

Abstract
Neuronal growth cones are the highly motile structures at the tip of axons that can detect guidance cues in the environment and transduce this information into directional movement towards the appropriate target cell. To fully understand how guidance information is transmitted from the cell surface to the underlying dynamic cytoskeletal networks, one needs a model system suitable for live cell imaging of protein dynamics at high temporal and spatial resolution. Typical vertebrate growth cones are too small to quantitatively analyze F-actin and microtubule dynamics. Neurons from the sea hare Aplysia californica are 5-10 times larger than vertebrate neurons, can easily be kept at room temperature and are very robust cells for micromanipulation and biophysical measurements. Their growth cones have very defined cytoplasmic regions and a well-described cytoskeletal system. The neuronal cell bodies can be microinjected with a variety of probes for studying growth cone motility and guidance. In the present protocol we demonstrate a procedure for dissection of the abdominal ganglion, culture of bag cell neurons and setting up an imaging chamber for live cell imaging of growth cones.
Protocol
Solutions
- L15-ASW cell culture medium (1l)
- 1 bag of L15 powder
- add 800 ml of H2O ultrapure
- NaCl 400 mM
- MgSO4 27 mM
- MgCl2 28 mM
- L-Glutamine 4 mM
- Gentamicin 50 µg/ml
- HEPES 5 mM
- Adjust to pH 7.9
- Add drop by drop CaCl2 9.3 mM (stop if precipitates)
- Add H2O ultrapure to 1 l
- Check osmolarity (950-1000 mmol/kg) with osmometer (vapor pressure, Wescor # 5520)
- Filter 0.22 µm using a positive pressure filtration unit (Filters: Millipore SVGV010RS)
- Store at 4°C for up to 1 month
Poly-L-lysine (70-150 kD) 200 µg/ml stock solution in sterile H2O ultrapure kept at -20oC
0.5 M MgCl2 solution
Day 1: Dissection of Aplysia abdominal ganglion
Preparation of the enzymatic dissociation solution
Weigh 9-10 mg of Dispase enzyme (Worthington, Cat: LS02111) in an Eppendorf tube
Add 900 µl of L15-ASW culture medium, and 100 µl of sterile H2O ultrapure
Mix and place at room temperature (RT)
Dissection of abdominal ganglion
Fill a 50 ml syringe with 0.5 M MgCl2 solution.
Take one animal from tank, handle gently to avoid animal's inking defense response.
Place the Aplysia on its side on a dissection board (Styrofoam board), rostral side to the right, caudal side to the left.
Insert the needle into animal just behind the head and inject all MgCl2 solution into the body cavity.
Gently rub the animal to disperse the MgCl2 solution throughout body cavity, wait for a few minutes for the animal to be completely anaesthetized.
Pin the head and tail of the animal to the dissection board with two needles.
Use forceps to lift skin, cut through skin and muscle layer with large scissors towards the head and tail to make a large opening on the side.
Find the abdominal ganglion under the brush-like ovary. Use forceps to hold the connecting nerves about 1 cm in front of the abdominal ganglion, cut the nerves with dissection scissors in front of the forceps holding position and behind the abdominal ganglion.
Place ganglion into the Dispase solution and incubate for 15-16 h at 22°C using a temperature-controlled water bath.
Day 2: Plating of Aplysia bag cells
Preparing poly-lysine-coated coverslips
In a flow bench, prepare three 35mm petri dishes as working dishes for dissection of the ganglion, filling them with 4 ml L15-ASW medium each. Transfer the abdominal ganglion from the dispase solution to one of the working dishes after 15.5 hours of digestion.
Using forceps take acid-cleaned #1.5 coverslips (22x22 mm) stored in ethanol, remove alcohol and sterilize coverslips by flaming them briefly over Bunsen burner. Let them briefly cool before placing the coverslips into the culture dishes.
Prepare a curved plating tip by bending a yellow tip over the Bunsen burner (flame only briefly to avoid melting of plastic!).
Prepare appropriate amount of 20 µg/ml poly-lysine solution by diluting 200 µg/ml stock solution with sterile H2O ultrapure.
Cover each coverslip with 500 µl of 20 µg/ml poly-lysine and incubate for 20 mins at RT.
Wash each coverslip 3-5 times with 500 µl sterile H2O ultrapure each.
Remove water from coverslips; fill each dish with 4 ml of L15-ASW culture medium.
Dissociation of bag cells
Under a dissection microscope, cut the abdominal ganglion in half using dissection scissors and forceps previously cleaned with 95% ethanol (Figure 1 line 1).
On one half of the ganglion, cut between bag cell cluster and the hemiganglion cells (Figure 1 line 2), and cut away remaining nerve extensions on the other side of bag cells (Figure 1 line 3). Work on one half of ganglion at a time; leave the other half in the dish while cells from first cluster are being plated.
Using two forceps (or one forceps and dissection scissors), release bag cell cluster by pushing aside the connective tissue sheath surrounding the bag cell cluster. Cut away connective tissue with dissection scissors if needed. Beware not to squeeze and touch the bag cell cluster too much during this process.
When most of the connective tissue is removed, transfer the bag cell cluster into a new working dish with the bent yellow tip prepared earlier. Be careful not to let the bag cell cluster be trapped inside the yellow tip or stuck at the air/medium interface.
Triturate the cluster with yellow tip to remove individual bag cells. Do this by pipetting the cluster in and out of the tip. Start with low shear forces and increase force as needed. Always use the lowest force needed to remove neurons without killing too many cells.
Collect 3-5 healthy bag cells and transfer them to a dish with a poly-lysine-coated coverslip. Avoid picking up dead cells (they are black/transparent in the center) and connective tissue debris.
Repeat step 6. and place about 15-25 live neurons into the center of each coverslip. If dead cells or debris are transferred as well, remove the unwanted material by picking it up or blowing it away from the healthy cells. This process takes about 2-3 hours for both clusters.
Once finished keep the culture dishes on the flow bench for at least 2 hours at RT to allow cells to attach (keep flow bench blower off to avoid unwanted vibration).
Place the dishes into an incubator at 14°C overnight, or until further use. Growth cones may develop in 4-10 hours.
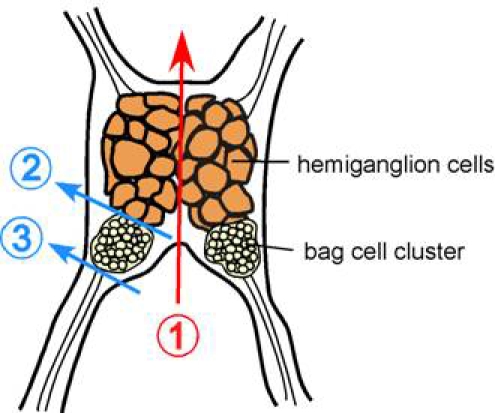 Figure 1. Schematic of Aplysia californica abdominal ganglion. Color lines indicate where to cut to obtain bag cell clusters.
Figure 1. Schematic of Aplysia californica abdominal ganglion. Color lines indicate where to cut to obtain bag cell clusters.
Day 3: Setting up chamber for live imaging of Aplysia bag cells
Assembly of imaging chamber
Before using bag cells for live cell imaging experiments, inspect cells on a low power microscope. If cells have not developed healthy growth cones, supply fresh medium and leave at RT for 1-2 hours.
To supply plates with fresh L15-ASW medium, remove half (~2ml) of old medium with a vacuum line, and add 2 ml of new L15-ASW medium per petri dish. It is important not to completely remove all medium from the dish all at once, as this will remove cells from the coverslip.
For bag cell imaging, we assemble two coverslips and two plastic spacers in a sandwich-like imaging chamber. The top coverslip in the sandwich is cut shorter to allow easy exchange of medium and reagents. During assembly, coverslips with bag cells attached should be submerged in L15-ASW medium at all times to prevent cells from dislodging.
To prepare the chamber, cut off ~2mm from right side of a #1 coverslips 22x22 mm with diamond pen (glass cutter) and the ruler.
Using a 1 ml syringe, apply high vacuum grease to two sides of the shorter coverslip, and attach plastic spacers to each side. Apply vacuum grease to the top of both spacers.
Invert the coverslip and spacer assembly, and place into petri dish containing bag cells on a coverglass. Align and gently press down to stick top coverslip to bottom coverslip with the reverse end of two Q-tips.
Using forceps, gently lift sandwich assembly out of the petri dish without the medium leaking out. Align the two coverslips, if needed, and wipe off excess medium with a Kimwipe. Place the sandwich onto the bottom part of the microscope slide holder and attach the cover with care using clips.
Clean the center of the top side of sandwich with Q-tips dabbed in glass cleaner (Sparkle is used as glass cleaner). Quickly dry off the glass cleaner with a second, dry Q-tip to avoid streak lines. Do the same for the bottom coverslip. Clean coverslips free of streak lines are important for high quality imaging.
Bag cells on the microscope
Since Aplysia bag cell neuronal cultures are at low density, it is best if your microscope setup has stage positioning functions to facilitate switching between cells. Bag cells can be easily kept at RT for several hours on stage during imaging experiments. Exchange the medium periodically to avoid evaporation.
Discussion
Aplysia bag cell neurons provide a serum-free neuronal cell culture system with very few non-neuronal cells. These neurons form very large growth cones suitable to address a number of important cell biological questions. Bag cell neurons can easily be manipulated and imaged at room temperature over several hours. Using Fluorescent Speckle Microscopy (FSM) one can quantitatively analyze the various parameters of F-actin and microtubule polymerization and translocation dynamics. These imaging tools together with the recently released Aplysia genome information as well as improved expression techniques make these neurons a powerful model system for studying molecular and cellular mechanisms of neuronal growth cone motility and guidance.
Acknowledgments
We would like to thank Ryan Maneri (Oystercatcher Productions, LLC) for filming our procedure and Rodney McPhail (Department of Biological Sciences, Purdue University) for assistance with editing the dissection video. Research in the Suter lab is supported by grants from the NIH (R01 NS049233) and the Bindley Bioscience Center at Purdue University to D.M.S.
References
- Forscher P, Kaczmarek LK, Buchanan JA, Stephen SJ. Cyclic AMP induces changes in distribution and transport of organelles within growth cones of Aplysia bag cell neurons. J. Neurosci. 1987;7:3600–3611. doi: 10.1523/JNEUROSCI.07-11-03600.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter DM, Errante LD, Belotserkovsky V, Forscher P. The Ig superfamily cell adhesion molecule, apCAM, mediates growth cone steering by substrate-cytoskeletal coupling. J. Cell. Biol. 1998;141:227–240. doi: 10.1083/jcb.141.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer AW, Kabir N, Forscher P. Filopodia and actin arcs guide the assembly and transport of two populations of microtubules with unique dynamic parameters in neuronal growth cones. J. Cell. Biol. 2002;158:139–152. doi: 10.1083/jcb.200203038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter DM, Schaefer AW, Forscher P. Microtubule dynamics are necessary for SRC family kinase-dependent growth cone steering. Curr. Biol. 2004;14:1194–1199. doi: 10.1016/j.cub.2004.06.049. [DOI] [PubMed] [Google Scholar]