

Abstract
Changes in genes encoding transcriptional regulators can alter development and are important components of the molecular mechanisms of morphological evolution. MADS-box genes encode transcriptional regulators of diverse and important biological functions. In plants, MADS-box genes regulate flower, fruit, leaf, and root development. Recent sequencing efforts in Arabidopsis have allowed a nearly complete sampling of the MADS-box gene family from a single plant, something that was lacking in previous phylogenetic studies. To test the long-suspected parallel between the evolution of the MADS-box gene family and the evolution of plant form, a polarized gene phylogeny is necessary. Here we suggest that a gene duplication ancestral to the divergence of plants and animals gave rise to two main lineages of MADS-box genes: TypeI and TypeII. We locate the root of the eukaryotic MADS-box gene family between these two lineages. A novel monophyletic group of plant MADS domains (AGL34 like) seems to be more closely related to previously identified animal SRF-like MADS domains to form TypeI lineage. Most other plant sequences form a clear monophyletic group with animal MEF2-like domains to form TypeII lineage. Only plant TypeII members have a K domain that is downstream of the MADS domain in most plant members previously identified. This suggests that the K domain evolved after the duplication that gave rise to the two lineages. Finally, a group of intermediate plant sequences could be the result of recombination events. These analyses may guide the search for MADS-box sequences in basal eukaryotes and the phylogenetic placement of new genes from other plant species.
Keywords: MEF2, SRF, homeotic genes, MADS, development
Changes in genes encoding transcriptional regulators may represent the most important determinants of morphological evolution in plants and animals (1), and phylogenetic analyses provide a historical framework to identify such changes. The MADS-box genes encode a eukaryotic family of transcriptional regulators involved in diverse and important biological functions, ranging from cardiac muscle development in animals to pheromone response in yeast (2). In plants, MADS-box genes encode the three floral homeotic functions predicted by the genetic ABC model of flower organ identity (3, 4). In addition, plant MADS-box genes regulate the timing of flower initiation and flower meristem identity, as well as various aspects of ovule, fruit, leaf, and root development (4, 5).
Previously identified plant MADS-box genes encode proteins that share a stereotypical MIKC structure (Fig. 1), with the highly conserved DNA-binding MADS domain at the amino terminus. The moderately conserved K domain in the central portion of these proteins has been shown to be important for protein–protein interactions and likely forms a coiled-coil structure. The MADS and K domains are linked to one another by a weakly conserved I domain, whereas a poorly conserved carboxyl-terminal (C) region may function as a trans-activation domain (4). In animals and fungi, two distinct types of MADS-box genes have been identified, the SRF-like and MEF2-like classes (ref. 2; see Fig. 1).
Figure 1.
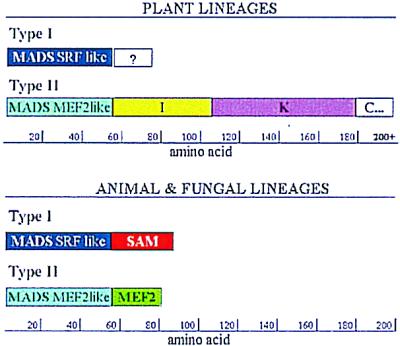
Schematic representation of the protein domains of plant, animal, and fungal TypeI (SRF-like) and TypeII (MEF2-like) MADS-domain proteins. The scale indicates the number of amino acids along the protein. Plant TypeII-like proteins have carboxyl-terminal domains that go beyond 200 amino acids. In plant TypeI-like proteins the “?” indicates carboxyl-terminal domains not well defined yet and of variable lengths.
This paper provides a hypothesis on the evolutionary history of the eukaryotic MADS-box gene family. Previous studies of eukaryotic MADS-box gene evolution, which included plant and animal sequences, provided unrooted trees useful to infer the phylogenetic relationships of the MADS-box lineages (6). These previous studies suggested that at least one MADS-box gene was present in the common ancestor of plants, animals, and fungi, and that probably the duplication that gave rise to the animal MEF2- and SRF-like genes occurred after animals diverged from plants but before fungi diverged from animals (6). However, previous plant and eukaryotic studies were based on a relatively small sampling of plant MADS-box sequences for a particular species (6–9). To test whether all Arabidopsis MADS-box sequences group in a monophyletic clade distinct from all animal and fungal MADS-box sequences, we performed phylogenetic analyses. We used 45 Arabidopsis MADS domain sequences, including 26 new ones, 9 sequences representative of the MEF2-like class from animals, and 8 sequences from the animal SRF-like group.
We present a rooted phylogenetic tree of the eukaryotic MADS domain lineages and postulate new hypotheses on the evolutionary history of this gene family. Our results suggest that a duplication ancestral to the divergence of plants and animals gave rise to two lineages (herein called TypeI and TypeII MADS), and that the protein motifs that define each group were fixed in the common ancestors of plants, animals, and fungi. Our analyses also identify new monophyletic clades of plant MADS-box sequences. Most plant MADS-box genes including all of the ones that have been characterized functionally in previous studies, group with the animal MEF2-like sequences in what we have named the TypeII MADS-box lineage. But we have identified a group of Arabidopsis MADS-box sequences that seems to be more closely related to the animal SRF-like genes forming the group that we herein call TypeI MADS. This finding suggests that both lineages are present in plants, animals, and fungi. Finally, we show that the K domain, typical of plant MADS-domain proteins, is found only in the TypeII MADS domain sequences of plants, suggesting that this domain evolved after this lineage diverged from the TypeI MADS. These results have enabled us to put forward a model for the evolution of this important family of regulatory genes in eukaryotes (see Fig. 4).
Figure 4.
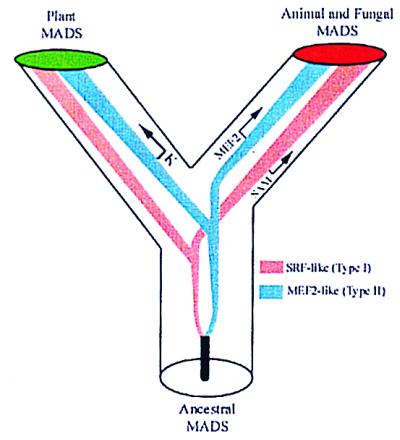
Model for the evolution of the MADS-box gene family in eukaryotes. At least one duplication of the ancestral MADS-box gene is postulated to have occurred before the divergence of plants and animals. The K domain was probably added to the plant TypeII (MEF2-like) lineage. Similarly, animal MADS-domain proteins evolved specific domains (SAM and MEF2) in SRF-like and MEF2-like lineages, respectively. Pink, TypeI (SRF-like) lineage; blue, TypeII (MEF2-like) lineage.
Materials and Methods
Sequence Sources and/or Accession Numbers.
Sequence sources or GenBank accession numbers are as follows: AGAMOUS (10), APETALA3 (11), PISTILLATA (12), AGL1–6 (13), APETALA1 (14), AGL8 (15), AGL9 (16), CAULIFLOWER (17), AGL11, AGL12, AGL13, AGL14, AGL15 and AGL17 (18), AGL16 (AL137080, S.L. and M.Y., unpublished data), AGL18 (AL137080, S.G. and M.Y., unpublished data), AGL19 (AL161558, S.G. and M.Y., unpublished data), AGL20 (AC003680, S.G. and M.Y., unpublished data), AGL21 (ATF20D10), AGL22 (AC006592), AGL23 (AC004512), AGL24 (AF005158), AGL25 (AF116527), AGL26 (AF007270), AGL27 (AC002291/cDNA sequence, S.P. and M.Y., unpublished data), AGL28 (Y12776), AGL29 (AC004077), AGL30 (AC004138), AGL31 (T45787/cDNA sequence, S.P. and M.Y., unpublished data), AGL32 (AB007648), AGL33 (AC004484), AGL34 (AF058914), AGL35 (AF058914), AGL36 (AF058914), AGL37 (AC00451), AGL38 (AC004512), AGL39 (AF007271), AGL40 (Z99708), ANR1 (19). AGL23, AGL26, and AGL28–38 were recently identified by the Arabidopsis Genome Sequencing project. Although we lack cDNA clones for these genes, their predicted MADS-box domain sequences, on which our analyses are based, are unequivocal, because no introns have ever been found in this region.
GenBank accession numbers for the animal and fungal sequences are as follows. The MEF2-like genes used are: Homo sapiens MEF2C (L08895), Caenorhabditis elegans CEMEF2 (U36198), H. sapiens MEF2A (S25831), H. sapiens MEF2D (Q14814), Halocynthia roretzi ASMEF2 (D49970), H. sapiens MEF2B (X68502), Drosophila melanogaster DMEF2 (U03292), Saccharomyces cerevisiae SMP1 (P38128), and S. cerevisiae RLM1 (D63340). The SRF-like genes used are: H. sapiens SRF (J03161), Xenopus laevis SRF (S15018), D. melanogaster DSRF (X77532), S. cerevisiae MCM1 (P11746), S. cerevisiae ARG80 (X05327), and Schizosaccharomyces pombe PLN (D78483). The bacterial Universal Stress Protein (USP) family sequences that served as outgroup for some of the analyses are: Escherichia coli EcuspA (X67639), E. coli Ecyiit (P32132), Coxiella burnetti Coxyfmu (P45680), and Bacillus subtilis Bsyxie (P42297).
Alignment and Phylogenetic Analyses.
We used 65 amino acid sequences for the analyses. These cover the 57–60 amino acids that different authors (2, 6) have defined as the MADS domain plus a few additional conserved amino acids. These sequences were aligned by using clustal x; the alignment generated was unambiguous (complete alignment available from authors on request, and see Fig. 2). Phylogenetic analyses were conducted with unweighted maximum parsimony (MP), neighbor joining (NJ), and quartet puzzling (QP), by using the test version 4d64 of paup* (D. L. Swofford, Laboratory of Molecular Systematics, Smithsonian Institution, Washington, D.C.). For MP analyses, 100 replicates of random addition sequences keeping all optimal trees in each replicate, TBR branch swapping, and no maxtrees limit were used. Gaps were treated as missing data. NJ analyses were done by using the default factory settings and the p-distance (proportion of different amino acids between two sequences) as a distance estimator. This is the recommended distance measure when comparing distantly related sequences, because it has a smaller variance than other estimates (20).
Figure 2.

Amino acid alignment of the MADS-domain (amino acids 1 to 60) for some representative members of the plant, animal, and fungal TypeI (SRF-like) and TypeII (MEF2-like) lineages. We also show representative sequences of the genes that are not clearly assigned to either one (MADS-domains Type?). One gene from each monophyletic clade identified in MP and NJ was selected. Conserved amino acids within each group and not found in any (or in no more than two) of the MADS domains of the other group are in red. Green names indicate plant sequences and red names, animal or fungal ones (see Materials and Methods).
Nonparametric bootstrap (100 pseudoreplicates) was used to assess the reliability of individual branches. Bootstrap proportions are considered here as an index of support for a particular clade and not a statement about probability or confidence limit in the statistical sense (21). QP trees were based on 1,000 replicates by using the factory default settings. The phylogenetic relationships inferred from the trees presented here do not depend on specific sequences used to estimate phylogeny; by using subsamples of protein sequences, the same relationships were inferred (data not shown). Trees were examined with treeview (22).
To study the branching order of MADS-box gene lineages and the timing of duplications relative to the divergence of the main groups of eukaryotes (plants, animals, and fungi), we need a rooted tree. An unambiguous root location depends on using an outgroup MADS-box domain sequence. We have attempted this rooting by using four bacterial sequences that belong to the USP family as outgroup. These share very few conserved amino acids with known eukaryotic MADS-box sequences but have been defined as MADS-domain homologues based on these few conserved residues and other functional criteria (23). A better outgroup could come from a taxon representative of a sister clade of plants, animals, and fungi, such as Euglena, but this is not yet available.
As an alternative way to objectively root the MADS-box tree, we used a parsimony-based approach from Page and Charleston (24, 25). This method reconciles the gene tree to the species tree and finds the rooted gene tree that minimizes the number of gene sorting events (which could include gene losses or insufficient sampling of genomes) and duplications. This is the MADS domain tree that we put forward as a polarized phylogenetic hypothesis for this gene family. We used the species tree proposed by Baldauf and Palmer (26), in which animals and fungi are each other's closest relatives. We used groups of sequences that were shared by the NJ and MP trees, which were supported by high bootstrap values and were bona fide subfamilies, as possible outgroups to be tested. We tested seven alternative outgroups from the NJ and MP searches. The reconciled trees' method requires completely resolved trees. Therefore, one tree from each island sampled in the MP search was used. Trees from each island were very similar and differed only in some of the terminal branches. To avoid a bias because of the excessive number of possible losses and duplications found in the terminal branches where only taxa from either plants (only Arabidopsis) or one of the animal or fungal groups used were represented, we repeated this analysis by counting only basal duplications (i.e., those that are at the base of clades that combine sequences of plants, animals, and fungi).
Protein Structure Prediction.
The predictions of coiled-coil regions within the protein sequences were performed with the programs paircoil and multicoil (27, 28) and were based on the presence in the sequences of heptat-repeat signature motifs. In all cases, both programs used yielded the same result. A K domain was predicted to be present when the probability cutoff of finding coiled-coils downstream of the MADS-box domain was >0.35. The default value of 0.5 has been determined empirically to work well. However, to avoid false negatives, we decreased the cutoff value by 20%. Additionally, we predicted possible protein secondary structures using discrete state-space probability models, as implemented by the program psa (http://bmerc-www.bu.edu/psa; ref. 29). These predictions identified α-helices for the same sequences and were used to confirm results obtained from the coiled-coil prediction programs.
Results and Discussion
Ancient Duplications of Eukaryotic MADS-Box Sequences.
We present molecular evolutionary analyses of plant, animal, and fungal MADS-domain sequences, including 26 newly identified MADS-domain sequences from Arabidopsis, along with 19 previously analyzed members of this extensive gene family. The most striking result of our analyses is the discovery that animal and fungal MEF2-like sequences are more closely related to most plant MADS-domain sequences than to animal SRF-like sequences. Some conserved amino acids put the MEF2-like animal and most plant sequences in a clear monophyletic clade (hereafter referred to as TypeII MADS domains), suggesting that at least one gene-duplication event occurred before the divergence of plants and animals. In addition, a group of Arabidopsis MADS-domain sequences (AGL34-like) seem to share a more closely related ancestor with the SRF-like sequences of animals and fungi than with other plant MADS-domain sequences. The clade formed by these two related groups is referred to hereafter as the lineage of TypeI MADS domains. However, the monophyly of this group is not as well supported as that of the TypeII MADS domains, because it is supported by very few shared and unique amino acids (Fig. 2). Finally, we found a group of intermediate plant sequences that could be the result of recombination between TypeI and II MADS-box genes. These results are based on NJ, QP, and MP phylogenies, described below.
The NJ tree rooted with the putative MADS-domain sequences from bacteria is well resolved (Fig. 3a) and is similar to the one obtained by the rooting method described below (Fig. 3b). In the tree of Fig. 3a, the TypeII MADS domains that group the animal MEF2-like and most plant sequences form a well-supported monophyletic clade. However, the rest of the clades that in Fig. 3b are grouped into the TypeI lineage do not form a monophyletic group in Fig. 3a. Results in Fig. 3a suggest that AGL39-like sequences were lost or have not been found in animals and fungi. Both of the latter possibilities are unlikely, because yeast and C. elegans, whose genomes are completely sequenced, have both TypeI and TypeII MADS domains and no other types. It would be highly improbable that in both organisms the same genes were lost. We also performed MP analyses using the bacterial sequences as outgroup (not shown), but the strict consensus MP tree for these sequences does not resolve any basal branching other than that of the bacterial sequences. In the rest of the analyses, we have included only the eukaryotic MADS-domain sequences.
Figure 3.

Phylogeny of the eukaryotic MADS-box gene family. Animal and fungal sequences (H. sapiens: MEF2A_Hs, MEF2C_Hs, MEF2D_Hs, MEF2B_Hs, SRF_Hs; X. laevis: SRF_Xl; C. elegans: CEMEF2, H. roretzi: ASMEF2; D. melanogaster: DMEF2; DSRF; S. cerevisiae: SMP1_Sc, RLM1_Sc, MCM1, ARG80; S. pombe: PLNYEAST) are red; plant sequences (all from A. thaliana) are green; bacterial USP family sequences (E. coli: EcuspA, Ecyiit; Coxiella burnetti: CoxYfmu; B. subtilis: Bsyxie) (23) are blue. TypeI (SRF-like) and TypeII (MEF2-like) lineages are indicated by blue and pink brackets, respectively. (a) The NJ tree rooted with the bacterial USP family (see ref. 23) is shown in a, and the NJ tree rooted by minimizing the reconciliation cost (see Materials and Methods) is shown in b. Branch lengths are proportional to the number of amino acid substitutions. Bootstrap values shown on branches; in b, values in parentheses correspond to analyses done without the underlined sequences. Branches with bootstrap values <50% are collapsed. Sequences within purple square are those for which a coiled-coil structure downstream of the MADS-domain (K domain) was predicted.
An alternative way to root the MADS-domain protein tree objectively is to use Page and Charleston's (24) approach to find the root position that minimizes the number of duplications and sorting events in the protein tree, when this is reconciled to the species tree (see Materials and Methods). We show the rooted NJ tree that minimized the reconciliation cost (49 total or 3 basal duplications and 17 sorting events) as the polarized phylogenetic hypothesis for this gene family. The bootstrap NJ tree reveals two well supported (>50%) clades. The first one is constituted by the TypeI MADS-domain sequences and groups the animal SRF-like genes with two newly identified plant lineages, AGL34- and AGL23-like, plus AGL30, AGL33, and AGL39. The second, TypeII MADS-domain sequences, includes the rest of the plant sequences and the animal MEF2-like sequences.
Using MP analyses, we obtained a total of 647 most parsimonious trees (consistency index = 0.544, retention index = 0.695, rescaled consistency index = 0.378) of a length of 700 steps. The strict consensus-rooted MP tree resolves the monophyletic clade that includes animal SRF-like and plant AGL34-like sequences plus AGL30, AGL33, and AGL39, but with a low bootstrap support (<50%). In contrast to the NJ tree, the strict consensus MP tree identifies the AGL-23 plant MADS-domain clade as a sister branch of the animal MEF2-like sequences, but with a very low bootstrap support (<20%). The MP tree also resolves the AGL25 clade as sister to the monophyletic group formed by the rest of the plant TypeII and the animal MEF2-like sequences, also with a very low bootstrap support (<20%). MP groups the animal and fungal MEF2-like sequences with the plant MADS-domain sequences in a monophyletic clade and places the animal and fungal SRF-like sequences as sister group with a good bootstrap support (>50%).
When reconciled to the species tree, the least costly MP gene tree still requires a greater number of basal gene duplications and losses (49 total or 8 basal duplications and 22 sorting events) than the NJ tree shown (Fig. 3b). This MP tree also defined TypeI and TypeII groups as sister to each other. These results confirm that the most parsimonious root location among all trees tested is between the TypeI and TypeII lineages that we have identified. We compared the length of the Bootstrap NJ topology with the MP strict consensus tree using macclade (Ver. 3.0) and found that they are of equal length. Therefore, based on the data at hand, we propose the tree shown in Fig. 3b as the most parsimonious hypothesis on the polarized evolutionary history of the eukaryotic MADS-box gene family. Finally, the QP tree also resolved the same TypeI and TypeII clades formed by the same family members as in the NJ tree shown (frequency value equal to 40%).
The inconsistent placement of the AGL23 clade between the NJ/QP and MP topologies, as well as the low bootstrap value for the TypeI clade in the MP strict consensus tree, suggests that some plant sequences cannot be unambiguously associated to either the TypeI or TypeII lineages. In fact, if AGL30, AGL33, AGL39, and the AGL23-like genes are removed, NJ, MP, and QP analyses yield resolved and well supported trees (bootstrap values of >90% and 50% for both lineages in NJ and MP analyses, respectively; see Fig. 3b; and 89% frequency in QP). These problematic sequences could be the result of recombination between TypeI and TypeII sequences. This possibility is suggested because they share some of the synapomorphies that define each of the two lineages (see Fig. 2). The fact that these sequences group in a clearly monophyletic clade suggests an ancient recombination event that would have been followed by several duplications. To unambiguously resolve the origin and phylogenetic position of these genes, more information is required.
In an effort to explore further the monophyly of the TypeI groups that we propose, we did MP and NJ phylogenetic analyses of this clade by using only one sequence of the MEF-2 sequences as outgroup (not shown). In these analyses, the plant AGL34-like, plus AGL30 and AGL33, plus the animal SRF-like sequences, form a well supported (bootstrap = 63%) monophyletic group, and AGL23-like and AGL39 sequences group in a clade sister to that formed by the former sequences. Both of these clades form a monophyletic lineage with 76% of bootstrap support.
The results presented here imply that features shared by proteins within the MEF2-like and SRF-like clades were present in the ancestral eukaryotes and have remained practically unchanged during the evolution of animal, fungal, and plant lineages. The TypeII MADS-domain sequences share some conserved amino acids that are found in none of the TypeI MADS domains (synapomorphies; see Fig. 2). In contrast, the TypeI MADS have only one synapomorphy that defines this clade and some that are shared by all but one or a few sequences. This suggests that there has been a stronger functional constraint within the TypeII than the TypeI MADS-domain lineages. TypeI MADS domains are conserved within animals and within plants, but they differ between these two species' lineages. MADS domains from yeast from both TypeI and TypeII lineages are the most divergent ones.
It will be interesting to determine whether the plant TypeI MADS-box sequences represent expressed genes or are instead pseudogenes. But the fact that at least one of these sequences, AGL39, is represented as an EST clone (GenBank accession no. C99890), as well as the high conservation among AGL34-like sequences, suggests that these members are indeed expressed. Future studies should be devoted to characterizing functionally these genes in Arabidopsis.
The conserved MADS-domain motifs within each lineage may serve as the basis of the common functional properties of all proteins within the TypeI and TypeII clades. Indeed, in vitro DNA-binding assays revealed that chimeric proteins with either the SRF or MEF2A amino-terminal region of the MADS domain and the rest of the AP1, AP3, PI, and AG plant proteins, acquired the respective and distinct DNA-binding specificity of SRF or MEF2A. However, in vivo assays did not distinguish between chimeric and full-length wild-type proteins' functions. Both results put together suggest that DNA-binding specificity, which must underlie functional specificity of MADS-domain proteins, is determined not solely by sequences within the MADS-domain but also by sequences within other domains that may affect dimerization with protein partners (30).
Additional in vivo experiments show that although chimeric genes with the amino terminus of either the SRF or MEF2A MADS-domain and the rest of AP1 may rescue ap1–1 mutant plants when expressed under the wild-type AP1 promoter, the chimera with the MEF2A MADS-domain amino terminus (i.e., within-lineage chimera) rescued mutant phenotypes more effectively than those harboring crosslineage constructs (i.e., from SRF; ref. 31). Our phylogenetic results support, as suggested by these functional analyses, that differences between TypeII and TypeI MADS domains have a role in defining function. Indeed, ectopic expression experiments of chimeric proteins suggest that the MADS and I domains define functional specificities of APETALA1 and AGAMOUS (32, 33), both TypeII (MEF2-like) plant members. However, the conservation of MADS-domain sequences within each lineage and additional functional studies (see below) also suggest that domains outside the MADS domain are important for functional specificity. The K domain, typical of previously characterized plant TypeII proteins, is one such domain.
Evolution of the Plant K Domain.
The K domain is an ≈70-aa domain located downstream of the DNA-binding MADS domain, typically spanning positions 110 to 180 of plant MADS proteins. It has a regular spacing of hydrophobic amino acids, and it is assumed to adopt a coiled-coil structure (see Fig. 1). This structural motif has been described for the great majority of previously identified plant MADS-domain proteins (4). To investigate the origin and evolution of the K domain, we used protein-structure programs to predict whether the AGL34 and AGL23 clade members, as well as the other plant and animal MADS-domain sequences analyzed, contain a K domain. In Fig. 3b, we boxed the sequences with a predicted coiled-coil structure downstream of the MADS domain.
Coiled-coil structures were not predicted for any of the animal sequences, any of the plant AGL34 or AGL23-like, or for AGL30, AGL33, and AGL39. These sequences also lack any significant sequence similarity to other plant MADS-domain sequences outside of the MADS domain. Interestingly, whereas protein–structure prediction programs clearly identify a coiled-coil domain for most plant members of the TypeII lineage (MEF2-like), they fail to predict such a structure for a few members of this group (the AGL25-like and AGL12) that seem to lack some of the conserved hydrophobic amino acids. This result suggests that the absent amino acids might be critical for the formation of the coiled-coil structure. Both methods used here have been reported to identify positively all of the sequences that form coiled coils in Protein Data Bank structures containing this type of helical structure (27). Thus, the coiled-coil predictions presented in this work have a high level of reliability (>95%), well above standard secondary structure prediction methods.
Animal SRF- and MEF2-like proteins contain additional conserved regions, referred to as SAM and MEF2 domains (2). These and the K domain could be the regions involved in the functional divergence among members of each MADS-domain lineage. Ectopic expression experiments of chimeric proteins suggest that functional specificities of APETALA3 and PISTILLATA MADS-domain proteins in organ determination rely on the I and K domains of these genes (31, 32). Recent experiments for two plant MADS-domain proteins (APETALA1 and CAULIFLOWER) suggest that differences between the K domains of these two recently duplicated genes explain at least part of the functional differences between these paralogous loci (E.R.A.-B. and M.F.Y., unpublished results).
Evolution of MADS-Domain Proteins in Eukaryotes: A Synthesis.
The results described here suggest a hypothetical scenario for the evolution of the MADS-box gene family in eukaryotes (Fig. 4). From our analyses, it appears that at least one ancestral MADS-box gene duplicated in the common ancestor of the major eukaryotic kingdoms more than a billion years ago to give rise to the distinct TypeI (SRF-like) and TypeII (MEF2-like) lineages found in plants, fungi, and animals today. In yeast and C. elegans genomes, MADS-box sequences of both TypeI and TypeII have been found (several of each in yeast and one of each in C. elegans). These results support our proposition that eukaryotic MADS-box sequences can be assigned to either of two main lineages that are both present at least in fungi and animals. The Arabidopsis genome will be sequenced to completion soon, and we will then be able to test unambiguously the presence of these and additional lineages in plants. Phylogenetic analyses that include MADS domains from basal eukaryotes and TypeI sequences from other plants will help confirm the uniqueness of the ancestral duplication and the monophyly of the TypeI clade.
The evolution of additional domains beyond the MADS domain could have occurred independently along the animal and plant lineages after their divergence from each other, as suggested in our model (Fig. 4), or these could have been present in the ancestral MADS-box genes and then lost along different lineages. In plants, the K domain evolved within the TypeII (MEF2-like) lineage but not the TypeI (SRF-like) lineage. Because most of the TypeII class of plant MADS-box genes are predicted to encode a K domain, this plant-specific domain probably evolved before the extensive duplications that generated this particular lineage. Interestingly, some of the recently cloned MADS-box genes from ferns (33) are predicted to contain K domains (data not shown), indicating that this domain was present at least 395 million years ago in the common ancestors of ferns and seed plants.
We can use parsimony to argue that the K domain originated after the duplication that led to the MEF2- and SRF-like animal MADS-box genes. However, based on the phylogeny of Fig. 3b, we cannot distinguish whether it evolved along the plant lineage after it diverged from the animal one, or whether it was present in the ancestral TypeII-like gene and then lost in animal and some plant lineages. A recent phylogenetic analysis of the M, I, and K domains of all plant protein sequences, (E.R.A.-B., S.L., S.P., S.G., C.B., G.D., and M.Y., unpublished work) suggests that AGL12 and the AGL25-like sequences are basal to the rest of the Arabidopsis TypeII AGLs. This result supports the hypothesis that the K domain evolved along the plant lineage after it diverged from animals and fungi (Fig. 4). Identification of MADS-box genes within the most basal extant green plant lineages (including green algae and the bryophytes) and in one of the extant common ancestors of plants and animals (e.g., Euglena) should provide experimental tests for the hypotheses postulated in this model of MADS-box gene family evolution. Animal SRF- and MEF2-like domains (see Figs. 1 and 4) may have evolved within animal lineages (as suggested in Fig. 4), or they could have been present also before the divergence of plants and animals and subsequently lost and replaced in plants.
MADS-box genes probably played key roles in the early evolution of flowering plants and in plant evolution in general, perhaps analogous to the roles played by homeobox genes in the evolution of animal form (34, 35). This scenario is suggested by the fact that MADS-box gene mutations, as those of homeobox genes in animals, also produce homeotic conversions in flowers, suggesting that they occupy similar places in the regulatory networks that control development (36). Like homeobox genes, MADS-box genes are also highly conserved among distantly related plants, and orthologous genes form monophyletic clades (6–9). To test the long-suspected parallel between the molecular evolution of the MADS-box gene family and the evolution of plant form, a polarized gene phylogeny is necessary. We have proposed here a hypothesis for the evolutionary history of the MADS-domain protein family, including the nearly complete Arabidopsis MADS-box sequence complement, which suggests that eukaryotic MADS-box sequences can be assigned to two main lineages and locates the root of the whole family between them. These analyses may be used to guide the search for MADS-box sequences in basal eukaryotes and the assignment of newly cloned genes from other plant species to one of the clades proposed in this study. Further phylogenetic and population genetic studies (e.g., ref. 37) as well as functional analyses of the MADS-box family and other important transcriptional regulators should lead to a better understanding of the molecular evolution of developmental mechanisms. These mechanisms underlie the morphological evolution of plants and animals, the understanding of which is still elusive to evolutionary biologists.
Acknowledgments
We thank C. Ferrándiz, W. Crosby, C. Gustafson-Brown, and S. Rounsley for providing unpublished sequences. Special thanks to A. Chaos and F. Vergara for illuminating phylogenetic conversations and for help during figure preparation. Two anonymous reviewers put important effort into helping us improve this paper. Many thanks to A. Cortés for help in various tasks and to R. Salas for help in Fig. 4. C. Ferrándiz, S. Kempin, M. Ng, and A. Sessions provided useful suggestions. This work was supported by a grant from the National Science Foundation to M.F.Y. and a CONACYT (Consejo Nacional de Ciencia y Tecnología, México) grant to E.R.A.-B. Also, E.R.A.-B. was a Pew Foundation Fellow during the completion of this work. S.P. had a long-term postdoctoral fellowship from the Human Frontiers Science Program Organization, S.L. had a Lucille P. Markey predoctoral fellowship, and C.B. and L.M.C. had a Ph.D. scholarship from CONACYT and Universidad Nacional Autónoma de México.
Abbreviations
- MP
maximum parsimony
- NJ
neighbor joining
- QP
quartet puzzling
- USP
Universal Stress Protein
References
- 1.Doebley J, Lukens L. Plant Cell. 1998;10:1075–1082. doi: 10.1105/tpc.10.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shore P, Sharrocks A D. Eur J Biochem. 1995;229:1–13. doi: 10.1111/j.1432-1033.1995.tb20430.x. [DOI] [PubMed] [Google Scholar]
- 3.Coen E S, Meyerowitz E M. Nature (London) 1991;353:31–37. doi: 10.1038/353031a0. [DOI] [PubMed] [Google Scholar]
- 4.Riechmann J L, Meyerowitz E M. J Biol Chem. 1997;378:1079–1101. [PubMed] [Google Scholar]
- 5.Liljegren S J, Ferrándiz C, Alvarez-Buylla E R, Pelaz S, Yanofsky M F. Flowering Newsletter. 1998;25:9–19. [Google Scholar]
- 6.Theissen G, Kim J T, Saedler H. J Mol Evol. 1996;43:484–516. doi: 10.1007/BF02337521. [DOI] [PubMed] [Google Scholar]
- 7.Doyle J J. Syst Biol. 1994;43:307–328. [Google Scholar]
- 8.Purugganan M D, Rounsley S D, Schmidt R J, Yanofsky M F. Genetics. 1995;140:345–356. doi: 10.1093/genetics/140.1.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Purugganan M D. J Mol Evol. 1997;45:392–396. doi: 10.1007/pl00006244. [DOI] [PubMed] [Google Scholar]
- 10.Yanofsky M F, Ma H, Bowman J L, Drews G N, Feldmann K A, Meyerowitz E M. Nature (London) 1990;346:35–39. doi: 10.1038/346035a0. [DOI] [PubMed] [Google Scholar]
- 11.Jack T, Brockman L L, Meyerowitz E M. Cell. 1992;68:683–697. doi: 10.1016/0092-8674(92)90144-2. [DOI] [PubMed] [Google Scholar]
- 12.Goto K, Meyerowitz E M. Genes Dev. 1994;8:1548–1560. doi: 10.1101/gad.8.13.1548. [DOI] [PubMed] [Google Scholar]
- 13.Ma H, Yanofsky M F, Meyerowitz E M. Genes Dev. 1991;5:484–495. doi: 10.1101/gad.5.3.484. [DOI] [PubMed] [Google Scholar]
- 14.Mandel M A, Gustafson-Brown C, Savidge B, Yanofsky M F. Nature (London) 1992;360:273–277. doi: 10.1038/360273a0. [DOI] [PubMed] [Google Scholar]
- 15.Mandel M A, Yanofsky M F. Plant Cell. 1995;7:1763–1771. doi: 10.1105/tpc.7.11.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandel M A, Yanofsky M F. Sexual Plant Reprod. 1998;11:22–28. [Google Scholar]
- 17.Kempin S A, Savidge B, Yanofsky M F. Science. 1995;267:522–525. doi: 10.1126/science.7824951. [DOI] [PubMed] [Google Scholar]
- 18.Rounsley S D, Ditta G S, Yanofsky M F. Plant Cell. 1995;7:1259–1269. doi: 10.1105/tpc.7.8.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang H, Forde B G. Science. 1998;279:407–409. doi: 10.1126/science.279.5349.407. [DOI] [PubMed] [Google Scholar]
- 20.Burke W D, Eickbush D G, Xiong Y, Jacubczak J, Eickbush T H. Mol Biol Evol. 1993;10:163–185. doi: 10.1093/oxfordjournals.molbev.a039990. [DOI] [PubMed] [Google Scholar]
- 21.Hillis D M, Bull J J. Syst Biol. 1993;42:182–192. [Google Scholar]
- 22.Page R D M. CABIOS. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 23.Mushegian A R, Koonin E V. Genetics. 1996;144:817–828. doi: 10.1093/genetics/144.2.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Page R D M, Charleston M A. Mol Phyl Evol. 1997;7:231–240. doi: 10.1006/mpev.1996.0390. [DOI] [PubMed] [Google Scholar]
- 25.Page R D M, Holmes E C. Molecular Evolution. A Phylogenetic Approach. Oxford: Blackwell Scientific; 1998. [Google Scholar]
- 26.Baldauf S L, Palmer J D. Proc Natl Acad Sci USA. 1993;90:11558–11562. doi: 10.1073/pnas.90.24.11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berger B, Wilson D B, Wolf E, Tonchev T, Milla M, Kim P S. Proc Natl Acad Sci USA. 1995;92:8259–8263. doi: 10.1073/pnas.92.18.8259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wolf E, Kim P S, Berger B. Protein Sci. 1997;6:1179–1189. doi: 10.1002/pro.5560060606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stultz C M, White J V, Smith T F. Protein Sci. 1993;2:305–314. doi: 10.1002/pro.5560020302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riechmann J L, Meyerowitz E M. Mol Biol Cell. 1997;8:1243–1259. doi: 10.1091/mbc.8.7.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krizek B A, Riechmann J L, Meyerowitz E M. Sex Plant Reprod. 1999;12:14–26. [Google Scholar]
- 32.Krizek B A, Meyerowitz E M. Proc Natl Acad Sci USA. 1996;93:4063–4070. doi: 10.1073/pnas.93.9.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Münster T, Pahnke J, DiRosa A, Kim J, Martin W, Saedler H, Theissen G. Proc Natl Acad, Sci USA. 1997;94:2415–2420. doi: 10.1073/pnas.94.6.2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Raff R A. The Shape of Life: Genes, Development, and the Evolution of Animal Form. Chicago: Univ. of Chicago Press; 1996. [Google Scholar]
- 35.Carroll S B. Nature (London) 1995;376:479–485. doi: 10.1038/376479a0. [DOI] [PubMed] [Google Scholar]
- 36.Mendoza L, Alvarez-Buylla E R. J Theor Biol. 1998;193:307–319. doi: 10.1006/jtbi.1998.0701. [DOI] [PubMed] [Google Scholar]
- 37.Purugganan M D, Suddith J I. Proc Natl Acad Sci USA. 1998;95:8130–8134. doi: 10.1073/pnas.95.14.8130. [DOI] [PMC free article] [PubMed] [Google Scholar]