

1. INTRODUCTION
To date twelve loci have been associated with the autosomal dominant form of Retinitis Pigmentosa (adRP), The genes at eleven of these have now been identified (http://www.sph.uth.tmc.edu/Retnet/). One such locus (RP10) is on chromosome 7q and was original identified as a result of a large linkage study undertaken at this laboratory (Jordan et al., 1993). Affected individuals of this family show the classic clinical symptoms of RP, including bone spicule pattern pigmentary deposits, optic disc pallor and retinal vascular attenuation (for a detailed clinical description of this family see Jordan et al., 1993). An unrelated American adRP family, UTAD045, exhibiting a later onset and a slower progression of symptoms, was also subsequently revealed to have a disease gene on the same region of 7q (McGuire et al., 1995).
Recently, we have shown that mutations within a gene of the de novo guanine nucleotide biosynthesis pathway, inosine monophosphate dehydrogenase type 1 (IMPDH1), cause the RP10 form of adRP (Kennan et al., 2002). IMPDH1 is the first protein of a nucleotide biosynthesis pathway to be associated with retinal disease and it shows a widespread pattern of expression. The enzyme catalyses the NAD-dependant conversion of inosine monophosphate (IMP) to xanthosine monophosphate (XMP), which is subsequently converted to GMP. GMP gives rise to one of the building blocks of DNA (dGTP) and guanine nucleotides also play an essential role in intracellular signalling pathways.
It is intriguing that a mutation in a gene with such an apparently critical function and widespread pattern of expression causes a disease with a clinical manifestation only in the retina. Detailed analysis of the expression of IMPDH1 in the retina has not previously been undertaken. In attempting to understand the tissue specific nature of the disease it is also important to consider that, in mammals, there are two alternative routes that may be used in the production of guanine. The first of these involves the isoenzyme of IMPDH1, IMPDH2. IMPDH2 is 84% homologous to the type 1 form of the gene and the two proteins are indistinguishable in terms of their catalytic activity and substrate affinities (Carr et al., 1993). Most tissues are also capable of synthesizing GMP from the degradation products of nucleic acids and catabolism by means of a salvage pathway. This salvage pathway is less energetically expensive than the de novo pathway and in fact exerts feedback control on the de novo pathway. The key enzyme of the purine salvage pathway is hypoxanthine phosphoribosyl transferase (HPRT) which phosphoribosylates the purine bases hypoxanthine and guanine for reutilisation by the ceil. The enzymes involved in purine metabolism and the control of these enzymes is not the same for all tissues and can in fact be cell type specific (Stone and Simmonds, 1991). The relative contributions of the de novo and salvage pathways to the synthesis of guanine nucleotides in the retina have not been previously determined. Analysis of the expression of IMPDH1, IMPDH2 and HPRT in the mouse retina, using in situ hybridisation and relative quantitative RT-PCR may shed some light on the pathology of RP10 and the results of such analysis are presented here.
2. MATERIALS AND METHODS
2.1. In situ Hybridisations
The in situ hybridisation technique employed in this study involved the use of digoxygenin (DIG)-labelled riboprobes on frozen cryosections. Sense and antisense DIG-labelled probes were generated from PCR templates which had incorporated T7 and T3 promoters. Sections were fixed in para-formaldehyde post cutting, treated with active DEPC and hybridisation with both sense and antisense probes allowed to proceed overnight at 58°C. Following stringent washes with SSC, incubation with an AP coupled, anti-DIG antibody was carried out. Binding of the probes was detected using NBT/BCIP solution. Sections were mounted and analysed using a Zeiss Axioplan 2 microscope.
2.2. Quantitative RT-PCR
Relative quantitative RT-PCR was used to quantify the levels of transcript in the retina for each of the three genes analysed in this study. First strand cDNA synthesis was carried out using 2μg of total RNA from the pooled wildtype and rho-/- RNA samples with 4 units Omniscript RT (Qiagen) and 1μM of random hexamers. Quantitative PCR was subsequently performed on a Roche LightCycler using the Quantitech PCR kit (Qiagen). Serial dilutions of cDNA were used to generate standard curves of crossing cycle number versus the logarithmus of concentration for each gene of interest. A linear regression line calculated from the standard curves allowed relative transcript levels in both RNA samples to be determined. Values were normalised to the relative amounts of GAPDH present in the same cDNA preparations.
3. RESULTS
RP-causing mutations were identified in the IMPDH1 gene following a microarray study which was carried out to compare levels of transcript for over 6,000 genes and ESTs in wild type and rhodopsin knockout (rho-/-) mouse retinas. When compared to wild type the IMPDH1 gene showed a down-regulation of 5.8-fold in 4 month rho-/- mouse samples, which have essentially no photoreceptors remaining (Kennan et al., 2002). Relative quantitative PCR. carried out to compare levels of IMPDH1 transcript in wild type and rho-/- retinas, also at 4 months of age, confirmed this result. Such a decrease in transcript levels of a gene is an indication that it may be highly expressed in photoreceptors and the pattern observed with IMPDH1 is similar to that of other genes known to be specifically expressed in photoreceptors, within the retina. In situ hybridisation carried out using IMPDH1 riboprobes on mouse retinal cryosections revealed a high level of expression of IMPDH1 in the photoreceptors, with only very low levels, if any, in other layers of the retina (Fig. 1A).
Figure 1.
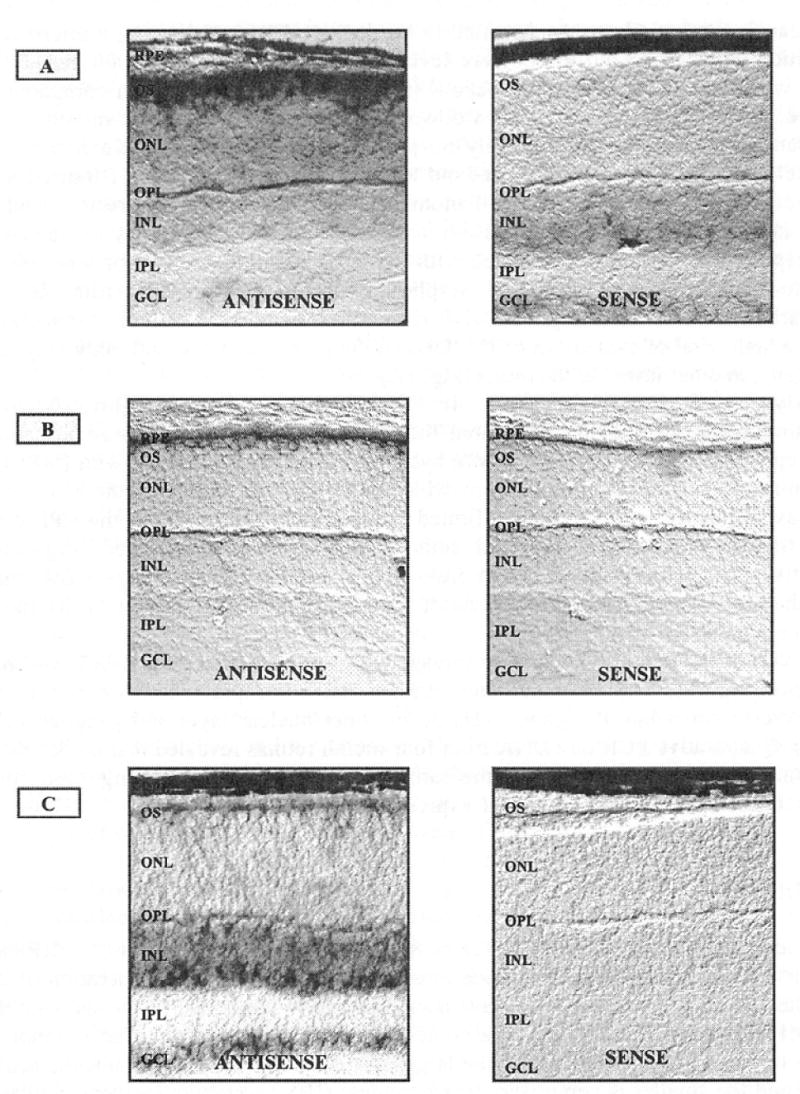
In situ hybridisations on mouse retinal cryosections using DIG-labelled riboprobes. Results for antisetise and sense probes for (A) IMPDH1, (B) IMPDH2 and (C) HPRT are shown. The IMPDH2 in situs are performed on CD1 mouse retinas which lack pigment in the RPE while the IMPDH1 and HPRT in situs are performed on wild type pigmented retinas. (RPE) retinal pigment epithelium, (OS) outer segments, (ONL) outer nuclear layer, (OPL) outer plexiform layer, (INL) inner nuclear layer, (IPL) inner plexiform layer, (GCL) ganglion cell layer. 200X Magnification.
Analysis of IMPDH2 ESTs in the UniGene database (http://www4.ncbi.nlm.nih.gov/UniGene) has indicated that this isoenzyme is expressed in retinal pigment epithelium (RPE) libraries. In situ hybridisations were carried out with IMPDH2 probes on retinal sections from CDl mice, which do not posess pigment in the RPE, so as not to mask a positive result. This confirmed that there are transcripts in the RPE cells and no transcription in the neuronal cells of the retina was detected (Fig. 1B), Quantitative PCR carried out on cDNA from retinas at four months of age shows that, despite the lack of photoreceptors, levels of transcript remained constant in the rho-/- retina in comparison to wild type.
Analysis of the guanine nucleotide salvage pathway gene encoding HPRT was also performed using in situ hybridisation and revealed that it is expressed only at low levels in the photoreceptors but at higher levels in the inner nuclear layer and ganglion cells (Fig. 1C). Quantitative PCR on cDNA from four month retinas revealed that the levels of HPRT transcript in rho-/- mice are the same as in wild type, providing supporting evidence for the lack of high level HPRT expression in photoreceptors.
4. DISCUSSION
The identification of IMPDH1 as the causitive gene in the RP10 form of adRP has, for the first time, implicated a nucleotide biosynthesis pathway in a degeneration of the retina. The IMPDH1 gene encodes a protein subunit of 514 amino acid residues, with the active IMPDH1 enzyme consisting of a homotetramer of these subunits. Each monomer of the protein possesses two domains; the larger forms a barrel which contains the active site loop and the smaller is comprised of two tandem CBS (cystathionine-beta-synthase) dimer domains (Nimmesgern et al., 1999). The established disease causing mutations identified to date lie in the CBS 2 domain of the protein (Kennan et al., 2002 and Bowne et al., 2002). CBS domains are motifs found in a number of different proteins which code for modules of unknown function. It has been suggested that these domains may attach to a wide range of other protein domains and in this way play a regulatory role (Sintchak et al., 2000). It is not clear at this stage how mutations in IMPDH1 bring about the degeneration of retinal photoreceptors. In addressing this however, there are two related questions that must be answered. Firstly, what effect are the mutations having on the IMPDH1 protein and secondly, why does this effect only reveal itself in the form of retinal disease? It is the latter question which we have attempted to address here.
The two IMPDH isoforms have previously been shown to exhibit considerable differences in their regulation and expression. The IMPDH2 gene exhibits inducible expression and is highly up-regulated in proliferating cells, whereas the IMPDH1 gene is more widely expressed but shows variability of expression in different tissues (Senda et al., 1994). Three different promoters have been identified in the human type 1 form of the protein which give rise to three distinct transcripts. This may be an indication that IMPDH1 expression is regulated in a complex cell type-specific manner (Gu et al., 1997). Investigations into the expression patterns of IMPDH1, IMPDH2 and HPRT within the retina have not been previously undertaken. The results from this study suggest that IMPDH1 is preferentially expressed in photoreceptor cells within the retina, as opposed to secondary retinal neurons. Conversely, no IMPDH2 expression was detected in photoreceptors and only low levels of HPRT were observed. The results therefore suggest that IMPDH1 is the enzyme primarily responsible for the production of guanine nucleotides within the photoreceptors. A previous study has shown that in mice lacking the IMPDH2 gene there is no observed compensatory mechanism for regulation of expression of IMPDH1 and HPRT (Gu et al., 2000). It is possible therefore, that the other guanine biosynthesis enzymes may not be able to substitute for a loss of IMPDH1 activity inRPl0.
As IMPDH1 is the first gene of its kind to be linked to a retinal degeneration it is difficult to speculate how precisely mutations within the gene may bring about the pathology. One possibility is that, while not in the active site of the enzyme, the mutation may cause a reduction in the levels of guanine nucleotides available to photoreceptors which it appears, may be relying almost solely on IMPDH1 for maintaining their guanine nucleotide reserves. Photoreceptors are among the most physiologically active cells of the body and require high levels of guanine. GDP is required for the activation of transducin during visual transduction and the circadian shedding of outer segment disc membranes requires a very high level of transcription. As the in situ hybridisations shown here have been performed on cyrosections it is difficult to say definitively which region of the photoreceptor cells are showing the higest levels of IMPDH1 transcription. It does appear however that the outer segments of the photoreceptors, where visual transduction takes place, are showing very high levels of staining. Large quantities of IMPDH1 transcript may be translocated to this region of the photoreceptors in order to satisfy the demand for guanine nucleotides during phototransduction.
A reduction in levels of guanine as a result of IMPDH1 haploinsufficiency in patients may, in theory, affect the viability of photoreceptors and result in apoptosis of these cells. Interestingly, perturbations in intracellular pools of dNTPs have been shown to result in DNA fragmentation and apoptosis in various systems (Oliver et al., 1998). Other tissues of the body may not be similarly affected due to a lower requirement for guanine nucleotides and/or a supply of guanine from IMPDH2 activity or the salvage pathway. Alternatively, the mutations may have a dominant negative effect on the IMPDH1 enzyme which is more detrimental to photoreceptors than other cells types due to the high level of IMPDH expression from the type 1 form of the gene. We are currently investigating the effect of the RP mutations on the activity and properties of the IMPDH1 enzyme in an attempt to futher expand our knowledge of the role of IMPDH1 in both normal and diseased photoreceptors.
Acknowledgments
The Ocular Genetics Unit of TCD is supported by grants from the Wellcome Trust, the Foundation Fighting Blindness (USA), the British RP Society, Fighting Blindness Ireland, the Health Research Board of Ireland and the European Union 5th Framework Programme. The Unit is a member of the HEA-Ireland-sponsored Biopharmaceutical Science Network. We thank the National Eye Institute (USA) and the Foundation Fighting Blindness (USA) for the travel award provided to AK to attend this meeting.
References
- Bowne SJ, Sullivan LS, Blanton SH, Cepko CL, Blackshaw S, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:559. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr SF, Papp E, Wu JC, Natsumeda Y. Characterization of human type I and type II IMP dehydrogenases. J Biol Chem. 1993;268:27286. [PubMed] [Google Scholar]
- Gu JJ, Spychala J, Mitchell BS. Regulation of the human inosine monophosphate dehydrogenase type I gene. Utilization of alternative promoters. J Biol Chem. 1997;272:4458. doi: 10.1074/jbc.272.7.4458. [DOI] [PubMed] [Google Scholar]
- Gu JJ, Stegmann S, Gathy K, Murray R, Laliberte J, Ayscue L, Mitchell BS. Inhibition of T lymphocyte activation in mice heterozygous for loss of the IMPDH2 gene. J Clin Invest. 2000;106:599. doi: 10.1172/JCI8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennan A, Aherne A, Palfi A, Humphries M, McKee A, Stitt A, Simpson DA, Demtroder K, Orntoft T, Ayuso C, Kenna PF, Farm GJ, Humphries P. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho(-/-) mice. Hum Mol Genet. 2002;11:547. doi: 10.1093/hmg/11.5.547. [DOI] [PubMed] [Google Scholar]
- Jordan SA, Farrar GJ, Kenna P, Humphries MM, Sheils DM, Kumar-Singh R, Sharp EM, Ayuso C, Benitez J, Humphries P. Localization of an autosomal dominant retinitis pigmentosa gene to chromosome 7q. Nat Genet. 1993;4:54. doi: 10.1038/ng0593-54. [DOI] [PubMed] [Google Scholar]
- McGuire RE, Gannon AM, Sullivan LS, Rodriguez JA, Daiger SP. Evidence for a major gene (RP10) for autosomal dominant retinitis pigmentosa on chromosome 7q: linkage mapping in a second, unrelated family. Hum Genet. 1995;95:71. doi: 10.1007/BF00225078. [DOI] [PubMed] [Google Scholar]
- Oliver FJ, Muñoz-Pinedo C, Collins MKL, López Rivas A. Regulation of apoptosis by survival factors: the role of metabolism. IV Metabolismo de la Metionina. Mecanismos moleculares e implicaciones clinicas. Boehringer Ingelheim. 1998 http:www.boehringer-ingelheim.es/workshop-methionina/anglesa/cap11.
- Nimmesgern E, Black J, Futer O, Fulghum JR, Chambers SP, Brummel CL, Raybuck SA, Sintchak MD. Biochemical analysis of the modular enzyme inosine 5’-monophosphate dehydrogenase. Protein Expr Purif. 1999;17:282. doi: 10.1006/prep.1999.1136. [DOI] [PubMed] [Google Scholar]
- Senda M, Natsumeda Y. Tissue-differential expression of two distinct genes for human IMP dehydrogenase (E.C.1.1.1.205) Life Sci. 1994;54:1917. doi: 10.1016/0024-3205(94)90150-3. [DOI] [PubMed] [Google Scholar]
- Stone TW, Simmonds HA. Purines: Basic and Clinical Aspects. Kluwer; London: 1991. [Google Scholar]