

Abstract
Cholestasis causes hepatocyte death, possibly due to mitochondrial injury. This study investigated whether NIM811, an inhibitor of the mitochondrial permeability transition (MPT), attenuates cholestatic liver injury in vivo. Cholestasis was induced in mice by bile duct ligation (BDL). NIM811 was gavaged (20 mg/kg) before BDL and daily (10 mg/kg) afterwards. Mitochondrial depolarization, cell death and MPT onset were assessed by intravital confocal/multiphoton microscopy of rhodamine 123 (Rh123), propidium iodide (PI) and calcein. After BDL, serum alanine aminotransferase (ALT), hepatic necrosis and apoptosis all increased. NIM811 decreased ALT, necrosis and apoptosis by 60 to 86%. In vehicle-treated mice at 6 h after BDL, viable hepatocytes with depolarized mitochondria were 18/high power field (hpf), and non-viable cells were ∼1/hpf, showing that depolarization preceded necrosis. Calcein entered mitochondria after BDL, indicating MPT onset in vivo. NIM811 decreased depolarization by 72%, prevented calcein entry into mitochondria and blocked release of cytochrome c. Hepatic TNFα, TGF-β1 and procollagen α1(I) mRNA, α-smooth muscle actin, and Sirius red staining for collagen increased after BDL but were not different in vehicle- and NIM811-treated mice. Taken together, NIM811 decreased cholestatic necrosis and apoptosis but did not block fibrosis, indicating that the MPT plays an important role in cholestatic cell death in vivo.
Introduction
Cholestasis represents an interruption of bile secretion developing either acutely or chronically. Cholestatic liver disease results from hepatitis, complications of surgery, trauma, intravenous feeding, tumors, gallstones, certain medications and idiopathic, genetic and metabolic diseases, such as primary biliary cirrhosis and sclerosing cholangitis (Poupon, et al., 2000). Accumulation of bile acids (BA) induces injury due to the toxicity of hydrophobic BA to hepatocytes and cholangiocytes (Kass, 2006). Toxic BA cause both necrotic and apoptotic cell death (Fickert, et al., 2005;Gujral, et al., 2004;Faubion, et al., 1999). Chronic cholestatic liver diseases eventually lead to liver cirrhosis, which is a leading indication for liver transplantation.
How cholestasis induces liver injury and fibrosis remains controversial. Cytotoxic bile salts trigger hepatocyte apoptosis partly by ligand-independent activation of CD95 (Fas) death receptors (Faubion, et al., 1999). In isolated hepatocytes, exposure to hydrophobic BA causes mitochondrial production of reactive oxygen species (ROS) (Rolo, et al., 2003;Sokol, et al., 1995). Cytotoxic BA also induce ROS formation by activation of NADPH oxidase (Reinehr and Haussinger, 2007). ROS can trigger opening of permeability transition (PT) pores in the mitochondrial inner membrane that nonspecifically transport aqueous solutes up to a molecular weight of about 1500 Da (Halestrap and Brennerb, 2003;Kantrow, et al., 2000). Onset of the MPT collapses the mitochondrial membrane potential, leading to failure of ATP synthesis, release of proapoptotic cytochrome c, and necrotic and apoptotic cell death (Zamzami, et al., 1996;Kim, et al., 2003). Antioxidants and cyclosporin A (CsA), a specific inhibitor of the MPT (Halestrap and Davidson, 1990), protect cultured hepatocytes from BA-induced loss of viability (Yerushalmi, et al., 2001;Gores, et al., 1998;Rolo, et al., 2003), implicating a role for a ROS-dependent MPT in BA hepatotoxicity. Mitochondria isolated from human livers also generate ROS and undergo MPT onset in response to BA (Sokol, et al., 2005). However, whether MPT pore opening occurs in vivo during cholestasis to contribute to cholestatic liver injury and liver fibrosis remains to be determined.
N-Methyl-4-isoleucine cyclosporin (NIM811) is a non-immunosuppressive derivative of CsA that inhibits the MPT in cultured hepatocytes, isolated liver mitochondria, and liver grafts after transplantation in mice and rats (Waldmeier, et al., 2002;Zhong, et al., 2007;Theruvath, et al., 2008). NIM811 is equipotent to CsA for inhibition of onset of the MPT in cultured cells and isolated mitochondria (Waldmeier, et al., 2002;Theruvath, et al., 2008). NIM811 also suppresses collagen production and proliferation of cultured hepatic stellate cells (HSC) (Kohjima, et al., 2007). Study of mitochondrial dysfunction and the MPT in vivo has been difficult. However, advances in intravital confocal/multiphoton microscopy now permit assessment of mitochondrial function and inner membrane permeability in livers of living animals (Theruvath, et al., 2008;Zhong, et al., 2007). Since the MPT plays an important role in both necrosis and apoptosis (Kim, et al., 2003), and since MPT onset occurs in hepatocytes after exposure to BA, we used intravital confocal/multiphoton microscopy to evaluate the role of the MPT in cholestatic liver injury in vivo and to determine whether blockade of the MPT with NIM811 prevents cholestatic cell death and liver fibrosis.
Methods
Animals
Male BALB/c mice (8 - 9 weeks) were gavaged with N-Methyl-4-isoleucine cyclosporin (NIM811, Novartis Pharma Ltd., Switzerland, 20 mg/kg) or an equal volume of vehicle containing 8.3% polyethoxylated castor oil (Sigma, St. Louis, MO) and 8.3% ethanol at 2 h before surgery. Mice underwent bile duct ligation (BDL) or sham-operation under ether anesthesia. Briefly, the common bile duct was double ligated near the liver and transected between ligatures. NIM811 (10 mg/kg) or vehicle was gavaged daily after surgery. All animals received humane care in compliance with institutional guidelines. Animal protocols were approved by the Institutional Animal Care and Use Committee.
Clinical Chemistry and Histology
At times indicated in the figure legends, mice were anesthetized with pentobarbital (80 mg/kg, i.p.), and blood was collected from inferior vena cava. Livers were harvested and slides were prepared as described elsewhere (Zhong, et al., 2003). In sections stained with hematoxylin and eosin (H+E), ten random fields per slide were captured in a blinded manner using a Universal Imaging Image-1/AT image acquisition and analysis system (West Chester, PA) with an Axioskop 50 microscope (Carl Zeiss, Inc., Thornwood, NY) and a 10× objective lens. Necrotic areas were quantified by image analysis using IP Lab 3.7v software (BD Biosciences, Rockville, MD) by dividing necrotic areas by total cellular area of the images. Some slides were stained with 0.1% Sirius red (Polysciences Inc., Warrington, PA) and fast green FCF (Sigma Chemical, St. Louis, MO) to evaluate liver fibrosis, and Sirius red-positive area was quantified as described (Zhong, et al., 2003). In other experiments, liver tissue was frozen and stored at -80°C for later mRNA analysis. Serum alanine transaminase (ALT) was measured using a kit from Pointe Scientific (Uncoln Park, MI).
Intravital Confocal and Multiphoton Microscopies
Rhodamine123 (Rh123, Sigma, St. Louis, MO) was used to monitor mitochondrial polarization after cholestasis. Rh123, a cationic fluorophore, is taken up by polarized mitochondria and is released when mitochondria depolarize (Emaus, et al., 1986). Propidium iodide (PI) was used to label nuclei of non-viable cells. At 6 h after BDL or sham-operation, mice were anesthetized with pentobarbital (80 mg/kg, i.p.) and connected to a small animal ventilator via a respiratory tube (20-gauge catheter) inserted into the trachea. Rh123 (2 μmol/mouse) and PI (0.04 μmol/mouse) were infused via polyethylene (PE10) tubing inserted into the carotid artery over 10 min. Preliminary studies showed that carotid injections yielded reliable and uniform labeling of hepatocytes by fluorophores without disturbance to hepatic blood supply.
MPT pore opening was assessed using calcein acetoxymethyl ester (calcein-AM), which is cleaved by esterases in the cytosol to form green-fluorescing calcein free acid. Since mitochondria are normally impermeant to calcein, calcein is retained in the cytosol, leaving mitochondria as nonfluorescent voids (Nieminen, et al., 1995). These voids disappear after onset of the MPT as calcein, a 623 Da solute, enters the mitochondrial matrix space through PT pores (Nieminen, et al., 1995). Due to the expense of calcein-AM and the potential for its de-esterification prior to entry into the liver, calcein-AM (1 mg/mouse) was injected slowly into the rectal vein at 6 h after BDL or sham-operation. To prevent biliary excretion of calcein, an anion channel inhibitor, bromosulphthalein (6.6 μmol/mouse), was injected into the rectal vein 5 min before calcein-AM.
For intravital confocal microscopy, laparotomized mice were put in a prone position over a coverslip mounted on the stage of a CARV spinning disk confocal microscopic system (ATTO Bioscience, Rockville, MD) with a 40× water immersion objective lens (Zeiss C-Apochromat, 1.2 NA) using excitation wavelengths of 488 and 555 nm, respectively, and a multiwavelength emission filter to detect Rh123 and PI fluorescence. The respirator was turned off for ∼5 sec to eliminate breathing movement artifacts during image acquisition. Ten random fields were imaged per mouse. Bright green punctate Rh123 fluorescence represented cells with polarized mitochondria, whereas dimmer diffuse Rh123 fluorescence represented cells with depolarized mitochondria. Nonviable PI positive cells, indicated by red nuclear fluorescence, were counted in the same fashion. Image analysis was performed in a blinded manner.
Green fluorescence of calcein was detected in some mice by intravital multiphoton microscopy using a Zeiss LSM 510 NLO laser scanning confocal/multiphoton microscope (Thornwood, NY) and a 63× C-apo N.A. 1.2 water-immersion objective lens. Two-photon excitation of calcein was performed with 720 nm light from a Coherent Chameleon Ultra laser and emission was collected through a 500-550 nm bandpass emission filter. Images of approximately 10 random liver fields were collected from each mouse.
Immunohistochemical Detection of α-Smooth Muscle Actin, and Cytochrome c
α-Smooth muscle actin (αSMA) was detected immunohistochemically as described elsewhere (Zhong, et al., 2003). Immunohistochemical staining of cytochrome c was performed using an InnoGenex™ Mouse-on-Mouse Iso-IHC kit (InnoGenex, San Ramon, CA) according to the manufacturer's instructions after applying primary antibody at 1:200 (Santa Cruz Biotec, Santa Cruz, CA) at room temperature for 30 min.
Detection of Apoptosis
Apoptosis was assessed by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) using an In Situ Cell Death Detection Kit (Zhong, et al., 2007). TUNEL-positive and negative cells were counted in a blinded manner in 10 randomly selected fields using a 40× objective lens.
Activation of caspase-3 was assessed by Western blotting using an antibody specific for cleaved caspase-3. Liver tissue was harvested at 3 days after BDL when TUNEL peaked and was homogenized in 0.1% Triton-X100 buffer containing protease and phosphatase inhibitors. The extract was centrifuged at 14,000 × g for 15 min at 4°C, and aliquots of supernatant (40 μg of protein) were separated on 4-12% SDS-PAGE gels, transferred onto nitrocellulose membranes and immunoblotted with primary antibodies (Affinity Bioreagents Golden, CO) specific for cleaved caspase-3 and actin (Cell Signaling Technology, Danvers, MA; 1:1000 and 1:3000, respectively, over night at 4°C). Horseradish peroxidase-conjugated secondary antibodies were applied, and detection was by chemiluminescence (ECL, Amersham).
Detection of TNFα, TGF-β1 and Procollagen α1(I) mRNA by Quantitative Real Time PCR
Total RNA from liver tissue was isolated with Trizol (Invitrogen, Grand Island, NY) according to the manufacturer's protocol. Single stranded cDNAs were synthesized from RNA (2 μg) isolated from liver tissue using a Bio-Rad iScript cDNA Synthesis kit (Bio-Rad, Hercules, CA). Primer sets were designed and synthesized by Integrated DNA technologies (IDT, Coralville, IA). The primer sequences are listed in Table 1. PCR reactions were performed in triplicate with a reaction mixture containing 2× IQ™SYBR Supermix (Bio-Rad), cDNA template, and 0.1 nM of the forward and reverse primers. Real-time PCR was conducted using an iCycler iQ Multi-Color Real-Time PCR Detection System (Bio-Rad, Hercules, CA). iTaq DNA polymerase was activated at 95°C for 10 min, and 40 cycles of amplification at 95°C for 30 sec and 62°C for 30 sec were performed. Data were analyzed with MyiQ software. The abundance of mRNAs was normalized against hypoxanthine phospho-ribosyl-transferase (HPRT) housekeeping gene using the ΔΔCt method.
Table 1. Primers for Real-Time PCR.
| Gene | Directions | Primer Sequences |
|---|---|---|
| TNFα | Forward | 5′-CATCTTCTCAAAATTCGAGTGACAA-3′ |
| Reverse | 5′-TGGGAGTAGACAAGGTACAA-CCC-3′ | |
| TGF-β1 | Forward | 5′-TGATACGCCTGAGTGGCTGTCTTT-3′ |
| Reverse | 5′-AAGCGAAAGCCCTGTATTCCGTCT-3′ | |
| Procollagen α1(I) | Forward | 5′-GAACAGGGTGTTCCTGGAGA-3′ |
| Reverse | 5′-GGAAACCTCTCTCGCCTCTT-3′ | |
| Hypoxanthine phospho-ribosyl-transferase | Forward | 5′-GCTTTCCCTGGTTAAGCAGTACA-3′ |
| Reverse | 5′-AAACTTGTCTGGAATTTCAAATC-3′ |
Statistical Analysis
Groups were compared using ANOVA plus Student-Newman-Keuls posthoc test. Data shown are means ± S.E.M. (4 to 5 livers per group). Differences were considered significant at p<0.05.
Results
NIM811 Decreases Liver Injury after Cholestasis
Histology revealed normal liver architecture after sham-operation (Fig. 1, upper left). After BDL, focal necrosis developed within 1 day (data not shown) and became more marked after 3 days (Fig. 1, middle left). Necrotic areas accounted for 11% of liver sections at 1 day after BDL, which increased to 20% after 3 days and 28% after 14 days (Fig. 2A). NIM811 decreased necrotic areas to 0.5%, 3% and 8% at 1, 3 and 14 days, respectively (Fig. 1, lower left and Fig. 2A).
Fig. 1. NIM811 Mitigates Hepatic Necrosis and Apoptosis after Bile Duct Ligation.

Livers were harvested 3 days after sham-operation (Sham) or BDL. H+E staining (left column) was performed to evaluate necrosis. TUNEL (right column) was performed to evaluate apoptosis. Arrows identify necrotic areas. The white bar is 100 μm and the black bar is 50 μm.
Fig. 2. NIM811 Decreases Liver Injury after Bile Duct Ligation.
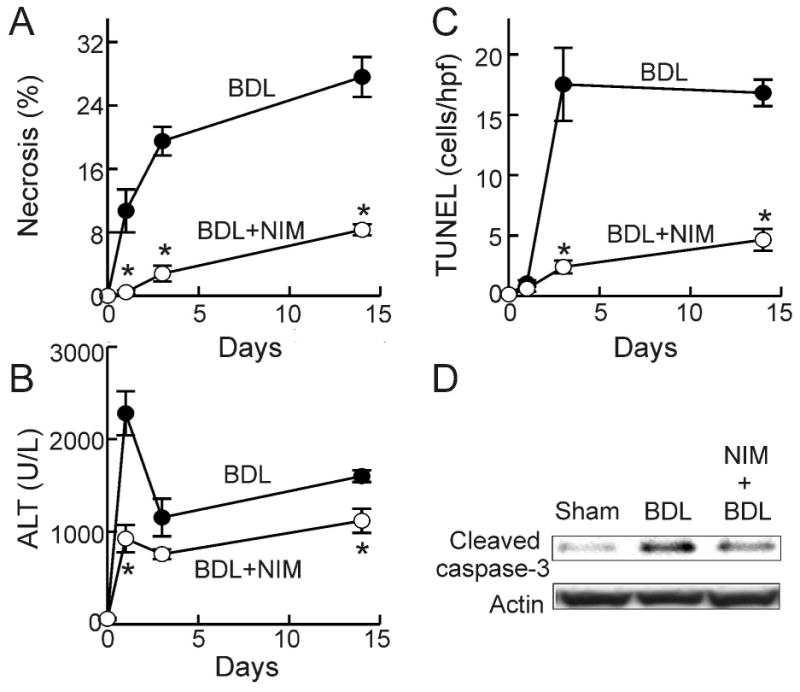
Livers and blood were collected at 0, 1, 3 and 14 days after BDL. Necrosis (A) was quantified by image analysis of 10 random fields per liver. Blood was collected for ALT measurement (B). TUNEL-positive cells were counted in a blinded manner in 10 randomly selected fields using a 40× objective lens (C). *, p<0.05 vs BDL of corresponding time point (n = 4-5 per group). Caspase-3 activation 3 days after BDL was evaluated by Western analysis of cleaved caspase-3, as illustrated in blot images representative of 4 experiments per group (D).
Serum ALT, an indicator of liver injury, averaged 58 U/L (Fig. 2B) after sham-operation and was not altered by NIM811 treatment. After BDL, ALT increased to 2280 U/L after 1 day, decreasing to 1150 U/L and 1600 U/L after 3 and 14 days (Fig. 2B). When mice were treated with NIM811, peak ALT after BDL was decreased by 60% and remained lower than the untreated group 2 weeks after BDL (Fig. 2B).
Apoptosis in liver sections was detected by TUNEL (Fig. 1, right column). TUNEL was rare in livers from sham-operated mice (0.13 cells/hpf, Fig. 1 upper right and Fig. 2C). TUNEL did not increase at 1 day after BDL but increased markedly to 18 cells/hpf and 17 cells/hpf at 3 and 14 days after BDL, respectively (Fig. 1, middle right and Fig. 2C). Increased TUNEL was almost completely blocked by NIM811 (Fig. 1, lower right and Fig. 2C). Occurrence of apoptosis was further confirmed by activation of caspase-3. Cleaved caspase-3 was barely detectable after sham-operation but increased markedly at 3 days after BDL (Fig. 2D). This effect was largely blocked by NIM811 (Fig. 2D).
Taken together, these data show that NIM811 protects against hepatocellular injury and death at the early and late stages of cholestasis. Cell killing after BDL involved both necrosis and apoptosis with necrosis occurring earlier than apoptosis. Specific inhibition of the MPT with NIM811 effectively prevented necrosis and apoptosis caused by cholestasis.
Mitochondrial Permeability Transition Occurs after Bile Duct Ligation: Prevention by NIM811
Previous studies showed that mitochondria of isolated hepatocytes generate ROS when exposed to hydrophobic BA, followed by MPT onset (Rolo, et al., 2003;Sokol, et al., 1995). Since mitochondrial depolarization is an immediate consequence of the MPT, we investigated if mitochondrial depolarization occurs after cholestasis in vivo. At 6 h after BDL, Rh123, a green-fluorescing fluorophore taken up by polarized mitochondria, and PI, a red-fluorescing fluorophore staining nuclei of non-viable cells, were infused, and intravital confocal fluorescent microscopy was performed. Overlays of green and red images allowed identification of viable cells with polarized mitochondria (no red nuclear fluorescence, intracellular punctate mitochondrial green fluorescence), viable cells with depolarized mitochondria (no red fluorescence, no or diffuse green fluorescence) and non-viable cells (red nuclear fluorescence). In non-viable cells, mitochondria were consistently depolarized.
In sham-operated mice, green Rh123 fluorescence was punctate in virtually all hepatocytes, indicating mitochondrial polarization (Fig. 3. upper left), and red PI labeling of nuclei was undetectable (Fig. 3. upper left). At 6 h after BDL, many hepatocytes contained depolarized mitochondria that did not accumulate Rh123 (Fig. 3, upper right,). Other hepatocytes showed diffuse but relatively bright Rh123 fluorescence, possibly indicating the recent depolarization of mitochondria and release of mitochondrial Rh123 into the cytosol. Occasionally, some parenchymal and non-parenchymal cells (NPC) were also labeled with PI (<1 cell/hpf, Fig. 3, upper right). Most hepatocytes with depolarized mitochondria excluded PI, whereas PI-labeled hepatocytes never contained polarized mitochondria. These results suggest that at 6 h after BDL, mitochondrial depolarization occurred in many hepatocytes and that mitochondrial depolarization preceded hepatocyte death. After NIM811 treatment, mitochondrial depolarization decreased from 18 cells/hpf after vehicle to 5 cells/hpf (Fig. 3, lower panels). PI-labeled cells were not different between the NIM811-treated and untreated groups, since cell death was rare at this early stage of cholestasis.
Fig. 3. Protection of NIM811 Against Mitochondrial Depolarization after Bile Duct Ligation.

At 6 h after surgery, Rh123 and PI were infused, and intravital confocal microscopy was performed. Bar is 20 μm. Viable cells with polarized mitochondria, viable cells with depolarized mitochondria and non-viable cells were counted in 10 random fields of each liver (low right panel). a, p<0.05 vs sham; b, p<0.05 vs BDL (n = 4-5 per group).
Calcein was used to monitor onset of the MPT by intravital multiphoton microscopy. Calcein loaded into the cytosol but did not permeate mitochondria. Thus, calcein fluorescence outlined individual mitochondria as dark voids in the hepatocytes of sham-operated mice (Fig. 4. upper left). These voids disappeared at 6 h after BDL, indicating movement of calcein into mitochondria through opened PT pores after onset of the MPT (Fig. 4. middle left). This loss of calcein holes cannot be due to premature cell death since cell death would cause loss of all calcein fluorescence. NIM811 effectively prevented calcein permeation into mitochondria in vivo after BDL (Fig. 4. lower left). These findings showed directly that NIM811-sensitive mitochondrial inner membrane permeabilization occurred in vivo due to cholestasis.
Fig. 4. NIM811 Prevents Onset of the Mitochondrial Permeability Transition and Cytochrome C Release after Bile Duct Ligation.

At 6 h after surgery, calcein-AM was infused, and intravital multiphoton microscopy was performed (left column). Bar is 5 μm. In the right column, cytochome c was detected by immunohistochemistry. Bar is 20 μm.
NIM811 Prevents Cytochrome c Release after Cholestasis
The MPT causes large amplitude mitochondrial swelling, outer membrane rupture and release of cytochrome c from the intermembrane space into the cytosol, which can trigger caspase activation and apoptosis (Kantrow, et al., 2000;Zamzami, et al., 1996). Accordingly, we investigated cytochrome c release after BDL with and without NIM811 treatment. Cytochome c detected immunohistochemically was punctate in hepatocytes of liver slides from sham-operated mice consistent with mitochondrial localization (Fig. 4, upper right). After BDL, cytochome c staining became diffuse, indicating release of cytochome c into the cytosol (Fig. 4, middle right). Release of cytochome c after BDL was largely prevented by NIM811.
NIM811 Does Not Prevent Inflammatory and Profibrogenic Cytokine Formation after Bile Duct Ligation
Cell death after cholestasis may cause inflammation that subsequently leads to liver fibrosis. The proinflammatory cytokine, TNFα, plays an important role in liver fibrosis (Friedman, 1999). Therefore, we investigated whether NIM811 prevents TNFα expression after BDL. TNFα mRNA increased 11-fold at 3 days after BDL and 38-fold at 2 weeks in vehicle-treated mice (Fig. 5A). Surprisingly, NIM811 did not prevent these increases of TNFα expression after BDL despite protection against cell death (Fig. 5A).
Fig. 5. NIM811 Does Not Prevent TNFα and TGF-β Expression after Bile Duct Ligation.
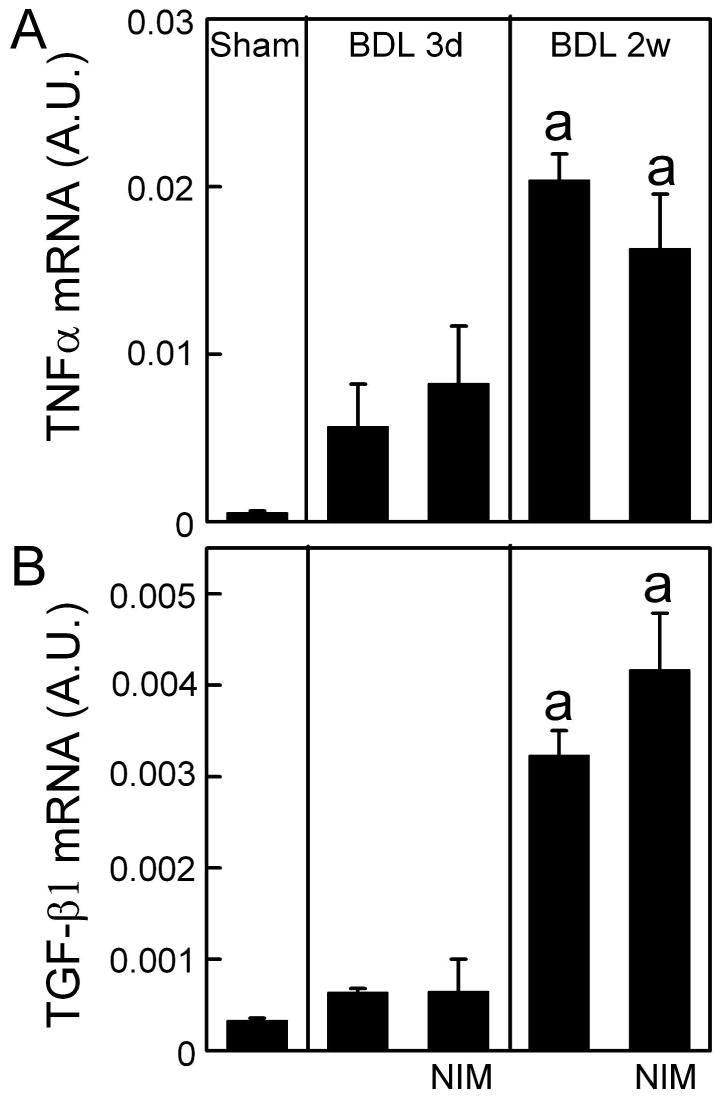
Livers were harvested 3 days and 2 weeks after sham-operation (Sham) or BDL. TNFα and TGF-β1 mRNAs were detected by quantitative real time PCR. The abundance of TNFα and TGF-β mRNAs was normalized against HPRT using the ΔΔCt method. A.U., arbitrary unit. a, p<0.05 vs sham; b, p<0.05 vs BDL (n = 4 per group).
TGF-β increases in a variety of inflammatory processes and is an important promoter of hepatic fibrosis (Friedman, 1999). Accordingly, we detected TGF-β expression after BDL. TGF-β1 mRNA increased 2-fold at 3 days after BDL and 10-fold after 2 weeks in vehicle-treated mice (Fig. 5B). Again, NIM811 did not prevent increases of TGF-β1 expression after BDL (Fig. 5B).
NIM811 Does Not Prevent Hepatic Fibrosis after Bile Duct Ligation
To evaluate the effects of NIM811 on liver fibrosis after BDL, liver sections were stained with Sirius red for collagen. No fibrosis was observed in livers from sham-operated mice (Fig. 6A, upper left). By contrast, hepatic fibrosis developed within 2 weeks after BDL (Fig. 6A, middle left). Sirius red-positive area increased from 1% in sham-operated mice to 21% at 2 weeks after BDL (Fig. 6B left). When mice treated with NIM811 were subjected to BDL, histology revealed similar fibrosis as vehicle treatment (Fig. 6A, lower left). Sirius red-positive area was 23% at 2 weeks after BDL (Fig. 6B left).
Fig. 6. NIM811 Does Not Prevent Hepatic Fibrosis and Stellate Cell Activation after Bile Duct Ligation.

Livers were harvested 2 weeks after sham-operation (Sham) or BDL. In A, Sirius red staining (left column) and immunohistochemical staining for α–smooth muscular actin (αSMA, right column) were performed. Rows are: upper, sham-operation; middle, BDL; lower, BDL plus NIM811 (NIM) treatment. The white and black bars are 200 μm and 50 μm, respectively. In B left, Sirius red-positive area was quantified by image analysis. In B right, procollagen α1(I) mRNA was detected by quantitative real time PCR. The abundance of procollagen α1(I) mRNA was normalized against HPRT using the ΔΔCt method. A.U., arbitrary unit. a, p<0.05 vs sham; b, p<0.05 vs BDL (n = 4 per group).
Hepatic collagen gene expression was evaluated by quantitative real time PCR for procollagen α1(I) mRNA. At 2 weeks after BDL, procollagen α1(I) mRNA increased 19-fold compared to sham-operated mice (Fig. 6B right). Treatment with NIM811 did not prevent the increase of procollagen α1(I) mRNA expression caused by cholestasis (Fig. 6B right).
NIM811 Does Not Prevent Stellate Cell Activation after Bile Duct Ligation
Activated HSCs are the major source of matrix proteins in diseased liver (Friedman, 2008). Accordingly, we evaluated αSMA, an indicator of HSC activation, by immunohistochemical staining after BDL. In livers from sham-operated mice, small amounts of αSMA were detected in the smooth muscle and endothelium of blood vessels (Fig. 6A, upper right). At 2 weeks after BDL, αSMA increased markedly in perisinusoidal cells (Fig. 6A, middle right), indicating activation of HSCs. After BDL, αSMA increased to the same extent in livers of NIM811-treated mice as after vehicle treatment (Fig. 6A, lower right). Taken together, these data show that NIM811 treatment did not prevent proinflammatory cytokine formation, fibrosis and HSC activation caused by cholestasis.
Discussion
Mitochondrial Permeability Transition Plays an Important Role in Cholestasis-Induced Liver Injury in vivo
In cholestasis, BA accumulate to cause hepatic injury. Mechanisms of BA-induced cell death are not fully understood. BA toxicity is not due to a detergent effect (Guicciardi and Gores, 2002). Several studies show that cytotoxic BA trigger hepatocyte apoptosis by activation of death receptors (Fickert, et al., 2005;Faubion, et al., 1999). Despite evidence of Fas and TRAIL-dependent apoptotic signaling in cholestatic liver injury, recent studies suggest that necrosis is the predominant form of cell death after BDL (Fickert, et al., 2005;Gujral, et al., 2004).
Oxidative stress and mitochondrial injury may also play a role in BA-induced cell death. Oxidative stress occurs in biliary obstructed rats (Pastor, et al., 1997;Parola, et al., 1996) and isolated hepatocytes and mitochondria exposed to hydrophobic BA (Rolo, et al., 2003;Sokol, et al., 1995). Antioxidants and overexpression of mitochondrial Mn-SOD decreased cell death after BDL (Zhong, et al., 2003;Zhong, et al., 2002). ROS not only directly damage macromolecules but also alter signal transduction. For example, BA cause activation of NADPH oxidase generating a ROS signal activating Yes-, JNK-, and EGFR-dependent CD95 (Fas) tyrosine phosphorylation and formation of the death-inducing signaling complex (Reinehr and Haussinger, 2007). ROS also trigger opening of PT pores (Kantrow, et al., 2000). Increased mitochondrial calpain-like protease activity during cholestasis might also induce MPT onset (Gores, et al., 1998).
The MPT is an important mechanism leading to mitochondrial dysfunction and cell death (Lemasters, 2005). Despite extensive work, the exact molecular composition of PT pores remains unclear. In one model, PT pores are composed of the voltage-dependent anion channel (VDAC), the adenine nucleotide translocator (ANT), cyclophilin D (CypD) and various ancillary proteins (Martinou and Green, 2001). However, recent studies using ANT and VDAC isoform knockout mice challenge this model (Kokoszka, et al., 2004). An alternative model is that PT pores are formed by aggregation of misfolded and damaged membrane proteins in association with CypD and other molecular chaperones (He and Lemasters, 2002), and recent studies confirm that CypD is an important regulator of PT pore activity (Baines, et al., 2005). MPT onset leads to collapse of the mitochondrial membrane potential, failure of ATP production, and necrotic cell death (Zamzami, et al., 1996;Kim, et al., 2003). Moreover, the MPT causes large amplitude swelling and release of cytochrome c into the cytosol, triggering apoptosis (Kantrow, et al., 2000;Zamzami, et al., 1996).
Both CsA and bongkrekic acid, inhibitors of the MPT, protect cultured hepatocytes from BA-induced cell death (Yerushalmi, et al., 2001;Gores, et al., 1998). Mitochondria isolated from cholestatic rats also exhibit disruption of mitochondrial calcium homeostasis by BA, which might increase susceptibility of mitochondria to calcium-induced MPT onset (Rolo, et al., 2002). In addition, human liver mitochondria undergo the MPT in response to hydrophobic BA (Sokol, et al., 2005). These data show that BA cause MPT onset in vitro. However, whether cholestasis-induced MPT onset occurs in live animals has never been determined. Using intravital confocal/multiphoton microscopy, here we show directly that mitochondrial depolarization and inner membrane permeabilization do occur in vivo after BDL (Fig. 3). This mitochondrial depolarization is caused by MPT onset, as indicated by entry of calcein into mitochondria after BDL (Fig. 4) and blockade of this permeabilization with the consequent depolarization by the specific MPT inhibitor, NIM811 (Fig. 3 and 4). Importantly, MPT onset and mitochondrial depolarization preceded necrosis and apoptosis (Fig. 3 and 4). Moreover, inhibition of MPT onset substantially attenuated cytochrome c release from mitochondria (Fig. 4). These data show clearly that the MPT plays an important role in cholestatic liver injury in vivo.
CsA at high therapeutic serum concentrations (0.17-0.8 μM) partially inhibits the canalicular bile salt excretion pump (BSEP) and the basolateral Na+/taurocholate co-transporting polypeptide (NTCP) (Mita, et al., 2006). Effects of NIM811 on BSEP and NTCP have not yet been reported, and a contribution of inhibition of bile salt transport to NIM811 cytoprotection after BDL cannot be excluded. However in the present work, neither partial inhibition of BSEP nor NTCP would be expected to prevent hepatocellular accumulation of toxic BA, since BSEP inhibition would only serve to increase intracellular BA concentration and since incomplete inhibition of NTCP would not prevent equilibration of BA accumulation inside hepatocytes with the plasmalemmal Na+ gradient. Future studies will be needed to better characterize the effects of NIM811 on hepatocellular BA transport.
Previous work establishes a role for death receptor activation in cholestatic liver injury (Fickert, et al., 2005;Gujral, et al., 2004;Faubion, et al., 1999), but controversy still exists as to how death receptor activation leads to mitochondrial dysfunction, cytochrome c release and cell death. Some studies support a mechanism in which caspase 8 activation causes tBid formation, which in turn activates Bax/Bak-dependent permeabilization of the outer membrane and cytochrome c release (Zhao, et al., 2003). Other findings indicate that death receptor activation causes the MPT, an inner membrane event, which leads to either ATP depletion-dependent necrosis or caspase-dependent apoptosis (Lemasters, 2005). Our findings here suggest that the MPT is the predominant event precipitating phenotypes of both apoptotic and necrotic cell death in cholestasis, at least in the mouse model of BDL. Future studies need to assess role of MPT in Fas-dependent injury, such as Jo-2 antibody stimulation.
MPT Inhibition Does Not Prevent Hepatic Fibrosis after Bile Duct Ligation
Chronic cholestatic liver diseases cause liver cirrhosis (Poupon, et al., 2000). Fibrogenesis in liver is likely an active wound-healing process regulated by cytokines, chemokines and additional signals like ROS (Friedman, 2008). Hepatocytes release fibrogenic lipid peroxides (Novo, et al., 2006), and lipid peroxidation associated with hepatocyte necrosis is considered a classical inflammatory and fibrogenic stimulus (Friedman, 2008). In addition, apoptotic hepatocytes can release cytoplasmic fragments that are fibrogenic towards cultured HSCs (Canbay, et al., 2003). Therefore, we expected that prevention of cell injury by NIM811 would alleviate subsequent liver fibrosis. Strikingly, although inhibition of the MPT by NIM811 effectively prevented cholestatic necrosis and apoptosis of hepatocytes through the whole cholestatic process (Figs 1-2), NIM811 did not attenuate liver fibrosis after BDL (Fig. 6).
The dissociation of cytoprotection of NIM811 from antifibrogenesis suggests the cell death may not play an essential role in cholestatic liver fibrosis. Although mechanisms of liver fibrosis are not well understood, the concept is generally accepted that prolonged liver injury triggers activation of HSCs and hepatic recruitment of inflammatory cells, effects likely mediated by inflammatory cytokines (Friedman, 1999). Therefore, we investigated if NIM811 altered inflammatory cytokine production. Expression of TNFα, a major proinflammatory cytokine that is important in liver fibrosis (Friedman, 1999), increased after BDL, but this effect was not attenuated by NIM811 (Fig. 5). Similarly, NIM811 did not block expression of TGF-β, a major profibrogenic cytokine (Friedman, 1999), after BDL (Fig. 5). These unexpected findings suggest that inflammatory processes in cholestasis are not due to cell death but elicited mainly by direct effects of BA or other excretion products accumulated during cholestasis on inflammatory cytokine-producing cells, such as Kupffer cells and HSCs.
Activation of HSCs is a critical step in hepatic fibrogenesis (Friedman, 2008). Activated HSCs produce pro-fibrogenic cytokines, such as TGF-β, synthesize collagen (Parsons, et al., 2007;Friedman, 1999;Kisseleva and Brenner, 2007) and secrete inhibitors of matrix-degrading enzymes (Kisseleva and Brenner, 2007). Therefore, we investigated whether NIM811 prevented HSC activation. As expected, αSMA, a marker of HSC activation, dramatically increased after BDL (Fig. 6). Although NIM811 inhibits proliferation of cultured hepatic HSCs in vitro (Kohjima, et al., 2007), NIM811 had no effect on αSMA production in vivo (Fig. 6). Thus, prevention of cell death did not block HSC activation after cholestasis consistent with the conclusion that excess BA or other excretion products accumulated in cholestasis either activate HSCs directly or stimulate other NPC (i.e., Kupffer cells, endothelial cells and cholangiocytes) to release inflammatory or fibrogenic cytokines and growth factors that activate HSCs. BA maintain HSC in an activated state (Brady, et al., 1996). Other substances that accumulate during cholestasis (e.g., bilirubin, cholesterol, phospholipids, metals) may also stimulate liver fibrosis. For example, accumulated iron might stimulate ROS production to cause HSC activation. Moreover, high cholesterol activates HSCs and induces liver fibrosis (Kainuma, et al., 2006). Future studies will be needed to better characterize these profibrotic mechanisms in cholestasis that are independent of hepatocellular death.
BDL represents acute and complete cholestasis that leads to overt cholangiocyte reactions. Whether independence of liver fibrosis from cell death as observed in acute BDL also occurs in cholestasis of more chronic onset or in other liver fibrosis models (e.g., CCl4, thioacetamide, ethanol) is still unknown and will also need to be the subject of future studies.
Taken together, the present study shows that the MPT occurs in vivo in hepatocytes during cholestasis and plays an essential role in necrotic and apoptotic hepatocellular death. NIM811, a specific inhibitor of the MPT, effectively prevents cell killing; but does not prevent subsequent liver fibrosis. Liver fibrosis is possibly elicited by direct effects of BA on HSCs, cholangiocytes and other NPC.
Acknowledgments
This study was supported, in part, by Grants DK70844, DK073336, DK037034 and C06 RR015455 from the National Institute of Health
Abbreviations used
- ALT
alanine transaminase
- BA
bile acid
- BDL
bile duct ligation
- CsA
cyclosporin A
- hpf
high power field
- HSC
hepatic stellate cells
- MPT
mitochondrial permeability transition
- NIM811
N-methyl-4-isoleucine cyclosporine
- NPC
non-parenchymal cells
- PI
propidium iodide
- Rh123
rhodamine 123
- ROS
reactive oxygen species
- SMA
smooth muscle actin
- TNFα
tumor necrosis factor-α
- TGF-β
transforming growth factor-β
Footnotes
Part of this research was reported as an abstract in Gastroenterology, 130; A-759, 2006.
References
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Brady LM, Beno DW, Davis BH. Bile acid stimulation of early growth response gene and mitogen-activated protein kinase is protein kinase C-dependent. Biochem J. 1996;316:765–769. doi: 10.1042/bj3160765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest. 2003;83:655–663. doi: 10.1097/01.lab.0000069036.63405.5c. [DOI] [PubMed] [Google Scholar]
- Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- Faubion WA, Guicciardi ME, Miyoshi H, Bronk SF, Roberts PJ, Svingen PA, Kaufmann SH, Gores GJ. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest. 1999;103:137–145. doi: 10.1172/JCI4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fickert P, Trauner M, Fuchsbichler A, Zollner G, Wagner M, Marschall HU, Zatloukal K, Denk H. Oncosis represents the main type of cell death in mouse models of cholestasis. J Hepatol. 2005;42:378–385. doi: 10.1016/j.jhep.2004.10.016. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19:129–140. doi: 10.1055/s-2007-1007105. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gores GJ, Miyoshi H, Botla R, Aguilar HI, Bronk SF. Induction of the mitochondrial permeability transition as a mechanism of liver injury during cholestasis: a potential role for mitochondrial proteases. Biochim Biophys Acta. 1998;1366:167–175. doi: 10.1016/s0005-2728(98)00111-x. [DOI] [PubMed] [Google Scholar]
- Guicciardi ME, Gores GJ. Bile acid-mediated hepatocyte apoptosis and cholestatic liver disease. Dig Liver Dis. 2002;34:387–392. doi: 10.1016/s1590-8658(02)80033-0. [DOI] [PubMed] [Google Scholar]
- Gujral JS, Liu J, Farhood A, Jaeschke H. Reduced oncotic necrosis in Fas receptor-deficient C57BL/6J-lpr mice after bile duct ligation. Hepatology. 2004;40:998–1007. doi: 10.1002/hep.20380. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Brennerb C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem. 2003;10:1507–1525. doi: 10.2174/0929867033457278. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial-matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J. 1990;268:153–160. doi: 10.1042/bj2680153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- Kainuma M, Fujimoto M, Sekiya N, Tsuneyama K, Cheng C, Takano Y, Terasawa K, Shimada Y. Cholesterol-fed rabbit as a unique model of nonalcoholic, nonobese, non-insulin-resistant fatty liver disease with characteristic fibrosis. J Gastroenterol. 2006;41:971–980. doi: 10.1007/s00535-006-1883-1. [DOI] [PubMed] [Google Scholar]
- Kantrow SP, Tatro LG, Piantadosi CA. Oxidative stress and adenine nucleotide control of mitochondrial permeability transition. Free Radic Biol Med. 2000;28:251–260. doi: 10.1016/s0891-5849(99)00238-5. [DOI] [PubMed] [Google Scholar]
- Kass GE. Mitochondrial involvement in drug-induced hepatic injury. Chem Biol Interact. 2006;163:145–159. doi: 10.1016/j.cbi.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Kim JS, He L, Qian T, Lemasters JJ. Role of the mitochondrial permeability transition in apoptotic and necrotic death after ischemia/reperfusion injury to hepatocytes. Curr Mol Med. 2003;3:527–535. doi: 10.2174/1566524033479564. [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22 Suppl 1:S73–S78. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- Kohjima M, Enjoji M, Higuchi N, Kotoh K, Kato M, Takayanagi R, Nakamuta M. NIM811, a nonimmunosuppressive cyclosporine analogue, suppresses collagen production and enhances collagenase activity in hepatic stellate cells. Liver Int. 2007;27:1273–1281. doi: 10.1111/j.1478-3231.2007.01560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasters JJ. Dying a thousand deaths: redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology. 2005;129:351–360. doi: 10.1053/j.gastro.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Green DR. Breaking the mitochondrial barrier. Nat Rev Mol Cell Biol. 2001;2:63–67. doi: 10.1038/35048069. [DOI] [PubMed] [Google Scholar]
- Mita S, Suzuki H, Akita H, Hayashi H, Onuki R, Hofmann AF, Sugiyama Y. Inhibition of bile acid transport across Na+/taurocholate cotransporting polypeptide (SLC10A1) and bile salt export pump (ABCB 11)-coexpressing LLC-PK1 cells by cholestasis-inducing drugs. Drug Metab Dispos. 2006;34:1575–1581. doi: 10.1124/dmd.105.008748. [DOI] [PubMed] [Google Scholar]
- Nieminen AL, Saylor AK, Tesfai SA, Herman B, Lemasters JJ. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t-butylhydroperoxide. Biochem J. 1995;307(Pt 1):99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo E, Marra F, Zamara E, Valfre di BL, Caligiuri A, Cannito S, Antonaci C, Colombatto S, Pinzani M, Parola M. Dose dependent and divergent effects of superoxide anion on cell death, proliferation, and migration of activated human hepatic stellate cells. Gut. 2006;55:90–97. doi: 10.1136/gut.2005.069633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parola M, Leonarduzzi G, Robino G, Albano E, Poli G, Dianzani MU. On the role of lipid peroxidation in the pathogenesis of liver damage induced by long-standing cholestasis. Free Radic Biol Med. 1996;20:351–359. doi: 10.1016/0891-5849(96)02055-2. [DOI] [PubMed] [Google Scholar]
- Parsons CJ, Takashima M, Rippe RA. Molecular mechanisms of hepatic fibrogenesis. J Gastroenterol Hepatol. 2007;22 Suppl 1:S79–S84. doi: 10.1111/j.1440-1746.2006.04659.x. [DOI] [PubMed] [Google Scholar]
- Pastor A, Collado PS, Almar M, Gonzalez-Gallego J. Antioxidant enzyme status in biliary obstructed rats: effects of N- acetylcysteine. J Hepatol. 1997;27:363–370. doi: 10.1016/s0168-8278(97)80183-3. [DOI] [PubMed] [Google Scholar]
- Poupon R, Chazouilleres O, Poupon RE. Chronic cholestatic diseases. J Hepatol. 2000;32:129–140. doi: 10.1016/s0168-8278(00)80421-3. [DOI] [PubMed] [Google Scholar]
- Reinehr R, Haussinger D. CD95 activation in the liver: ion fluxes and oxidative signaling. Arch Biochem Biophys. 2007;462:124–131. doi: 10.1016/j.abb.2006.12.019. [DOI] [PubMed] [Google Scholar]
- Rolo AP, Oliveira PJ, Seica R, Santos MS, Moreno AJ, Palmeira CM. Disruption of mitochondrial calcium homeostasis after chronic alpha-naphthylisothiocyanate administration: relevance for cholestasis. J Investig Med. 2002;50:193–200. doi: 10.2310/6650.2002.33433. [DOI] [PubMed] [Google Scholar]
- Rolo AP, Palmeira CM, Wallace KB. Mitochondrially mediated synergistic cell killing by bile acids. Biochim Biophys Acta. 2003;1637:127–132. doi: 10.1016/s0925-4439(02)00224-7. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Dahl R, Devereaux MW, Yerushalmi B, Kobak GE, Gumpricht E. Human hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J Pediatr Gastroenterol Nutr. 2005;41:235–243. doi: 10.1097/01.mpg.0000170600.80640.88. [DOI] [PubMed] [Google Scholar]
- Sokol RJ, Winklhofer-Roob BM, Devereaux MW, McKim JM., Jr Generation of hydroperoxides in isolated rat hepatocytes and hepatic mitochondria exposed to hydrophobic bile acids. Gastroenterology. 1995;109:1249–1256. doi: 10.1016/0016-5085(95)90585-5. [DOI] [PubMed] [Google Scholar]
- Theruvath TP, Zhong Z, Pediaditakis P, Ramshesh VK, Currin RT, Tikunov A, Holmuhamedov E, Lemasters JJ. Minocycline and N-methyl-4-isoleucine cyclosporin (NIM811) mitigate storage/reperfusion injury after rat liver transplantation through suppression of the mitochondrial permeability transition. Hepatology. 2008;47:236–246. doi: 10.1002/hep.21912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmeier PC, Feldtrauer JJ, Qian T, Lemasters JJ. Inhibition of the mitochondrial permeability transition by the nonimmunosuppressive cyclosporin derivative NIM811. Mol Pharmacol. 2002;62:22–29. doi: 10.1124/mol.62.1.22. [DOI] [PubMed] [Google Scholar]
- Yerushalmi B, Dahl R, Devereaux MW, Gumpricht E, Sokol RJ. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology. 2001;33:616–626. doi: 10.1053/jhep.2001.22702. [DOI] [PubMed] [Google Scholar]
- Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G. Mitochondrial control of nuclear apoptosis. J Exp Med. 1996;183:1533–1544. doi: 10.1084/jem.183.4.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ding WX, Qian T, Watkins S, Lemasters JJ, Yin XM. Bid activates multiple mitochondrial apoptotic mechanisms in primary hepatocytes after death receptor engagement. Gastroenterology. 2003;125:854–867. doi: 10.1016/s0016-5085(03)01066-7. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Froh M, Lehnert M, Schoonhoven R, Yang L, Lind H, Lemasters JJ, Thurman RG. Polyphenols from Camellia sinenesis attenuate experimental cholestasis-induced liver fibrosis in rats. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1004–G1013. doi: 10.1152/ajpgi.00008.2003. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Froh M, Wheeler MD, Smutney O, Lehmann TG, Thurman RG. Viral gene delivery of superoxide dismutase attenuates experimental cholestasis-induced liver fibrosis in the rat. Gene Ther. 2002;9:183–191. doi: 10.1038/sj.gt.3301638. [DOI] [PubMed] [Google Scholar]
- Zhong Z, Theruvath TP, Currin RT, Waldmeier PC, Lemasters JJ. NIM811, a Mitochondrial Permeability Transition Inhibitor, Prevents Mitochondrial Depolarization in Small-for-Size Rat Liver Grafts. Am J Transplant. 2007;7:1103–1111. doi: 10.1111/j.1600-6143.2007.01770.x. [DOI] [PubMed] [Google Scholar]