tetra-acetyl-chitotetraose (41) (Protein Data Bank code 1LZR) was used to model primate lysozymes. Hevamine with bound allosamidin (42) (Protein Data Bank code 1LLO) was used to model class III chitinase. Protein accession numbers are: Solanaceae class I, AF043247–48, A21091, S44869, U02605–7, X07130, X64518, X51599, X16939, Z15140, and class III, 3452147, 1076250, 116328, 167538, 167540, 116330, 486991, 116332, 116337, 1362073, 1362074, 1705812, 1839589, 2072742, 2293066, 2317270, 2425170, 2696227, 2696231, 116327, 2342433, 2342435, 2342439, 2342443, 2342447, 2342451, 2342453, 2342457, 2342459, 2342461, and 320555. For primate lysozyme see ref. 32.
Results
One common neutral rate test compares the number of synonymous (silent) substitutions per synonymous site (Ks) with the number of nonsynonymous (amino acid replacing) substitutions per nonsynonymous site (Ka). In the absence of selection on codon usage, Ks represents the rate of neutral substitution. In most proteins most nonsynonymous substitutions are deleterious, resulting in purifying selection and Ka << Ks. A rate of amino acid replacement significantly greater than the rate of neutral evolution, Ka > Ks, provides unambiguous evidence of adaptive sequence evolution (31, 32, 35, 38). We found Ka > Ks in 85 of 231 pairwise sequence comparisons, including several of the highest Ka/Ks ratios yet reported (Fig. 3A). Eleven of these comparisons—all those with Ka/Ks > 3—were significantly greater than 1 (t test, P < 0.01, Fig. 3B). There was no evidence of selection for codon usage (effective number of codons is 53–59). Ratios varied greatly among species—A. blepharophylla, both A. holboellii accessions, A. gunnisoniana, A. lemmonii, and A. lignifera each contained genes that exhibited Ka > Ks in nearly all pairwise comparisons. Comparisons with particularly high Ka/Ks include A. blepharophylla vs. A. holboellii Colorado (Ka/Ks = 9.2, 21 nonsynonymous substitutions (N) vs. only one synonymous substitution (S), A. holboellii Greenland vs. A gunnisoniana 2a (Ka/Ks = 7.2, N = 15, S = 1) and A gunnisoniana 2a vs. A. lemmonii 2 (N = 8, S = 0). In contrast, genes from A. parishii and A. fecunda exhibited only two comparisons with Ka > Ks. Ka > Ks was found on branches throughout the gene phylogeny (Fig. 2). Genes with significant comparisons are somewhat clustered, but clusters are found in three different clades (Fig. 2).
Figure 3.

(A) Distribution of 231 Ka/Ks ratios into intervals, where Ka and Ks are the number of nucleotide substitutions that cause amino acid replacements or are silent, respectively, divided by the number of possible substitutions of each type. Ka/Ks > 1 indicates comparisons where amino acid substitutions are more frequent than predicted by neutral substitution rates. (B) Ka vs. Ks. Ratios > 1 occur above the line Ka = Ks. ▴ denote significant comparisons.
In the group of Arabis with n = 7 chromosomes we take advantage of the two distinct gene clades, distinguished by intron sequences (Fig. 2), to compare the frequency of elevated Ka/Ks within intron groups (i.e., orthologues) to those between intron groups (i.e., paralogues). Taking Ka/Ks > 1.9 as an arbitrary cutoff, we find no difference in the frequency of positively selected pairs in paralogous vs. orthologous comparisons (15 cases of 84 comparisons, and seven cases of 88 comparisons, respectively), nor were intraspecific and interspecific comparisons for the n = 7 group disproportionate (one case in 12 vs. 21 cases in 159).
Our analysis of Ka/Ks provides strong evidence for positive selection, but further interpretation is difficult given that (i) many of the comparisons were not significant and (ii) the lineages are phylogenetically correlated. These problems can be circumvented by using maximum-likelihood substitution models to test the hypothesis that positive selection is operating across the entire data set (35). We compared a null model in which substitutions are assumed to be either neutral or deleterious with a positive selection model that includes a third category for advantageous substitutions. For all phylogenies tested, the positive selection model provided a better fit than the null model (χ2 range: 24–57, df = 2, P < 0.0001). The positive selection model estimated that substitutions at 64–77% of the amino acid residues are deleterious and 5–14% of substitutions are advantageous, with Ka/Ks = 6.8 for the latter. Bayesian analysis of the models (35) identified 15 sites that are likely (P < 0.05) to sustain advantageous amino acid replacements (Fig. 1). Seven of these sites involve only one alternative residue, but are identified as positively selected because that residue must evolve multiple times in the majority of the tested phylogenies.
Mapping the observed replacements onto the x-ray crystallographic structure of chitinase reveals that numerous replacements occur in the active site cleft (Fig. 4 Upper Left). Indeed, the mean number of replacements per site inside the cleft [defined as those residues within 0.6 nm of bound substrate (40)] approximately equals the mean number of replacements per site outside it (rate inside: rate outside = 0.28: 0.19, χ2 = 1.63, P > 0.05, assuming 59 cleft positions vs. 227 outside positions, as shown in Fig. 1), with positively selected sites more than twice as likely to reside within the cleft (0.11: 0.04, χ2 = 3.94, P < 0.05; Fig. 1). Physically adjacent to the two rigidly conserved catalytic residues Glu-141 and Glu-163, sites 191 and 164 sustain multiple advantageous replacements, as do sites 284 and 288, directly across the cleft from Glu-163 (Figs. 1 and 4 Upper Left). A similar pattern of amino acid replacements is observed for the class I chitinases of the Solanaceae (0.51: 0.55, χ2 = 0.11, P > 0.05; Fig. 4 Lower Left) and other dicots (results not shown). Neutral rate tests for the Solanaceae data reveal one instance in which Ka > Ks (Solanum tuberosum GenBank accession nos. AF043247–48; Ka/Ks = 4.32, P < 0.01, t test). These results suggest that rapid adaptive evolution of the active site is a general phenomenon in the class I chitinases of dicotyledonous plants.
Figure 4.

Crystal structures with bound polysaccharide ligands (dot surfaces) showing the location and frequency of amino acid replacements for Arabis class I chitinase (Upper Left), Solanaceae class I chitinase (Lower Left), primate lysozyme (Upper Right), and plant class III chitinase (Lower Right). Ligands are hexa-N-acetyl-d-glucosamine (class I chitinases), tetra-acetyl-chitotetraose (lysozyme), and allosamadin inhibitor (class III chitinase). Catalytic residues are colored yellow. Color-coded legends show the number of replacements occurring at positions illustrated in the corresponding color.
Discussion
Molecular evolution of class I chitinase is driven by selection for advantageous mutations, causing an excess of amino acid replacements in the active site and substrate binding cleft. We observed a significant excess of amino acid replacements, compared with neutral expectations, in 11 sequence comparisons in the genus Arabis and in one pair of sequences from S. tuberosum. Elevated Ka/Ks ratios have not been observed in ADH1 and chalcone synthase gene sequences from some of the same plant accessions (M. Koch and T.M.-O., unpublished work), indicating our results are not attributable to a genomewide elevation of Ka, as might occur under small effective population sizes. Codon substitution models confirm that Ka > Ks for the Arabis sequences considered simultaneously and taking into account their phylogenetic relationships. Ka/Ks significantly > 1 has been documented for only a few whole proteins (29) and the domains of a handful of additional proteins (35, 43, 44). In plants, such ratios have been documented only for the solvent-exposed domain of the leucine-rich repeat-containing R-gene family, involved in recognizing pathogenic elicitors (5–7), and in self-recognition genes (45). Chitinase sequences with Ka/Ks > 1 exhibited low levels of synonymous divergence (Fig. 3B), indicating that the occurrence of adaptive replacements saturates at relatively low levels of sequence divergence—i.e., only a limited number of sites respond to positive selection. This constrained response likely stems from the need to preserve catalytic function and may indicate more complex functional constraints than seen in receptor-ligand systems like the R-genes. In R-genes Ka/Ks > 1 at very high levels of synonymous divergence (Ks > 0.5) is consistent with continuous response to diversifying selection for recognition capability (5–7).
We observed an equitable distribution of replacements throughout the enzymic structure of chitinase and adaptive replacements physically adjacent to catalytic residues. Both observations are remarkable. Active sites are usually the subject of intense evolutionary constraint engendered by the need to preserve function. In primate lysozyme, for example, the mean number of replacements per site within the cleft is only half that without it (rate within/rate without = 0.23:0.45, χ2 = 5.32, P < 0.05) and not a single replacement contacts either catalytic residue (Fig. 4 Upper Right). A similar distribution of replacements is found in plant class III chitinase where, despite greater taxonomic diversity and 10 times as many replacements per site (1.86:4.27, χ2 = 60.37, P < 0.0001), few replacements occur in the active site cleft (Fig. 4 Lower Right). Primate lysozymes and class III chitinases are not unusual in this respect; active site residues in most enzymes are highly conserved.
The unusual pattern of replacements in class I chitinase, as compared with lysozyme and class III chitinase, is not simply a consequence of constraints imposed by differences in form and function. Although highly divergent, class I chitinases and lysozymes are homologous, sharing similar structures and catalytic mechanisms (46). Murein, the substrate of lysozyme, differs from chitin only by a lactyl side chain that embellishes the C3 position of alternate N-acetyl-d-glucosamine residues in the polymer; many chitinases, including hevamine (Fig. 4 Lower Right), display lysozyme activity and vice versa (42). Nor are the differences from lysozyme merely a consequence of strong positive selection. Recent analyses demonstrate adaptive protein evolution in primate lysozymes, featuring Ka/Ks ratios similar to those found here (32). Rather, the unusual pattern of adaptive amino acid replacements in the active site of plant class I chitinase is a direct consequence of the mode of natural selection operating at the molecular level.
The situation is reminiscent of the evolutionary consequences of therapeutic use of inhibitors on HIV protease where mutations in the active site reduce inhibitor binding by, for example, introducing a polar group close to a hydrophobic methyl on the inhibitor or removing a hydrophobic contact (47). Such mutations need not be chemically “radical”—for example two chemically “conservative” replacements, V82T and I84V, together reduce binding of Indinavir by a factor of 70, rendering the inhibitor ineffective in cell culture assays. Replacements Q164V and M191I in chitinase may have similar effects as they are very likely to contact any inhibitor bound in the active site cleft. Such mutations are also likely to reduce activity by disrupting substrate binding. Selected replacements at sites 167 and 169 in the mobile loop that includes the catalytic E163 may be compensatory, improving catalysis without necessarily affecting the newly acquired resistance. Indeed, in HIV protease a similarly disposed mutation in a flap that guards the entrance to the active site is believed to compensate for a loss in catalytic efficiency by affecting the dynamic behavior of the flap and hence accessibility to the active site (48).
The presence of positively selected substitutions within the active site cleft indicates rapid adaptive functional modification of class I chitinase. We suggest the observed substitutions are favored as a response to inhibition attributable to carbohydrate or protein inhibitors (12, 24, 25) or to resistance mediated by physical modification of the cell wall (49). Plants are likely to encounter novel resistance or inhibition mechanisms in the face of geographic variation in pathogen species, and chitinases are known to differ in their ability to inhibit different fungi (20–23) and in their substrate specificities (19). Although the precise selective agent remains unknown, the notion that amino acid replacements in class I chitinase may be a response to chitinolytic inhibition is supported by our finding that amino acid replacements in class III chitinase/lysozyme contact the bacterial inhibitor allosamidin (Fig. 3). We predict that enzymes involved in antagonistic enzyme-inhibitor interactions will be especially prone to positive molecular evolution. Interestingly, several candidate systems involve cell wall-attacking enzymes deployed by plants and their pathogens (12, 50).
More generally, rapid adaptive evolution in cell wall-attacking enzymes such as chitinase implies that the arms races and other coevolutionary interactions between plants and pathogens are not solely races for information among detection proteins, but also involve proteins that directly attack pathogens. Changes in these proteins thus may account for more variation in disease resistance than commonly realized. However, the ecological interactions in which novel forms of chitinase are advantageous remain to be elucidated. R-genes were named for their involvement in gene-for-gene resistance against strains of particular pathogen species, and all those characterized to date function to detect invading pathogens. In contrast our results indicate that PR proteins might not be involved in similar gene-for-gene interactions with a single pathogen species, but perhaps respond to geographic or temporal changes in the prevalence of different pathogen species.
Acknowledgments
We thank I. Dews, J. Neuhaus, B. Schaal, H. Stotz, J. Thompson, Z. Yang, and three anonymous reviewers for thoughtful comments on the manuscript. We also thank J. Dacy, D. Pedersen, B. Stranger, M. Tobler, D. Schnaubelrauch, S. Ring, M. Koch, J. Kroymann, and H. Stotz for technical assistance; P. Connors, M. Marler, and B. Roy for seed collections; C.-B. Stewart for providing aligned lysozyme sequences; and M. Koch for access to unpublished data. This work was supported by the Max Planck Institute for Chemical Ecology and National Science Foundation Grants BIR-9626079 to J.G.B. and DEB-9527725 to T.M.-O.
Abbreviations
- PR
pathogenesis response
- Ka
nonsynonymous substitution rate
- Ks
synonymous substitution rate
Footnotes
References
- 1.Thompson J N, Burdon J J. Nature (London) 1992;360:121–125. [Google Scholar]
- 2.Simms E L. BioScience. 1996;46:136–145. [Google Scholar]
- 3.Caicedo A L, Schaal B A, Kunkel B N. Proc Natl Acad Sci USA. 1999;96:302–306. doi: 10.1073/pnas.96.1.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stahl E A, Dwyer G, Mauricio R, Kreitman M, Bergelson J. Nature (London) 1999;400:667–671. doi: 10.1038/23260. [DOI] [PubMed] [Google Scholar]
- 5.Meyers B C, Shen K A, Rohani P, Gaut B S, Michelmore R W. Plant Cell. 1998;11:1833–1846. doi: 10.1105/tpc.10.11.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang G L, Ruan D, Song W Y, Sideris S, Chen L, Pi L Y, Zhang S, Zhang Z, Fauquet C, Gaut B S, et al. Plant Cell. 1998;10:765–779. doi: 10.1105/tpc.10.5.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McDowell J M, Dhandaydam M, Long T A, Aarts M G M, Goff S, Holub E B, Dangl J L. Plant Cell. 1998;10:1861–1874. doi: 10.1105/tpc.10.11.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frey M, Chomet P, Glawischnig E, Stettner C, Gruen S, Winklmair A, Eisenriech W, Bacher A, Meeley R B, Briggs S P, et al. Science. 1997;277:696–699. doi: 10.1126/science.277.5326.696. [DOI] [PubMed] [Google Scholar]
- 9.Bowyer P, Clarke B R, Lunness P, Daniels M J, Osbourn A E. Science. 1995;267:371–374. doi: 10.1126/science.7824933. [DOI] [PubMed] [Google Scholar]
- 10.Glazebrook J, Zooki M, Mert F, Kagan I, Rogers E E, Crute I R, Holub E B, Hammerschmits R, Ausubel F M. Genetics. 1997;146:381–392. doi: 10.1093/genetics/146.1.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bevan M, Bancroft I, Bent E, Love K, Goodman H, Dean C, Bergkamp R, Kirksse W, Van Staveren M, Stiekema W, et al. Nature (London) 1998;391:485–488. doi: 10.1038/35140. [DOI] [PubMed] [Google Scholar]
- 12.Ham K, Wu S, Darvill A G, Albersheim P. Plant J. 1997;11:169–179. [Google Scholar]
- 13.Golding G B, Dean A M. Mol Biol Evol. 1998;15:355–369. doi: 10.1093/oxfordjournals.molbev.a025932. [DOI] [PubMed] [Google Scholar]
- 14.Samac D A, Hironaka C M, Yallaly P E, Shah D M. Plant Physiol. 1990;93:907–914. doi: 10.1104/pp.93.3.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Collinge D B, Kragh K M, Mikkelsen J D, Nielsen K K, Rasmussen U, Vad K. Plant J. 1993;3:31–40. doi: 10.1046/j.1365-313x.1993.t01-1-00999.x. [DOI] [PubMed] [Google Scholar]
- 16.Broglie K, Chet I, Holliday M, Cressman R, Biddle P, Knowlton S, Mauvais C J, Broglie R. Science. 1991;254:1194–1197. doi: 10.1126/science.254.5035.1194. [DOI] [PubMed] [Google Scholar]
- 17.Jach G, Goerhardt B, Mundy J, Logemann J, Pinsdorf E, Leah R, Schell J, Maas C. Plant J. 1995;8:97–109. doi: 10.1046/j.1365-313x.1995.08010097.x. [DOI] [PubMed] [Google Scholar]
- 18.Grison R, Grezes-Besset B, Schneider M, Lucante N, Olsen L, Leguay J-J, Toppan A. Nat Biotechnol. 1996;4:643–656. doi: 10.1038/nbt0596-643. [DOI] [PubMed] [Google Scholar]
- 19.Brunner F, Stintzi A, Fritig B, Legrand M. Plant J. 1998;14:225–234. doi: 10.1046/j.1365-313x.1998.00116.x. [DOI] [PubMed] [Google Scholar]
- 20.Verburg J G, Huynh Q K. Plant Physiol. 1991;95:450–455. doi: 10.1104/pp.95.2.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neuhaus J-M, Ahl-Goy P, Hinz U, Flores U, Meins F. Plant Mol Biol. 1991;16:151–161. doi: 10.1007/BF00017924. [DOI] [PubMed] [Google Scholar]
- 22.Broekhaert W F, Van Parajs J, Allen A K, Peumans W J. Physiol Mol Plant Pathol. 1988;33:319–331. [Google Scholar]
- 23.Mauch F, Mauch-Mani B, Boller T. Plant Physiol. 1988;88:936–942. doi: 10.1104/pp.88.3.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hodge A, Gooday G W, Alexander I J. Phytochemistry. 1996;41:77–84. doi: 10.1016/0031-9422(95)00571-4. [DOI] [PubMed] [Google Scholar]
- 25.Sakuda S, Isogai A, Matsumoto S, Suzuki A. Tetrahedron Lett. 1986;27:2475–2478. [Google Scholar]
- 26.Koch M, Bishop J G, Mitchell-Olds T. Plant Biol. 1999;1:529–537. [Google Scholar]
- 27.Felsenstein J. phylipPhylogeny Inference Package. Seattle: Univ. of Washington; 1996. , Version 3.5. [Google Scholar]
- 28.Adachi J, Hasegawa M. molphy. Institute of Statistical Mathematics, Tokyo: Computer Science Monographs; 1996. [Google Scholar]
- 29.Li W-H. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- 30.Wolfe K. li93. Dublin: Trinity College; 1997. [Google Scholar]
- 31.Zhang J, Kumar S, Nei M. Mol Biol Evol. 1997;14:1335–1338. doi: 10.1093/oxfordjournals.molbev.a025743. [DOI] [PubMed] [Google Scholar]
- 32.Messier W, Stewart C-B. Nature (London) 1997;385:151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- 33.Wright F. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- 34.Lloyd A T, Sharp P M. J Hered. 1992;83:239–240. doi: 10.1093/oxfordjournals.jhered.a111205. [DOI] [PubMed] [Google Scholar]
- 35.Nielsen R, Yang Z. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang Z. paml. London: University College; 1999. [Google Scholar]
- 37.Yang Z, Nielsen R. J Mol Evol. 1998;46:409–418. doi: 10.1007/pl00006320. [DOI] [PubMed] [Google Scholar]
- 38.Yang Z. Mol Biol Evol. 1998;15:568–573. doi: 10.1093/oxfordjournals.molbev.a025957. [DOI] [PubMed] [Google Scholar]
- 39.Hart P J, Pfluger H D, Monzingo A F, Hollis T, Robertus J D. J Mol Biol. 1995;248:402–413. [PubMed] [Google Scholar]
- 40.Brameld K A, Goddard W A., III Proc Natl Acad Sci USA. 1998;95:4276–4281. doi: 10.1073/pnas.95.8.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song H, Inaka K, Maenaka K, Matsushima M. J Mol Biol. 1994;244:522–540. doi: 10.1006/jmbi.1994.1750. [DOI] [PubMed] [Google Scholar]
- 42.Terwisscha van Scheltinga A C, Armand S, Kalk K H, Isogai A, Henrissat B, Dijkstra B W. Biochemistry. 1995;34:15619–15623. doi: 10.1021/bi00048a003. [DOI] [PubMed] [Google Scholar]
- 43.Hughes A L, Nei M. Nature (London) 1988;335:167–170. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- 44.Endo T, Ikeo K, Gojobori T. Mol Biol Evol. 1996;13:685–690. doi: 10.1093/oxfordjournals.molbev.a025629. [DOI] [PubMed] [Google Scholar]
- 45.Richman A D, Kohn J R. Proc Natl Acad Sci USA. 1999;96:168–172. doi: 10.1073/pnas.96.1.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Monzingo A F, Marcotte E M, Hart P J, Robertus J D. Nat Struct Biol. 1996;3:133–140. doi: 10.1038/nsb0296-133. [DOI] [PubMed] [Google Scholar]
- 47.Schock H B, Garsky V M, Kuo L C. J Biol Chem. 1996;271:31957–31963. doi: 10.1074/jbc.271.50.31957. [DOI] [PubMed] [Google Scholar]
- 48.Collins J R, Burk S K, Erikson J W. Nat Struct Biol. 1995;2:334–338. doi: 10.1038/nsb0495-334. [DOI] [PubMed] [Google Scholar]
- 49.Van Pelt-Heerschap H, Sietsma J H. Mycol Res. 1990;94:979–984. [Google Scholar]
- 50.Stotz H U, Bishop J G, Bergmann C W, Koch M, Albersheim P, Darvill A G, Labavitch J M. Mol Physiol Plant Pathol. 2000;56:117–118. [Google Scholar]
- 51.Iseli-Gamboni B, Boller T, Neuhaus J-M. Plant Sci. 1998;134:45–51. [Google Scholar]
- 52.Allona I, Collada C, Casado R, Paz-Ares J, Aragoncillo C. Plant Mol Biol. 1996;32:1171–1176. doi: 10.1007/BF00041402. [DOI] [PubMed] [Google Scholar]
- 53.Swofford D L. paup*, Phylogenetic Analysis Using Parsimony. Sunderland, MA: Sinauer; 1998. , Version 4. [Google Scholar]