

Abstract
CKIP-1 is a pleckstrin homology domain-containing protein that induces alterations of the actin cytoskeleton and cell morphology when expressed in human osteosarcoma cells. CKIP-1 interacts with the heterodimeric actin-capping protein in cells, so we postulated that this interaction was responsible for the observed cytoskeletal and morphological effects of CKIP-1. To test this postulate, we used peptide “walking arrays” and alignments of CKIP-1 with CARMIL, another CP-binding protein, to identify Arg-155 and Arg-157 of CKIP-1 as residues potentially required for its interactions with CP. CKIP-1 mutants harboring Arg-155 and Arg-157 substitutions exhibited greatly decreased CP binding, while retaining wild-type localization, the ability to interact with protein kinase CK2, and self-association. To examine the phenotype associated with expression of these mutants, we generated tetracycline-inducible human osteosarcoma cells lines expressing R155E,R157E mutants of CKIP-1. Examination of these cell lines reveals that CKIP-1 R155E,R157E did not induce the distinct changes in cell morphology and the actin cytoskeleton that are characteristic of wild-type CKIP-1 demonstrating that the interaction between CKIP-1 and CP is required for these cellular effects.
CKIP-14 was identified in a yeast two-hybrid screen for novel interaction partners of protein kinase CK2 (1). The cDNA for CKIP-1 codes for a protein of ~46 kDa with an amino-terminal pleckstrin homology (PH) domain and a carboxyl-terminal leucine-rich region as well as five putative PXXP motifs. The PH domain of CKIP-1 is required for phospholipid binding in vitro and for plasma membrane localization in cells (1, 2). Furthermore, this domain is necessary for interactions with protein kinase CK2, because mutants lacking the PH domain fail to interact with the kinase. Additionally, we have demonstrated that a subpopulation of protein kinase CK2 is targeted to the plasma membrane by CKIP-1 in cells (2). This targeting of CK2 is lost when the PH domain of CKIP-1 is replaced by a myristoylation recognition sequence, even though the CKIP-1 mutant still localizes to the plasma membrane. These results suggest that CKIP-1 may function in an analogous manner to protein kinase A anchoring proteins, which target cAMP-dependent protein kinase A (3-6). In addition to this potential role as a CK2-targeting protein, CKIP-1 appears to have roles independent of CK2. Recent reports have shown that CKIP-1 functions in muscle cell differentiation (7) and AP-1 regulation and apoptosis (8).
To investigate the cellular functions of CKIP-1, we generated cell lines with tetracycline-regulated expression of FLAG-CKIP-1. Induction of FLAG-CKIP-1 in these cells caused changes in cellular morphology, as well as increases in F-actin and total cellular levels of actin (9). To determine the mechanistic basis for these observations, we performed a proteomic screen using Tandem affinity purification (10) and large-scale immunoprecipitations to identify CKIP-1 interaction partners. We identified the heterodimeric actin-capping protein as a novel CKIP-1-interacting protein (9). Moreover, we showed that CKIP-1 can partially inhibit the activity of CP at the barbed ends of actin filaments. Collectively, these observations suggested two hypothetical models. First, it is possible that the interaction of CP with CKIP-1 at the plasma membrane inhibits binding of CP to the barbed ends of the actin filament leading to increased actin polymerization and changes in cellular morphology. Alternatively, the effects of CKIP-1 on cell morphology may require interactions with CP to target CKIP-1 to the barbed ends of actin filaments.
To distinguish between these models, we used peptide “walking arrays” and alignments with the CP-binding protein, CARMIL (11), to identify Arg-155 and Arg-157 as residues of CKIP-1 potentially required for its binding with CP. To investigate the importance of these residues for interactions between CKIP-1 and CP, we employed mutants of CKIP-1 harboring substitutions at Arg-155 and Arg-157 as well as a synthetic peptide encompassing the putative CP-binding region of CKIP-1. Finally, to directly test whether changes in cell morphology and the actin cytoskeleton induced by CKIP-1 require its interactions with CP, we generated human osteosarcoma cell lines expressing CP-binding deficient mutants of CKIP-1 under the control of tetracycline.
MATERIALS AND METHODS
Antibodies
Monoclonal antibodies against the FLAG epitope and polyclonal antibodies directed against actin were purchased from Sigma-Aldrich. Polyclonal antibodies directed against green fluorescent protein (GFP) were purchased from Clontech (Palo Alto, CA). Monoclonal antibodies against CPα (mAb 5B12.3) and CPβ (mAb 3F2.3 and mAb 1E5.25.4) subunits of actin-capping protein (12) were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD, National Institutes of Health and maintained by the University of Iowa, Department of Biological Sciences (Iowa City, IA). Polyclonal antibodies against CKIP-1 and GST were described in a previous study (1). Monoclonal antibodies against β-tubulin were a generous gift from Dr. Lina Dagnino (Department of Pharmacology, University of Western Ontario).
Plasmid Constructs
FLAG-CKIP-1 was generated by PCR amplification and subcloned into pRc/CMV and pTRE as previously described (9). CKIP-1 mutations to disrupt interactions with CP were generated for FLAG-CKIP-1, GFP-CKIP-1, GST-CKIP-1, and/or FLAG-CKIP-1/pTRE using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) as described by the manufacturer.
FLAG-CKIP-1 R133,K135E was generated using the forward primer: 5′-CTC AAC TCT GCC ATC ACC GAG GCC GAG AAC CGT ATC TTG GAT GAG-3′ and the reverse primer: 5′-CTC ATC CAA GAT ACG GTT CTC GGC CTC GGT GAT GGC AGA GTT GAG-3′. FLAG-CKIP-1 R133A,K135A,R137A was similarly generated using the forward primer: 5′-CTC AAC TCT GCC ATC ACC GCT GCC GCT AAC GCT ATC TTG GAT GAG GTC ACC-3′ and the reverse primer: 5′-GGT GAC CTC ATC CAA GAT AGC GTT AGC GGC AGC GGT GAT GGC AGA GTT GAG-3′. CKIP-1 R155A,R157A constructs were generated using the forward primer: 5′-TCT TGC CCA TCC CAC TGC AGA CGC AGC AAA AAT CCA GCA CTC CCG C-3′ and the reverse primer: 5′-GCG GGA GTG CTG GAT TTT TGC TGC GTC TGC AGT GGG ATG GGC AAG A-3′. CKIP-1 R155E,R157E constructs were generated using the forward primer: 5′-TCT TGC CCA TCC CAC TGA GGA CGA GGC AAA AAT CCA GCA CTC CCG C-3′ and the reverse primer: 5′-GCG GGA GTG CTG GAT TTT TGC CTC GTC CTC AGT GGG ATG GGC AAG A-3′. Similarly, CKIP-1 R155A,R157A,K159A constructs were generated using the forward primer: 5′-CCC ACT GCA GAC GCA GCA GCA ATC CAG CAC TCC CGC CGC-3′ and the reverse primer: 5′-GCG GCG GGA GTG CTG GAT TGC TGC TGC GTC TGC AGT GGG-3′ using CKIP-1 R155A,R157A as a template. All mutations were confirmed by DNA sequencing.
Immunoprecipitations
For endogenous CKIP-1 immunoprecipitations, U2-OS cells (9 × 15-cm plates) or C2C12 cells (3 × 10-cm plate) were harvested in radioimmune precipitation assay lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholic acid, 0.l% SDS, 1% aprotinin and 0.1 mm phenylmethylsulfonyl fluoride). The lysates were sonicated on ice with four 4-s bursts and then centrifuged at 55,000 rpm in a Beckman TL120.2 rotor for 15 min at 4 °C. The cleared lysates were divided into three aliquots, and precleared with pansorbin and protein G beads. Immunoprecipitations were then performed with anti-CKIP-1 or as a control with anti-GST antibodies bound to protein A-Sepharose for 1 h at 4 °C. An additional control employed protein A-Sepharose in the absence of antibody. For FLAG and GFP immunoprecipitations, U2-OS cells were lysed on ice in 0.5 ml of Nonidet P-40 lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P-40, 1% aprotinin, and 0.1 mm phenylmethylsulfonyl fluoride) or radioimmune precipitation assay lysis buffer. The lysates were sonicated and cleared as described above. The cleared lysates were subjected to immunoprecipitation using anti-FLAG M2 antibodies bound to protein G-Sepharose or anti-GFP antibodies bound to protein A-Sepharose for 1 h at 4 °C. In all cases, the lysates were centrifuged briefly, and the protein A/G-Sepharose beads were washed four times with the appropriate lysis buffer. Following the last wash, bound proteins were eluted from the beads by the addition of sample buffer.
Immunoblot Analysis
Samples were separated by 12% SDS-PAGE using the method of Laemmli (13). Proteins were transferred to polyvinylidene difluoride membranes for 1 h at 15 V, 0.3 A using a semidry transfer apparatus (Bio-Rad, Hercules, CA). Anti-FLAG blots were performed according to the manufacturer’s instructions using anti-FLAG M2 at 1:2,500 and goat anti-mouse secondary antibody (1:20,000) conjugated to horseradish peroxidase. Anti-actin blots were performed according to the manufacturer’s instructions using anti-actin at 1:2,000 and goat anti-rabbit secondary antibody (1:20,000) conjugated to horseradish peroxidase. Anti-GFP immunoblots were performed by blocking for 1 h in 3% gelatin in TBST (Tris-buffered saline with 0.05% Tween-20), followed by incubation with primary antibody at 1:3,000. Immune complexes were detected with goat anti-rabbit secondary antibodies (1:20,000) conjugated to horseradish peroxidase. Immunoblotting with anti-β-tubulin antibodies was performed by blocking for 1 h in 5% skim milk powder in TBST, followed by incubation with primary antibody (1:100). Immune complexes were detected using goat anti-mouse secondary antibodies (1:5,000) conjugated to horseradish peroxidase. Anti-CPα and -CPβ blots were performed by blocking membranes for 1 h in 3% bovine serum albumin in TBST followed by incubation with primary antibodies at dilutions of 1:200 and 1:1,000, respectively. In both cases, secondary goat anti-mouse antibodies were used at a dilution of 1:5,000. Where indicated, membranes were stripped with 0.1 m NaOH for 30 min.
Peptide Array Generation
The SPOT method (14, 15) was employed in the synthesis of peptide arrays for use in determining the residues of CKIP-1 required for interaction with CP. Peptides were synthesized on Amino-PEG500-UC540 membranes derivatized with a polyethylene glycol spacer (Intavis AG). Stock solutions of amino acid derivatives (Peptides International) were prepared at a concentration of 0.33 m in a solution of 0.5 m 1-hydroxybenzotriazole/N-methyl-2-pyrrolidone. Immediately prior to each cycle of synthesis, amino acid solutions were activated with the addition of one part diisopropylcarbodiimide (DIC) solution (1.1 m DIC in N-methyl-2-pyrrolidone) to three parts of the amino acid stock solution. Each cycle of peptide synthesis consisted of (i) coupled spotting of amino acids on the membrane, (ii) capping of the membrane with a solution of 2% acetic anhydride in DMF (1 × 30 s, 1 × 5 min), (iii) wash with DMF (3 × 2 min), (iv) deprotection with 20% piperidine in DMF (10 min), (v) wash with DMF (4 × 2 min), (vi) wash with ethanol (2 × 2 min), and (vii) drying of membranes in a stream of cold air and preparation for the next cycle of synthesis. After the final cycle in the peptide synthesis, the membrane was thoroughly dried overnight in a fume hood to prepare the peptides for side-chain deprotection. Side-chain deprotection was carried out on the membrane in the following steps: (i) deprotection in 95% trifluoroacetic acid, 3% triisopropylsilane, 2% water for 2 h, (ii) wash with dichloromethane (4 × 2 min), (iii) wash with DMF (4 × 2 min), (iv) wash with ethanol (2 × 2 min), and (v) dry with a stream of cold air.
Purification of Recombinant CP
Recombinant CP was purified using a tandem bacterial expression vector (pET-3d/CP), allowing for co-translation of chicken α1- and β1-subunits or mouse α1- and β2-subunits from a single plasmid (16). CP was expressed and purified to homogeneity from BL21(DE3) Escherichia coli, as described (17). Chicken CP was used for GST pull-down assays and peptide array assays. Mouse CP was used for in vitro actin polymerization assays.
Peptide Overlay Assay
Prior to the overlay assay, the membrane was wetted with 95% ethanol and water. The peptide filter was then blocked for 1 h in 2% bovine serum albumin in Tris-buffered saline followed by incubation with CP (1 μm) overnight at 4 °C with rocking. Following washing, the filter was incubated with antibodies against native CP (anti-CPβ 1E5.25.4) at a dilution of 1:200. Bound CP was detected with goat anti-mouse secondary antibodies (1:5000) conjugated to horseradish peroxidase.
Isothermal Titration Calorimetry
ITC experiments were carried out on a MicroCal VP-ITC isothermal titration calorimeter (18). The heterodimeric actin-CP was purified as described (17). Purified CP and CKIP-1 peptide (SYLAHPTRDRAKIQHSRRPPTR) were dialyzed into 10 mm potassium phosphate, pH 7.2, 100 mm KCl, 1 mm Tris (2-carboxyethyl)phosphine hydrochloride and 5% glycerol. For titration, purified CP and CKIP-1 peptide were diluted to 18 and 180 μm, respectively, using the equilibrating solution obtained after dialysis was complete, and each was degassed prior to titration. CP protein was loaded into the cell (~1.4 ml), and CKIP-1 peptide was loaded into the syringe. Titration of the CKIP-1 peptide was performed at 25 °C starting with an initial 1-μl injection, followed by 65 5-μl injections, with a spacing of 300 s. The sample cell was stirred at 300 rpm throughout the experiment. The buffer blank performed under the same conditions showed negligible heats of binding (<0.015 μcal/s). The dissociation constant (KD) and stoichiometry of binding (n) were obtained by non-linear least-squares fitting of the CP versus CKIP-1 peptide data to a one-site model provided with the data analysis software (Origin). Baseline was subtracted automatically using Origin software. The results of two independent experiments are shown.
Spectrin-F-actin-seeded Actin Polymerization Assay
Phalloidin-stabilized F-actin seeds were prepared as described in Harris et al. (19). Polymerization of 1 μm actin (5% Pyrene-labeled) in F-buffer was measured in the presence of F-actin seeds, 4 nm CP, and different amounts of CKIP-1 protein and CKIP-1 and human CARMIL peptides. Pyrene fluorescence (Ex = 368 nm; Em = 386 nm) was monitored for 500 s at 25 °C. The sequences of the CKIP-1 and CARMIL peptides were SYLAHPTRDRAKIQHSRRPPTR and RRLEHFTKLRPKRNKKQQPTQA, respectively.
In Vitro Translation Assays and GST Pull-down Assays
Radiolabeled (35S-lableled) FLAG-CKIP-1 and FLAG-CKIP-1 R155E,R157E were produced by in vitro transcription and translation using a TnT kit (Promega, Madison, WI) with T7 polymerase according to the manufacturer’s recommendations. GST fusion proteins encoding CK2α or GST alone were expressed in bacteria and purified using glutathione-agarose as described previously (20). Following in vitro transcription and translation, GST pull-down assays were performed by diluting 5 μl of the TnT reactions with 50 μl of Nonidet P-40 buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet P-40) and adding 11 μl of GST or GST-CK2α beads. Incubation was at 4 °C for 1 h with rocking. The beads were collected by centrifugation, the supernatant was removed, and the beads were washed three times with 400 μl of Nonidet P-40 buffer. Bound proteins were eluted by the addition of sample buffer and were subjected to SDS-PAGE on a 12% gel. After drying, the radiolabeled proteins were visualized using a PhosphorImager (Amersham Biosciences) and quantitated for two independent experiments using ImageQuaNT software.
Generation of Cell Lines
UTA6 cells were derived from the human osteosarcoma cell line U2-OS and express the tetracycline-regulated transcriptional activator protein (generous gift from Dr. Christoph Englert, Forschungszentrum, Germany (21)). Generation of a cell line with tetracycline-regulated expression of FLAG-CKIP-1 has been previously described (9). Cell lines expressing FLAG-CKIP R155E,R157E were generated by transiently transfecting UTA6 cells with mutant FLAG-CKIP-1/pTRE and pTK-Hyg in the presence of tetracycline (1.5 μg/ml). Drug selection with hygromycin (500 μg/ml) and G418 (460 μg/ml) began 4 days after transfection. Once stably transfected colonies were formed, they were picked and transferred to 96-well plates. Individual colonies of FLAG-CKIP-1 R155E,R157E-expressing cells were grown up and screened for highly regulated inducible expression by immunoblot analysis.
Immunofluorescence
Cells (~200,000) grown on sterile coverslips were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Invitrogen) at 37 °C in an atmosphere of 5% CO2. Coverslips were washed three times with PBS and fixed for 20 min in 3.7% paraformaldehyde at 37 °C. Cells were permeabilized for 5 min with 0.1% Triton X-100 in PBS and then treated with for 5 min with 0.1 m glycine in PBS. Coverslips were washed with PBS and then incubated with anti-FLAG M2 at 1:250 for 1 h at 37 °C. Following washing with PBS, secondary antibodies conjugated to TRITC (1:1,000) were added for 1 h in the dark. Coverslips were washed, mounted with Gelvatol (Celvol 205, Celanese Chemicals, Dallas, TX), and examined with a Zeiss LSM 510 confocal microscope.
Phalloidin Staining, Extraction, and Quantitation
For TRITC-phalloidin staining of cells, coverslips were washed three times with PBS and fixed for 20 min in 3.7% paraformaldehyde at 37 °C. Following permeabilization with 0.1% Triton X-100, coverslips were stained with TRITC-phalloidin (1 μg/ml) for 40 min in the dark, washed with PBS, and mounted with Gelvatol. Slides were visualized using a Zeiss Axiovert inverted fluorescence microscope and Northern Elite software. F-actin quantitation experiments were performed by the method of Machesky et al. (22). Briefly, triplicate plates of cells were washed 2× with PBS and scraped into 400 μl of F-actin stabilizing extraction buffer (10 mm Pipes, pH 7.0, 20 mm potassium phosphate, 5 mm EGTA, 2 mm MgCl2, 0.1% Triton X-100) with 3.7% paraformaldehyde and 2 μm TRITC-phalloidin and rocked for 1 h at room temperature. The cells were pelleted by brief centrifugation and washed 2× with 200 μl of 0.1% saponin, 20 mm potassium phosphate, 10 mm Pipes, pH 7.0, 5 mm EGTA, and 2 mm MgCl2. The bound phalloidin was extracted in 1 ml of methanol by rocking 1 h at room temperature, and TRITC levels were determined by reading the emission at 563 nm following excitation at 542 nm. Values were normalized to the overall protein concentration and represent the average of three 10-cm plates ± S.D. from two independent experiments.
RESULTS
Co-immunoprecipitation of Endogenous CP with Endogenous CKIP-1
We have previously shown that elevated expression of CKIP-1 induces alterations in cell morphology and the actin cytoskeleton. The discovery of the heterodimeric actin-CP in complexes with Tandem affinity purification-CKIP-1 and FLAG-CKIP-1 suggested that CKIP-1-CP interactions could have a role in the observed morphological and cytoskeletal alterations (9). Examination of Fig. 1A illustrates co-immunoprecipitation of endogenous CKIP-1 and endogenous CP in mouse myoblast (C2C12) cells where CKIP-1 has been shown to be induced during differentiation and in human osteosarcoma (U2-OS) cells where CKIP-1 has also been observed. In this respect, although CPβ is detected in anti-CKIP-1 immunoprecipitates from extracts of U2-OS and C2C12 cells, CPβ is not detected following immunoprecipitation with control anti-GST (IgG) antibodies or with protein A beads alone. These observations reinforce the physiological relevance of CP-CKIP-1 interactions.
FIGURE 1. Identification of two putative CP-binding sites on CKIP-1 by peptide array analysis and alignment with CARMIL.
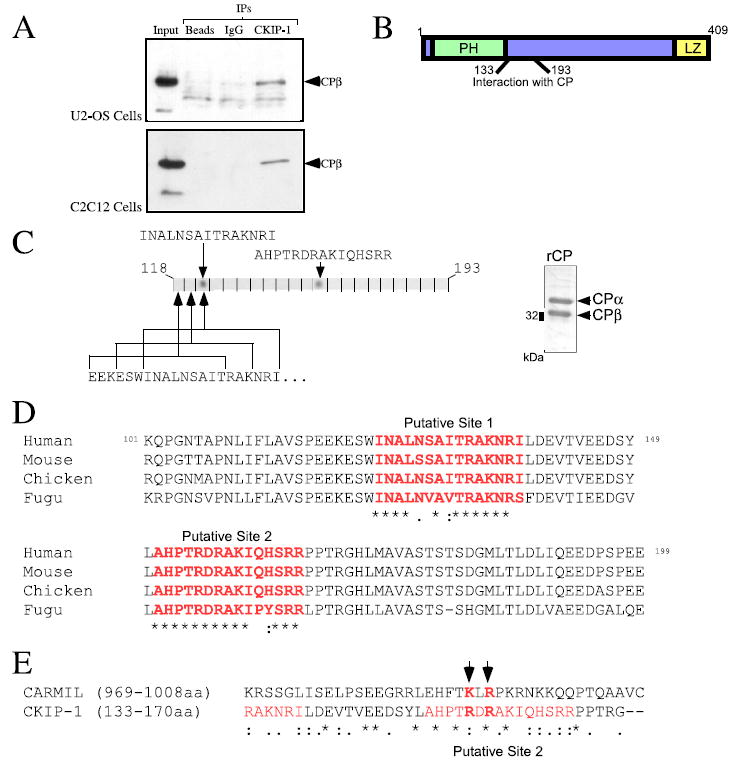
A, endogenous CKIP-1 and CP interact in U2-OS and C2C12 cells. Lysates derived from U2-OS and C2C12 cells were immunoprecipitated with anti-CKIP-1 antibodies and immunoblotted with anti-CPβ antibodies. Control immunoprecipitations were performed with anti-GST antibodies (IgG) or protein A-Sepharose beads alone. B, schematic illustrating the 60-amino acid domain of CKIP-1 required for interaction with CP. C, peptide walking array analysis of the region of CKIP-1 required for CP binding. CKIP-1 peptides (16-mers, frame shifted by three amino acids) were synthesized using the SPOT method as described under “Materials and Methods.” An overlay assay was performed using recombinant CP (1 μm, see inset) followed by detection with antibodies against native CPβ. Two peptides found to mediate interactions with CP are indicated. D, the region spanning these two peptides (101–199 amino acids) was aligned with sequences of CKIP-1 from various species using ClustalW. Completely conserved residues (*), highly conserved residues (:), and conserved residues (.) are indicated. Both CP-binding sites are well conserved between species. E, alignment of the putative CP-binding region on CKIP-1 with the CP-binding site on CARMIL. The residues highlighted in red in the CARMIL sequence were shown to be necessary for its interactions with CP.
Identification of CP-binding Sites on CKIP-1
We have previously demonstrated using deletion analysis that interaction between CKIP-1 and CP is dependent on amino acids 133–193 of CKIP-1 (9) (highlighted in Fig. 1B). To identify residues required for this interaction more precisely, we generated a CKIP-1 peptide filter composed of 16-mers “walking” from the amino to carboxyl termini of this region, with frame shifts of three amino acids between each peptide (Fig. 1C). The resulting filter was incubated with recombinant CP (see inset) followed by detection with antibodies against native CPβ. As shown in Fig. 1C, two peptides exhibited interactions with CP. A multiple sequence alignment of the CP-binding domain of CKIP-1 reveals that both peptides, designated site 1 and site 2, appear to be very well conserved between species (Fig. 1D).
Recently, a study by Yang et al. (11) demonstrated that the interaction of CARMIL with CP requires two conserved basic residues, Lys-991 and Arg-993. It was therefore intriguing that both site 1 and site 2 (Fig. 1D) contain basic-X-basic motifs. Consequently, we performed an alignment of CARMIL (residues 969–1008) with the region of CKIP-1 encompassing sites 1 and 2. As illustrated in Fig. 1E, the sequence encompassing site 2 could be aligned with the CARMIL sequence suggesting that the residues necessary for interactions between CARMIL and CP might be present in CKIP-1. Moreover, the equivalent residues in CKIP-1 (Arg-155 and Arg-157) are highly conserved between species (Fig. 1D).
Disruption of Interactions between CKIP-1 and CP
Based on the data in Fig. 1, we performed mutagenesis to determine if disruption of one or both of the CP-binding sites on CKIP-1 could result in loss of CKIP-1-CP interactions. To disrupt site 2, which could be aligned with the CP-binding region of CARMIL, we generated constructs in which residues Arg-155 and Arg-157 were mutated to alanine or glutamic acid. Similarly, we mutated Lys-159, because this residue was also conserved between CKIP-1 and CARMIL. The resulting constructs were designated CKIP-1 R155A,R157A, R155E,R157E, and R155A,R157A, K159A. Because site 1 contained similar positively charged residues, we also generated CKIP-1 R133,K135E and R133A,K135A,R137A mutants. These constructs are shown schematically in Fig. 2A.
FIGURE 2. Identification of the residues of CKIP-1 required for interaction with CP.

A, schematic representation of the CKIP-1 constructs generated to disrupt interaction with CP. B, U2-OS cells were transfected with FLAG-CKIP-1 constructs or empty vector as indicated. Lysates were immunoblotted with anti-FLAG M2 antibodies to demonstrate equivalent expression (left panel). Immunoprecipitations were performed with anti-FLAG M2 antibodies, followed by immunoblotting with anti-CPβ antibodies (right panel). The positions of FLAG-CKIP-1, CPβ, and the heavy and light chains (HC and LC) of IgG are indicated.
To distinguish between these two binding sites, we tested if mutations within these regions affected interactions with CP by immunoprecipitation assays. U2-OS cells were transfected with FLAG-CKIP-1 constructs as indicated, and the derived lysates were immunoblotted with anti-FLAG M2 antibodies to demonstrate equal loading (Fig. 2B, left panel). Immunoprecipitations were performed on these lysates using anti-FLAG M2 antibodies followed by immunoblotting with anti-CPβ antibodies (Fig. 2B, right panel). As shown, mutation of R155E,R157E in putative site 2 resulted in greatly diminished binding to CP as did R155A,R157A or R155A,R157A,K156A (data not shown). By comparison, CKIP-1 harboring R133A,K135A,R137A substitutions (Fig. 2B) or R133,K135E substitutions (data not shown) in site 1 had no effect on binding to CP. These results indicate that disruption of Arg-155 and Arg-157 within site 2 on CKIP-1 is sufficient to prevent interaction with CP, whereas similar substitutions within site 1 do not affect CKIP-1-CP interactions.
To further characterize CKIP-1 with substitutions within site 2, we next examined the effects of various mutations on binding to CP using immunoprecipitation and GST pull-down assays (Fig. 3). For immunoprecipitations, U2-OS cells were transfected with FLAG-CKIP-1 constructs, and the lysates derived from these cells were immunoblotted with anti-FLAG M2 antibodies to demonstrate equal loading (Fig. 3A, left panel). The lysates were immunoprecipitated with anti-FLAG M2 antibodies and immunoblotted with anti-CPβ antibodies (Fig. 3A, right panel). Compared with wild-type CKIP-1, all three mutants exhibit greatly reduced binding to CP. Similar experiments were performed with GFP-tagged constructs (Fig. 3B). Again, the three CKIP-1 mutants exhibit greatly diminished binding to CP.
FIGURE 3. Disruption of the interaction of CKIP-1 with CP.

A, U2-OS cells were transfected with FLAG-CKIP-1 constructs or empty vector as indicated. Lysates were immunoblotted with anti-FLAG M2 antibodies to demonstrate equivalent expression (left panel). Anti-FLAG M2 immunoprecipitations were performed followed by immunoblotting with anti-CPβ antibodies (right panel). B, U2-OS cells were transfected with GFP-CKIP-1 constructs or GFP alone as indicated. Cell extracts were immunoblotted with anti-GFP antibodies to demonstrate equivalent expression (left panel). Immunoprecipitations were performed with anti-GFP antibodies, followed by immunoblotting with anti-CPβ antibodies (right panel). C, GST and GST-CKIP-1 were purified as described under “Materials and Methods” and visualized using GelCode Blue to demonstrate equivalent protein expression (left panel). Recombinant CP (0.4 μg) was incubated with GST or GST-CKIP-1 constructs bound to glutathione beads. Pull-down assays were immunoblotted with anti-CPα antibodies (right panel). The positions of CPα, CPβ, and the heavy and light chains (HC and LC) of IgG are indicated. The designation of CKIP-1 mutants is as in Fig. 2.
To complement the results of the immunoprecipitation assays, we employed GST pull-down assays. GST and GST-CKIP-1 were purified on glutathione beads and stained with GelCode Blue to demonstrate equivalent protein expression (Fig. 3C, left panel). The GST fusions were used to perform pull-down assays with purified recombinant CP (Fig. 3C, right panel). In good agreement with the results described above, both mutants exhibit greatly reduced binding to CP. Collectively, these data suggest that Arg-155, Arg-157, and possibly Lys-159 of CKIP-1 are required for interaction with CP and imply that CKIP-1 and CARMIL may share similar CP-binding residues.
Dissociation Constant for CP Interactions with CKIP-1 Peptide
To further characterize interactions between CKIP-1 and CP, we employed a synthetic 22-residue peptide corresponding to the area of CKIP-1 responsible for interactions with CP as determined in Fig. 1. Based on two independent measurements, the average dissociation constant for this interaction was determined to be 9.5 × 10−8 m, fitting the data to a one-site binding model (Fig. 4). Notably, this value compares favorably with measurements recently made for the interactions between CP and V-1 (23) as well as interactions between CP and CD2AP, another CP-binding protein with a similar basic-X-basic motif to that found in CKIP-1 and CARMIL (24). It is also notable that in the study by Bruck et al. (24), the CKIP-1 peptide performed at least as well as analogous peptides derived from CD2AP and CARMIL-1 in inhibiting interactions between CD2AP and CP, an observation consistent with the conclusion that CKIP-1 shares a CP-binding motif with these other proteins. Collectively, these data extend the biochemical characterization of interactions between CKIP-1 and CP and further reinforce the physiological relevance of these interactions.
FIGURE 4. Isothermal titration calorimetry of CP versus CKIP-1 peptide.
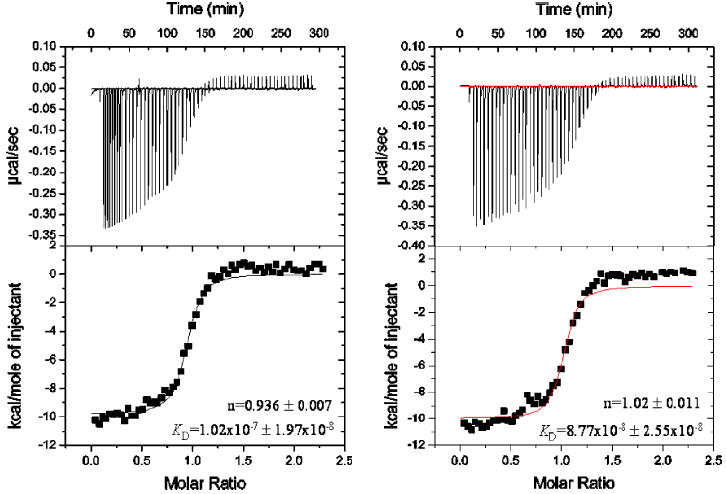
ITC was performed at 25 °C using purified CP (18 μm) and CKIP-1 peptide (180 μm). Data were fitted to a one-site binding model using Origin software. The results of two independent experiments are shown, with the estimated number of binding sites (n) and the dissociation constant (KD) indicated.
Functional Analysis of the Proposed CP-binding Region of CKIP-1
CKIP-1 inhibits the ability of CP to cap the barbed ends of actin filaments (9). As illustrated above (Figs. 2 and 3), point mutations of CKIP-1 in the proposed CP-binding region showed decreased physical binding. We asked whether the mutations would also decrease the ability of CKIP-1 to inhibit the capping activity of CP in actin polymerization assays for barbedend growth (Fig. 5). Pyrene actin polymerization was nucleated with F-actin seeds (Fig. 5). CP-inhibited the actin polymerization, due to capping of barbed ends. Increasing concentrations of wild-type CKIP-1 inhibited the capping activity of CP (Fig. 5, A and B), reaching saturation at ~50% inhibition, consistent with our previous results (9). In contrast, the R155E,R157E and R155A,R157A,K159A CKIP-1 mutants had no effect on the capping activity of CP (Fig. 5B). Thus, the functional assays agree that these CKIP-1 mutants do not interact with CP.
FIGURE 5. Functional analysis of the critical conserved region of CKIP-1 on the capping activity of CP.

Actin polymerization from seeds is measured over time. The addition of 4 nm CP blocks actin polymerization by capping the growing barbed ends, and CKIP-1 inhibits the capping activity of CP. A, CKIP-1 (80 nm) inhibits CP activity by ~50%. B, point mutations of the critical conserved residues of CKIP-1 abolish its ability to inhibit CP. Two different CKIP-1 point mutants have no effect at 200 nm, under conditions where 20 nm and 80 nm of wild-type CKIP-1 inhibit CP. C, peptides corresponding to the critical conserved region of CKIP-1 inhibit CKIP-1 interaction with CP. Addition of the CKIP-1 peptide lessens the inhibitory effect of whole CKIP-1 on CP. The peptide alone has a small effect on CP. The concentration of the peptide was 500 nm, and the CKIP-1 concentration was 80 nm. In each panel, a control without seeds is shown. The designation of CKIP-1 mutants is as in Fig. 2.
We extended the functional analysis of the CP-binding region of CKIP-1 in similar assays with the 22-amino acid CKIP-1 peptide spanning site 2 (Fig. 5C). First, we found that addition of the peptide had a small inhibitory effect on the capping activity of CP. We also attempted to use a wider range of peptide concentrations, but we were only successful at inhibiting CP activity at concentrations up to 500 nm. We do not know the precise reason for the inability to use higher peptide concentrations; however, we anticipate that this may be a limitation of the polymerization assay and/or the solubility of the peptide. In the presence of full-length CKIP-1, addition of the peptide suppressed the inhibitory effect of CKIP-1 on the activity of CP. Both full-length CKIP-1 and the peptide were present at concentrations near saturation in this assay, so the results are consistent with the peptide competing with CKIP-1 for binding CP. If the peptide and CKIP-1 interacted with different regions of CP, one would have expected an additive effect from the combination. These results are consistent with the proposed CP-binding region of CKIP-1 being sufficient for binding CP and having a direct role in the interaction of CKIP-1 with CP.
Localization of CKIP-1 Mutants in U2-OS Cells
To test if loss of CP-binding altered the cellular localization of CKIP-1, we examined the localization of the CKIP-1 mutants in cells (Fig. 6). U2-OS cells were transiently transfected with FLAG-CKIP-1 constructs as indicated, fixed and labeled with anti-FLAG M2 antibodies for visualization by confocal microscopy (Fig. 6A). Consistent with our previous results, wild-type CKIP-1 localizes predominantly to the plasma membrane in U2-OS cells (1, 2, 9). Importantly, all three mutants that show reduced CP binding appear to have comparable cellular localization to wild-type CKIP-1. Similar results were obtained when U2-OS cells were transiently transfected with GFP-CKIP-1 constructs (Fig. 6B). These results demonstrate that mutation of residues to disrupt interactions with CP has no overt effect on the cellular localization of CKIP-1. Of the three mutants examined above, we selected one, CKIP-1 R155E,R157E, to be used to extend our studies.
FIGURE 6. Localization of CKIP-1 mutants in U2-OS cells.
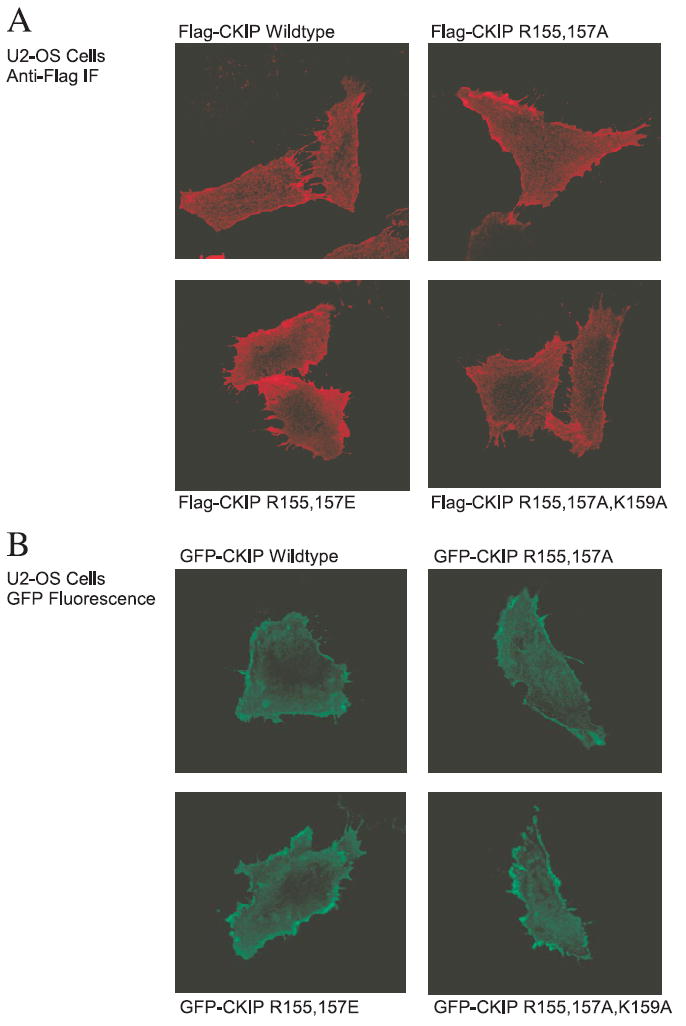
FLAG- and GFP-tagged CKIP-1 mutants, designated as in Fig. 1, exhibit comparable membrane localization to wild-type CKIP-1. A, U2-OS cells transfected with FLAG-CKIP-1 constructs were subjected to anti-FLAG M2 immunofluorescence and visualized by confocal microscopy. B, U2-OS cells transfected with GFP-CKIP-1 constructs were fixed to visualize GFP fluorescence by confocal microscopy.
CKIP-1 R155E,R157E Retains the Ability to Interact with Protein Kinase CK2 and to Self-associate
To further characterize the properties of this CKIP-1 mutant, we designed experiments to examine interactions between CKIP-1 and CK2 as well as self-association of CKIP-1. Because the PH domain of CKIP-1 is responsible for interactions with protein kinase CK2 (2), mutation of Arg-155 and Arg-157, which lie outside of this domain, would not be expected to influence interactions with CK2. Nevertheless, to test that the R155E,R157E mutations have not disrupted interactions between CKIP-1 and protein kinase CK2, plasmids encoding wild-type FLAG-CKIP-1 and FLAG-CKIP-1 R155E,R157E were subjected to in vitro transcription and translation in the presence of [35S]methionine (Fig. 7A). Translated products were utilized in pull-down assays with GST-CK2α or GST alone, and bound proteins were separated by SDS-PAGE and visualized using a PhosphorImager (Fig. 7A, top panel). As shown, both wild-type and mutant CKIP-1 retain the ability to interact with protein kinase CK2. To quantitate these results, the data for two independent experiments were analyzed using ImageQuaNT software and expressed as percentage of bound radiolabeled protein (Fig. 7A, lower panel). These data suggest that the two CKIP-1 constructs bind protein kinase CK2 to the same degree.
FIGURE 7. CKIP-1 R155E,R157E retains the ability to interact with protein kinase CK2 and to oligomerize.

A, constructs expressing FLAG-CKIP-1, FLAG-CKIP-1 R155E,R157E, or empty vector were subjected to in vitro transcription and translation and GST pull-down assays as described under “Materials and Methods.” Radiolabeled proteins bound to the GST fusion proteins were visualized using a PhosphorImager (top panel). The positions of FLAG-CKIP-1 and CKIP-1 are indicated. Data were analyzed using ImageQuaNT software and expressed as the percentage of bound radiolabeled product for two independent experiments (lower panel). B, U2-OS cells were co-transfected with GFP-CKIP-1 along with FLAG-CKIP-1, FLAG-CKIP-1 R155E,R157E, or empty vector as indicated. Cell extracts were immunoblotted with anti-GFP antibodies to demonstrate equivalent expression (left panel). Lysates were then subjected to immunoprecipitations with anti-FLAG M2 antibodies and immunoblotting with anti-GFP antibodies.
Previous studies by our laboratory and others have suggested that CKIP-1 can oligomerize to form dimers or higher order structures in cells (2, 8). To examine if the R155E,R157E mutations have disrupted CKIP-1-CKIP-1 interactions, we performed immunoprecipitations to assess self-association of CKIP-1 (Fig. 7B). U2-OS cells were co-transfected with GFP-CKIP-1 along with empty vector, FLAG-CKIP-1, or FLAG-CKIP-1 R155E,R157E, and the lysates derived from these cells were immunoblotted with anti-GFP antibodies to demonstrate equal expression (Fig. 7B, left panel). The lysates were immunoprecipitated with anti-GFP antibodies, and immunoblotted with anti-FLAG M2 antibodies (Fig. 7B, right panel). As shown, both wild-type FLAG-tagged CKIP-1 and the R155E,R157E mutant are able to interact with GFP-tagged CKIP-1. Collectively, these studies indicate that disruption of interaction between CKIP-1 and CP does not affect the ability of CKIP-1 to interact with protein kinase CK2 or with itself validating that disruption of interactions between CP and CKIP-1 can be achieved without affecting other functional properties of CKIP-1.
Inducible Expression of FLAG-CKIP-1 R155E,R157E
Our results in Figs. 2-7 indicate that the CKIP-1 R155E,R157E mutant is an ideal candidate to distinguish between our working models for the effects of CKIP-1 on cell morphology, because it fails to interact with CP yet retains other known characteristics of wild-type CKIP-1. Therefore, we generated U2-OS cell lines with tetracycline-regulated expression of FLAG-CKIP-1 R155E,R157E. For these experiments, FLAG-CKIP-1 R155E,R157E was ligated into the pTRE vector immediately downstream of the tet-responsive promoter. Following transfection and drug selection, stably transfected colonies were picked and screened for highly regulated inducible expression of mutant FLAG-CKIP-1 in the absence of tetracycline. U2-OS cells expressing wild-type CKIP-1 (DC1.4) or CKIP-1 R155E,R157E (DC3.20) were grown in the presence (+) or absence (−) of tetracycline, fixed, and labeled with anti-FLAG M2 antibodies and visualized by confocal microscopy (Fig. 8A). Both cell lines exhibit strong induction and tight regulation of CKIP-1 expression. To examine protein expression, cells grown in the presence (+) of tetracycline or induced (−) for 24 h were harvested. These lysates were subjected to immunoblotting with anti-FLAG M2 antibody (Fig. 8B, upper panel). Again, induction is tightly regulated, and both cell lines appear to express similar levels of FLAG-CKIP-1. To ensure equal protein loading, the membrane was stripped and reprobed with anti-β-tubulin antibodies (Fig. 8B, lower panel).
FIGURE 8. Tetracycline-regulated expression of FLAG-CKIP-1 R155E,R157E.

A, anti-FLAG M2 immunofluorescence of DC1.4 and DC3.20 cells grown in the presence or absence of tetracycline. B, DC1.4 and DC3.20 cells were grown in the presence or absence of tetracycline for 24 h to induce expression of wild-type FLAG-CKIP-1 or FLAG-CKIP-1 R155E,R157E. Total cell lysate (40 μg) was immunoblotted with anti-FLAG M2 antibodies (top panel). To ensure equal loading, the membrane was stripped and reprobed with anti-β-tubulin (lower panel). C, DC1.4 and DC3.20 cells were grown in the presence or absence of tetracycline and microscopic images were captured after 24-h induction. D, perimeters of DC1.4 and DC3.20 cells were measured and quantitated using Northern Elite software. Results shown represent the average of ~100 cells in 6 fields ± S.D.
To determine the phenotype associated with expression of CKIP-1 R155E,R157E, we used phase-contrast microscopy to examine cells grown in the presence or absence of tetracycline for 24 h. As we have previously documented, expression of wild-type CKIP-1 in the absence of tetracycline causes cells to become elongated and exhibit morphology consistent with fibroblastic cells (Fig. 8C) (9). By contrast, induction of CKIP-1 R155E,R157E expression fails to elicit any change in morphology. To quantify these observations, we performed morphometric analysis of the cell lines grown in the presence or absence of tetracycline by measuring the perimeter of ~100 cells from numerous fields of view (Fig. 8D). The results from this experiment clearly show that cells induced to express wild-type CKIP-1 have a significantly greater perimeter than non-induced cells, whereas induction of CKIP-1 R155E,R157E expression has no effect on cell shape.
FLAG-CKIP-1 R155E,R157E Does Not Increase Phalloidin Binding or Cellular Actin Levels
Previously, we documented a 1.5-fold increase in phalloidin staining and modest increases in cellular actin levels associated with CKIP-1 expression (9). Based on these results, we asked whether induction of CKIP-1 R155E,R157E would elicit the same response by examining the F-actin profile of these cells using fluorescently-labeled phalloidin. The wild-type and mutant CKIP-1 cell lines were grown in the presence or absence of tetracycline and then stained with TRITC-phalloidin (Fig. 9A). Induction of FLAG-CKIP-1 caused an increase in overall phalloidin staining, along with increased numbers of transverse actin stress fibers and actin aggregates, consistent with our previous report (9). In contrast, induction of FLAG-CKIP-1 R155E,R157E failed to cause any apparent change in the actin cytoskeleton. To extend these results, we performed quantitative measurements of TRITC-phalloidin binding (Fig. 9B). In accordance with our previous study, the data for wild-type CKIP-1 expression, normalized to extract protein, clearly shows a 1.5-fold increase in phalloidin binding. On the other hand, increased phalloidin staining was not observed following induction of FLAG-CKIP-1 R155E,R157E. These data demonstrate that induction of wild-type FLAG-CKIP-1 results in an increase in the overall amount of cellular F-actin, whereas induction of a CKIP-1 mutant that fails to interact with CP does not.
FIGURE 9. Expression of FLAG-CKIP-1 R155E,R157E does not increase phalloidin binding or cellular actin levels.

A, DC1.4 and DC3.20 cells grown in the presence of tetracycline or induced for 24 h were stained with TRITC-phalloidin. B, F-actin quantitation of DC1.4 and DC3.20 cells grown in the presence or absence of tetracycline for 24 h was performed as described under “Materials and Methods.” Data represent the average of two independent experiments each using three 10-cm plates of cells normalized for protein concentration (±S.D.). C, DC1.4 and DC3.20 cells were grown in the presence or absence of tetracycline for 18 h. Lysates prepared with radioimmune precipitation assay buffer were immunoblotted with anti-actin antibodies (left panel). To ensure equal loading, the membrane was stripped and reprobed with anti-β-tubulin (right panel).
Finally, we examined the total cellular levels of actin in these cell lines, grown in the presence of tetracycline or induced for 18 h. As reported, induced expression of FLAG-CKIP-1 results in a modest increase in the cellular levels of actin (Fig. 9C, left panel) (9). However, induction of FLAG-CKIP-1 R155E,R157E expression failed to cause a similar increase. The membrane was stripped and reprobed with anti-β-tubulin antibodies to ensure equal protein loading on the gel (Fig. 9C, right panel). Quantification of actin levels by densitometry and normalization to β-tubulin levels from two independent experiments illustrated that actin levels are increased by ~30% in cells expressing FLAG-CKIP-1, whereas there was no corresponding increase in FLAG-CKIP-1 R155E,R157E cells. Taken together, these data suggest that interaction with CP is necessary for induced CKIP-1 to increase levels of total and F-actin in cells.
DISCUSSION
In cells, actin filaments extend predominantly by the addition of actin monomers to barbed (fast growing) ends of the filament. To stop filament growth, barbed ends are capped by actin-CP in a Ca2+-independent (25-27) and phosphatidylinositol 4,5-bisphosphate-dependent manner (28-31). According to the dendritic nucleation model (32), free barbed ends of actin filaments are capped relatively quickly, which limits actin polymerization to free barbed ends near the membrane. Capping keeps the filament short, the degree of branching high, and thus increases the strength of the filament network. This model is supported by the observations that depleting Dictyostelium discoideum of capping protein caused an increase in F-actin but a decrease in motility (33), and studies with Listeria monocytogenes and purified proteins demonstrating that when capping protein is removed, motility of the bacterium was completely inhibited (34).
With respect to the regulation of actin polymerization, an additional level of control is provided by proteins that can interact with and modulate the activity of CP. To date, proteins found to interact with CP include: V-1 (23, 35, 36), CARMIL (11, 37, 38), Twinfilin (39), CD2AP (40), CapZIP (41), and CKIP-1 (9). Like V-1 and CARMIL, our studies indicate that CKIP-1 inhibits the activity of CP in a dose-dependent manner (11, 35). The fact that the level of inhibition by CKIP-1 is not complete at saturation indicates that the complex of CKIP-1 with CP has a decreased affinity for barbed ends but that binding is not completely abolished. In this respect, CARMIL shows a similar effect with less than complete inhibition of CP activity at saturation (11). Whether the complexes of CP with CKIP-1 or CARMIL do in fact bind to barbed ends of actin filaments in cells or not will depend on the concentration of the molecules in a given location, which may be influenced by scaffolding effects from other interactors.
Given that expression of CKIP-1 affects cell morphology and the cytoskeleton, it is interesting that expression of CARMIL in human glioblastoma cells was shown to increase the number and size of lamellipodia (11). Moreover, small interference RNA-induced depletion of CARMIL caused a decrease in F-actin and in lamellipodia that could be rescued by expression of wild-type CARMIL but not deletions lacking a region implicated in binding CP. These data suggest that CARMIL, through interactions with CP, can alter both cell morphology and the actin cytoskeleton. Our results also reinforce the conclusion that CKIP-1 and CARMIL contain conserved CP-interaction residues, or a possible CP-binding motif as recently suggested by Bruck et al. (24). This observation suggests that CKIP-1 is a member of a family of proteins that can modulate the activity of capping protein, and therefore actin polymerization and cell morphology.
We proposed two working models for the role of CKIP-1 in cell morphology. Our first model suggested that the interaction of CP with CKIP-1 at the plasma membrane inhibits binding of CP to the barbed ends of the actin filament leading to the generation of a “capping-free” zone at the plasma membrane and to increased actin polymerization and changes in cellular morphology. However, the inhibition of CP by CKIP-1 is only partial (9) raising the alternative model in which the effects of CKIP-1 on cell morphology involve targeting of CKIP-1 to the barbed ends of actin filaments by CP. Our results indicate that disruption of binding to CP did not affect the localization of CKIP-1 in cells, arguing against the second model. In addition, the disruption of CP-binding did cause CKIP-1 to lose the ability to change the actin cytoskeleton and the shape of cells, favoring the first model. This hypothetical model is illustrated on the left hand side of Fig. 10. Inhibition of CP activity would lead to an increase in free barbed ends and, therefore, an increase in F-actin. This model is also consistent with reports in the literature. For example, expression of CP mutants that fail to interact with actin in Saccharomyces cerevisiae led to an increase in F-actin (42). In a similar vein, as noted earlier, depletion of CP from D. discoideum or L. monocytogenes resulted in affects on the actin cytoskeleton as did short hairpin RNA-induced depletion of the CP-inhibitory protein CARMIL in human glioblastoma cells (11). Taken together, these studies suggest that inhibition of CP leads to polymerization of actin filaments in vivo. As illustrated in Fig. 10, our previous studies demonstrated that CK2 phosphorylates CPα at S9 and that CK2 increases the inhibitory activity of CKIP-1 on CP, although this inhibitory activity appears to be phosphorylation-independent (9). In the future, it will clearly be of interest to define the precise role of CK2 in the regulation of CP and the role of CKIP-1 in the control of this event.
FIGURE 10. Hypothetical model illustrating the opposing effects of wild-type and mutant CKIP-1 on cell morphology and the actin cytoskeleton.
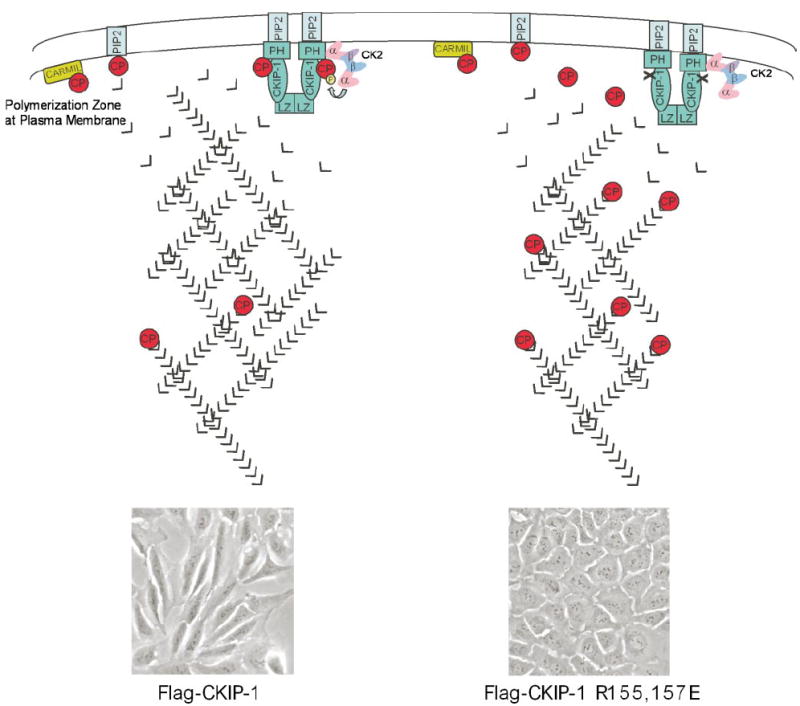
Interactions between CKIP-1 and CP at the plasma membrane inhibit binding of CP to the barbed ends of actin filaments leading to increased actin polymerization and changes in cellular morphology (left side). CK2 has been shown to phosphorylate CPα at serine 9. The presence of CK2 increases the inhibitory effect of CKIP-1 on CP; however, this inhibition is phosphorylation-independent. In cells, CP is also inhibited by other cellular components, including PIP2 and CARMIL. Disruption of the interactions between CKIP-1 and CP rescues the ability of CP to bind to the barbed ends of actin filaments (right side). This results in basal actin polymerization and parental U2-OS cell morphology. The inset illustrates microscopic images of cells expressing wild-type CKIP-1 or CKIP-1 R155E,R157E. Actin monomers are shown as the “v” symbols. CP is shown as a monomer for simplicity. CKIP-1 is shown as a dimer, but its exact oligomeric structure is unknown.
In addition to providing new insights regarding the role of CKIP-1 and its interactions with CP in the regulation of cell morphology and the cytoskeleton, this study establishes a foundation for further investigation. We have shown that both the morphology change and the increase in F-actin are dependent on interactions between CKIP-1 and CP, because these changes are prevented by disrupting the interactions CKIP-1 and CP. In this respect, these studies have led us closer to an understanding of how CKIP-1 regulates the actin cytoskeleton through interactions with CP.
Acknowledgments
Confocal microscopy was performed at the London Regional Molecular Imaging Facility within the Schulich School of Medicine and Dentistry at the University of Western Ontario. Peptide SPOT synthesis was performed in the laboratory of Dr. Shun-Cheng Li. ITC was performed in the Biomolecular Interactions and Conformations Facility and protein purification in the Functional Proteomics Facility at the University of Western Ontario. We are grateful to Dr. Eric Ball for helpful discussions during the course of this work, to Lee-Ann Briere for assistance with ITC, Victoria Clark for technical assistance, and to Serawit Bruck and Dr. Nandini Bhattacharya for providing the CKIP-1 peptide and recombinant CP, respectively.
Footnotes
This work was supported in part by operating grants from the Canadian Institutes of Health Research (CIHR, Grant MOP 37854 to D. W. L.) and by National Institutes of Health Grant GM38542 (to J. A. C.).
The abbreviations used are: CKIP-1, CK2-interacting protein 1; CK2, protein kinase CK2 or casein kinase II; CP, actin-capping protein; GFP, green fluorescent protein; mAb, monoclonal antibody; GST, glutathione S-transferase; DMF, N,N-dimethylformamide; ITC, isothermal titration calorimetry; PBS, phosphate-buffered saline; TRITC, tetramethylrhodamine isothiocyanate.
References
- 1.Bosc DG, Graham KC, Saulnier RB, Zhang C, Prober D, Gietz RD, Litchfield DW. J Biol Chem. 2000;275:14295–14306. doi: 10.1074/jbc.275.19.14295. [DOI] [PubMed] [Google Scholar]
- 2.Olsten ME, Canton DA, Zhang C, Walton PA, Litchfield DW. J Biol Chem. 2004;279:42114–42127. doi: 10.1074/jbc.M407628200. [DOI] [PubMed] [Google Scholar]
- 3.Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD. Neuron. 2000;27:107–119. doi: 10.1016/s0896-6273(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 4.Dodge K, Scott JD. FEBS Lett. 2000;476:58–61. doi: 10.1016/s0014-5793(00)01671-9. [DOI] [PubMed] [Google Scholar]
- 5.Alto N, Carlisle Michel JJ, Dodge KL, Langeberg LK, Scott JD. Diabetes. 2002;51(Suppl 3):S385–S388. doi: 10.2337/diabetes.51.2007.s385. [DOI] [PubMed] [Google Scholar]
- 6.Wong W, Scott JD. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- 7.Safi A, Vandromme M, Caussanel S, Valdacci L, Baas D, Vidal M, Brun G, Schaeffer L, Goillot E. Mol Cell Biol. 2004;24:1245–1255. doi: 10.1128/MCB.24.3.1245-1255.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang L, Xing G, Tie Y, Tang Y, Tian C, Li L, Sun L, Wei H, Zhu Y, He F. EMBO J. 2005;24:766–778. doi: 10.1038/sj.emboj.7600532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canton DA, Olsten ME, Kim K, Doherty-Kirby A, Lajoie G, Cooper JA, Litchfield DW. Mol Cell Biol. 2005;25:3519–3534. doi: 10.1128/MCB.25.9.3519-3534.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 11.Yang C, Pring M, Wear M, Huang M, Cooper JA, Svitkina TM, Zigmond SH. Dev Cell. 2005;9:209–221. doi: 10.1016/j.devcel.2005.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schafer DA, Jennings PB, Cooper JA. J Cell Biol. 1996;135:169–179. doi: 10.1083/jcb.135.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 14.Frank R. J Immunol Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
- 15.Tegge WJ, Frank R. Methods Mol Biol. 1998;87:99–106. doi: 10.1385/0-89603-392-9:99. [DOI] [PubMed] [Google Scholar]
- 16.Soeno Y, Abe H, Kimura S, Maruyama K, Obinata T. J Muscle Res Cell Motil. 1998;19:639–646. doi: 10.1023/a:1005329114263. [DOI] [PubMed] [Google Scholar]
- 17.Palmgren S, Ojala PJ, Wear MA, Cooper JA, Lappalainen P. J Cell Biol. 2001;155:251–260. doi: 10.1083/jcb.200106157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pierce MM, Raman CS, Nall BT. Methods. 1999;19:213–221. doi: 10.1006/meth.1999.0852. [DOI] [PubMed] [Google Scholar]
- 19.Harris ES, Li F, Higgs HN. J Biol Chem. 2004;279:20076–20087. doi: 10.1074/jbc.M312718200. [DOI] [PubMed] [Google Scholar]
- 20.Bosc DG, Slominski E, Sichler C, Litchfield DW. J Biol Chem. 1995;270:25872–25878. doi: 10.1074/jbc.270.43.25872. [DOI] [PubMed] [Google Scholar]
- 21.Englert C, Hou X, Maheswaran S, Bennett P, Ngwu C, Re GG, Garvin AJ, Rosner MR, Haber DA. EMBO J. 1995;14:4662–4675. doi: 10.1002/j.1460-2075.1995.tb00148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machesky LM, Hall A. J Cell Biol. 1997;138:913–926. doi: 10.1083/jcb.138.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhattacharya N, Ghosh S, Sept D, Cooper JA. J Biol Chem. 2006;281:31021–31030. doi: 10.1074/jbc.M606278200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bruck S, Huber TB, Ingham RJ, Kim K, Niederstrasser H, Allen PM, Pawson T, Cooper JA, Shaw AS. J Biol Chem. 2006;281:19196–19203. doi: 10.1074/jbc.M600166200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caldwell JE, Heiss SG, Mermall V, Cooper JA. Biochemistry. 1989;28:8506–8514. doi: 10.1021/bi00447a036. [DOI] [PubMed] [Google Scholar]
- 26.Casella JF, Maack DJ, Lin S. J Biol Chem. 1986;261:10915–10921. [PubMed] [Google Scholar]
- 27.Maruyama K, Kurokawa H, Oosawa M, Shimaoka S, Yamamoto H, Ito M. J Biol Chem. 1990;265:8712–8715. [PubMed] [Google Scholar]
- 28.Barkalow K, Witke W, Kwiatkowski DJ, Hartwig JH. J Cell Biol. 1996;134:389–399. doi: 10.1083/jcb.134.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.DiNubile MJ, Huang S. Biochim Biophys Acta. 1997;1358:261–278. doi: 10.1016/s0167-4889(97)00076-1. [DOI] [PubMed] [Google Scholar]
- 30.Haus U, Hartmann H, Trommler P, Noegel AA, Schleicher M. Biochem Biophys Res Commun. 1991;181:833–839. doi: 10.1016/0006-291x(91)91265-e. [DOI] [PubMed] [Google Scholar]
- 31.Heiss SG, Cooper JA. Biochemistry. 1991;30:8753–8758. doi: 10.1021/bi00100a006. [DOI] [PubMed] [Google Scholar]
- 32.Pollard TD, Borisy GG. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 33.Hug C, Jay PY, Reddy I, McNally JG, Bridgman PC, Elson EL, Cooper JA. Cell. 1995;81:591–600. doi: 10.1016/0092-8674(95)90080-2. [DOI] [PubMed] [Google Scholar]
- 34.Loisel TP, Boujemaa R, Pantaloni D, Carlier MF. Nature. 1999;401:613–616. doi: 10.1038/44183. [DOI] [PubMed] [Google Scholar]
- 35.Taoka M, Ichimura T, Wakamiya-Tsuruta A, Kubota Y, Araki T, Obinata T, Isobe T. J Biol Chem. 2003;278:5864–5870. doi: 10.1074/jbc.M211509200. [DOI] [PubMed] [Google Scholar]
- 36.Kitazawa M, Yamakuni T, Song SY, Kato C, Tsuchiya R, Ishida M, Suzuki N, Adachi E, Iwashita S, Ueno S, Yanagihara N, Taoka M, Isobe T, Ohizumi Y. Biochem Biophys Res Commun. 2005;331:181–186. doi: 10.1016/j.bbrc.2005.03.127. [DOI] [PubMed] [Google Scholar]
- 37.Jung G, Remmert K, Wu X, Volosky JM, Hammer JA., 3rd J Cell Biol. 2001;153:1479–1497. doi: 10.1083/jcb.153.7.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remmert K, Olszewski TE, Bowers MB, Dimitrova M, Ginsburg A, Hammer JA., 3rd J Biol Chem. 2004;279:3068–3077. doi: 10.1074/jbc.M308829200. [DOI] [PubMed] [Google Scholar]
- 39.Falck S, Paavilainen VO, Wear MA, Grossmann JG, Cooper JA, Lappalainen P. EMBO J. 2004;23:3010–3019. doi: 10.1038/sj.emboj.7600310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hutchings NJ, Clarkson N, Chalkley R, Barclay AN, Brown MH. J Biol Chem. 2003;278:22396–22403. doi: 10.1074/jbc.M302540200. [DOI] [PubMed] [Google Scholar]
- 41.Eyers CE, McNeill H, Knebel A, Morrice N, Arthur SJ, Cuenda A, Cohen P. Biochem J. 2005;389:127–135. doi: 10.1042/BJ20050387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim K, Yamashita A, Wear MA, Maeda Y, Cooper JA. J Cell Biol. 2004;164:567–580. doi: 10.1083/jcb.200308061. [DOI] [PMC free article] [PubMed] [Google Scholar]