

1. INTRODUCTION
In a project begun more than 15 years ago we used linkage mapping and positional candidate gene cloning to identify two genes causing autosomal dominant retinitis pigmentosa (adRP). The genes are RP1, which maps to chromosome 8q12.1, and RP10 (IMPDH1), which maps to 7q32.1. Although different families and different regions of the genome are involved, the studies were done in parallel because the methods used, and the underlying genetic concepts, are identical. Now that the genes have been identified, though, the functional properties of the two proteins are strikingly different, so divergent approaches are required to better understand the pathophysiology of mutations in each gene. This report is a summary of our current understanding of these two genes, contrasting the similar approaches to identifying the genes and mutations with the dissimilar strategies for functional analysis. In microcosm, this serves as a reminder that retinal disease genes associated with similar clinical phenotypes may have very different biological roles.
Research to identify and characterize disease-causing genes now follows a traditional methodology which has developed over the past 15 years. Although there are many nuances, the standard steps are …
ascertainment and characterization of families,
linkage mapping,
positional candidate gene cloning,
mutation screening,
gene sequence analysis, and
functional analysis.
The first steps are part of the process originally called “reverse genetics”, with emphasis on identification of the disease locus independent of the biological or clinical details of the disease. Subsequent steps are focused on a biological understanding of the gene and gene product. This summary describes the current status of these steps in understanding the RP1 and RP10 loci.
2. ASCERTAINMENT OF FAMILIES AND LINKAGE MAPPING
2.1 The RP1 locus
In 1982 Field et al. described a large, six-generation adRP family located predominantly in the Kentucky - West Virginia area (Figure 1). This was named the “RP1” locus based on preliminary linkage mapping to chromosome 1 (later shown to be incorrect). Subsequent linkage testing assigned the disease locus in this family to human chromosome 8q11-q12 (Blanton et al., 1991). Linkage testing in an Australian adRP family led to identification of a second RP1 family. Combining data from the two families reduced the maximum non-recombinant region to 4 cM, or roughly 4 Mb (Xu et al., 1996). An additional British RP1 family was also identified by linkage mapping (Inglehearn et al., 1999). Data from this family did not help refine the critical region for the RP1 locus.
Figure 1.

RP01 family with the RP1 form of autosomal dominant retinitis pigmentosa.
All of the RP1 families so far described have relatively late onset retinitis pigmentosa, usually described as type 2 or “R” type with regional loss of both rod and cone photoreceptor activities (Field et al., 1982; Inglehearn et al., 1999; Xu et al., 1996). There is wide variation in severity and age of onset among affected members of large RP1 families, with a few reported instances of unaffected “carriers” over age 50. One notable clinical feature of the Kentucky family (RP01) is the presence of two affected individuals who are homozygous for an RP1 mutation as indicated by haplotype analysis and, more recently, by mutation testing (Sullivan et al., 1999). These individuals have severe, congenital or early-onset retinal degeneration, but are otherwise apparently unaffected by these mutations.
2.2 The RP10 locus
The RP10 locus was first assigned to chromosome 7q in 1993 by Jordan et al. based on linkage mapping in a Spanish adRP family. Identification of a second RP10 family by linkage mapping localized the RP10 gene to 7q31-q35 (McGuire et al., 1995), later refined to a 5 cM (4.9 Mb) region at 7q32.1 (McGuire et al., 1996). The second RP10 family is of American origin, with at least 6 known, affected generations (Figure 2). Two additional RP10 families were identified by linkage mapping, one of Spanish origin (Millán et al., 1995) and the second of Scottish origin (Mohamed et al., 1996).
Figure 2.
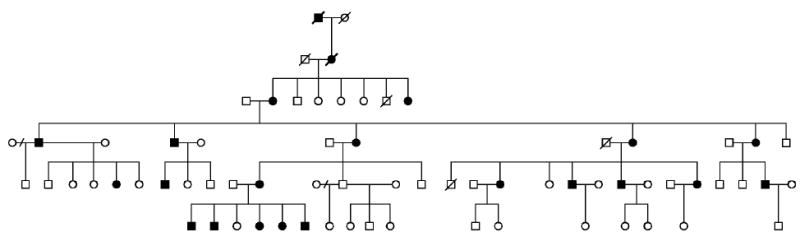
UTAD045 family with the RP10 form of autosomal dominant retinitis pigmentosa.
The clinical findings in RP10 families are similar to RP1 families, except that RP10 has earlier onset and is more consistent with type 1or “diffuse” retinitis pigmentosa (Bowne et al., 2002). Like RP1, patients with the RP10 form of adRP have equal reduction in both rod and cone ERGs, though with earlier onset. There is also considerable variation in clinical findings among affected members of families with the RP10 form of adRP but “skipped generations” (unaffected carriers) have not been reported.
3. POSITIONAL CANDIDATE CLONING
3.1 The RP1 gene
Fine structure linkage mapping and physical mapping of flanking markers reduced the RP1 genetic region to 4.0 Mb (Sullivan et al., 1999). Candidate genes were screened by sequencing in heterozygous and homozygous members of the RP01 family, with emphasis on genes preferentially expressed in photoreceptors. Among these, a large gene mapping to 8q12.1 was found to contain a nonsense substitution in codon 677 producing an Arg677ter mutation tracking in phase with the disease haplotype in all affected family members (Sullivan et al., 1999). The transcribed gene produces a 6,471 bp mRNA composed of 4 exons, 3 of which code for a novel protein 2,156 amino acids in length (Figure 3). Published ESTs of the RP1 gene are exclusively from retinal cDNA libraries suggesting expression is limited to the retina. Because sequence analysis did not immediately suggest a function for the RP1 gene product, the gene symbol and protein name have remained “RP1”.
Figure 3.

Structure of the RP1 gene.
Two other research groups independently identified the RP1 gene at the same time as Sullivan et al. Pierce et al. (1999) used differential display to identify mouse retinal genes that show significant changes in expression following retinal hypoxia. The human homolog of one of these, oxygen response protein 1 (ORP1), was subsequently shown to map to the RP1 critical region on chromosome 8. Sequencing of members of the RP01 family showed this to be the RP1 gene. Additionally, Guillonneau et al. (1999) identified a retinal-expressed gene in the mouse chromosomal region (proximal 1) syntenic to human 8q. A disease-causing mutation was identified in the human homolog in an adRP family previously linked to 8q and, subsequently, the mouse gene proved to be identical to ORP1.
Since these initial reports, the research groups who independently identified the RP1 gene have joined to form the RP1 Consortium (see footnote 4, page 1). The Consortium members meet regularly to share research results and plan future RP1-related projects.
3.2 The RP10 gene
Two laboratories also reported independent identification of the RP10 gene. Bowne et al. (2002) identified additional RP10 families by linkage mapping and used this information to reduce the physical region containing the gene to three non-contiguous regions totaling 3.45. Dr. C. L. Cepko provided information on retinal genes whose expression is reduced in crx-/crx- knockout mice in comparison to normal mice, that is, genes whose retinal expression might be limited to photoreceptors (Blackshaw et al., 2001). The human homologs of three of these genes map within the RP10 physical region. Sequencing of one of these genes, IMPDH1, identified a missense mutation, Asp226Asn, segregating with disease in the UTAD045 family (Bowne et al., 2002). Missense mutations were also found in two other RP10 families identified earlier by linkage mapping and in 3 of 60 other, unrelated adRP families. The RP10 gene thus identified codes for the enzyme inosine monophosphate dehydrogenase 1, which catalyzes the rate limiting step in guanine biosynthesis. Because the RP10 gene has been known by the symbol “IMPDH1” for many years, this has become the new symbol for the adRP locus on 7q32.1.
Kennan et al. (2002) also identified IMPDH1 as the RP10 gene using a similar approach. Retinal cDNAs from rho-/rho- knockout mice were compared with retinal cDNAs from wild-type retinas using microarray analysis. As was true for the crx-/crx- mice, the IMPDH1 transcript was significantly reduced in the rho-/rho- knockout retinas. Sequencing in the original, Spanish RP10 family (Jordan et al., 1993) revealed a missense mutation, Arg224Pro, segregating with disease. Thus two different approaches to identifying photoreceptor transcripts led to identification of the same RP10 gene, confirming the power of expression analysis to prioritize potential candidate genes.
The IMPDH1 gene spans 18 kb and is comprised of 17 exons, of which 14 are coding exons (Figure 4) (Gu et al., 1997). The gene uses alternate 5’ sites for initiation of transcription but the variant mRNAs produce identical proteins.
Figure 4.
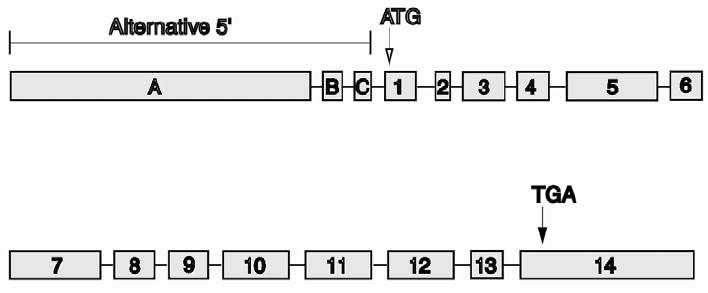
Structure of the IMPDH1 gene.
4. MUTATION SCREENING
4.1 RP1
At least 20 distinct disease-causing mutations have been identified in RP1 in families with adRP (Table 1). To date, mutations in RP1 appear to cause from 6 to 8% of adRP cases among Americans of European origin and Europeans (Berson et al., 2002; Bowne et al., 1999; Dietrich et al., 2002; Payne et al., 2000; Pierce et al., 1999; Sullivan et al., 1999). All of the mutations with convincing evidence of pathogenicity cause premature termination of translation, either by introduction of a premature stop codon or a frame shift leading to early termination. Interestingly, these mutations all fall within a relatively narrow region of exon 4, and apparently escape nonsense mediated mRNA decay because they are located within the terminal exon. Although many missense changes have been reported, both in isolation and as polymorphic variants, none have been shown to cause retinal disease, at least not in heterozygotes. Finally, mutation screening of RP1 in patients with other forms of retinopathy has failed to identify non-adRP mutations
Table 1.
Disease-causing mutations in RP1.
| Mutation | No. of families | Reference |
|---|---|---|
| Met500 ins 2 | 1 | Payne et al., 2000 |
| Pro658 ins 1 | 1 | Berson et al., 2002 |
| Arg677ter | 24 | Berson et al., 2002; Bowne et al., 1999; Payne et al., 2000; Sullivan et al., 1999 |
| Arg677 del 1 | 1 | Bowne et al., 1999 |
| Gln679ter | 1 | Sullivan et al., 1999 |
| Glu700ter | 2 | Bowne et al., 1999; Payne et al., 2000 |
| Gly723ter | 1 | unpublished |
| Gly724 del 14 | 3 | Bowne et al., 1999; Payne et al., 2000 |
| Ile725 ins 1 | 1 | Bowne et al., 1999 |
| Glu729 del 1 | 1 | unpublished |
| Thr736 ins 1 | 1 | Bowne et al., 1999 |
| Cys744ter | 2 | Bowne et al., 1999; Payne et al., 2000 |
| Ser747 del 1 | 1 | Berson et al., 2002 |
| Leu762 del 5 | 9 | Berson et al., 2002; Bowne et al., 1999; Payne et al., 2000; Pierce et al., 1999 |
| Asn763 del 4 | 3 | Payne et al., 2000; Pierce et al., 1999 |
| Ser768 del 1 | 1 | Bowne et al., 1999 |
| Lys778ter | 1 | Dietrich et al., 2002 |
| Thr865 del 2 | 1 | Payne et al., 2000 |
| Arg872 ins 1 | 1 | Payne et al., 2000 |
| Tyr1053 del 1 | 1 | Berson et al., 2002 |
4.2 RP10
Mutation screening of IMPDH1 in adRP families has been less extensive than RP1 screening but, nonetheless, at least 6 distinct pathogenic mutations have been reported, including unpublished data from S.J.B. (Table 2) (Bowne et al., 2002; Kennan et al., 2002). Mutations in IMPDH1 account for from 3 to 5% of adRP cases among Americans of European origin. In contrast to RP1, all disease-causing variants of IMPDH1 are missense mutations, and IMPDH1 displays no polymorphic amino acid variation. Pathogenic mutations in IMPDH1have not been reported in other forms of retinopathy.
Table 2.
Disease-causing mutations in IMPDH1.
| Mutation | No. of families | Protein domain | Reference |
|---|---|---|---|
| Thr116Met | 1 | CBS | Bowne et al., 2002 |
| Arg224Pro | 1 | CBS | Kennan et al., 2002 |
| Arg226Asn | 5 | CBS | Bowne et al., 2002 |
| Val268Ile | 1 | unknown | unpublished |
| Gly324Asp | 2 | active site | unpublished |
| His372Pro | 1 | active site | unpublished |
5. GENE SEQUENCE AND FUNCTIONAL ANALYSIS
5.1 RP1
Analysis of the human RP1 coding sequence demonstrates very little sequence similarity to other human and non-human genes (Sullivan et al., 1999). The first 10% of the amino-terminus of the protein, residues 1 through 228, shares 25% identity (39% similarity) to the human doublecortin gene (DCX) and 15 to 40% identity to other members of the doublecortin family. Mutations in DCX, an X-linked gene in humans, cause lissencephaly, a congenital malformation of the brain resulting from impaired neuronal migration. The DCX protein interacts with microtubules so the cognate region of the RP1 gene may do likewise. Unfortunately, this sheds little light on the functional properties of the RP1protein because the remaining 90% contains no extended protein motifs, at least none conserved across mammalian species.
Northern analysis and in situ hybridization suggest that RP1 is expressed in the retina only and, specifically, in photoreceptors (Pierce et al., 1999; Sullivan et al., 1999). Immunolocalization indicates that the RP1 protein, 240 kDa in size, accumulates within the connecting cilium of rods and cones (Pierce et al., 1999). Thus the doublecortin-like domain of RP1 may interact directly with the cilium. Finally, two different lines of RP1 knockout mice have been developed (Gao et al., 2002; Liu et al., 2002). The heterozygous knockouts show subtle retinal abnormalities but homozygous animals have overt retinal degeneration at a young age, consistent with the human phenotypes.
There is one exception to the absence of genes similar to RP1 in the human genome. A second gene, with the symbol RP1L1 for “RP1-like protein 1”, has the same gene structure and overall length of RP1 and is 31% identical (52% similar) over the first 15%, that is, residues 28 through 355 (Figure 5) (unpublished). The remaining 85% of the two proteins are not statistically similar. Preliminary evidence suggests that RP1L1 is expressed exclusively in photoreceptors. Thus RP1 and RP1L1 appear to be true paralogs. It remains to be determined what roles the two proteins play in photoreceptors and how closely their biological functions are interconnected.
Figure 5.

Human RP1 vs. RP1LI sequence comparison (first 15%).
5.2 RP10 (IMPDH1)
In sharp contrast to the RP1 gene and protein, the IMPDH1 gene is expressed in all tissues, is highly conserved in all living organisms, and has functional properties that are well-understood (Bowne et al., 2002; Gu et al., 1997; Kerr and Hedstrom, 1997). Inosine monophosphate dehydrogenase (EC 1.1.1.205) is an essential enzyme, required for de novo synthesis of guanine nucleotides, found in all species, including bacteria. In humans there are two paralogs, IMPDH1 and IMPDH2, which are 84% identical at an amino acid level and share identical kinetic properties (Figure 6). Across species, IMPDH genes are 30 to 85% identical. Thus IMPDH1 is a member of a family of highly-conserved, widely-expressed enzymes which, taken together, are essential for life. The mystery, of course, is why dominant-acting missense mutations in IMPDH1 cause retinal degeneration, and retinal degeneration only (as far as is known). One possible explanation is suggested by the crx-/crx- and rho-/rho- knockout retinas: IMPDH1 is significantly downregulated but not IMPDH2, suggesting that IMPDH1is expressed in photoreceptors, but not IMPDH2.
Figure 6.
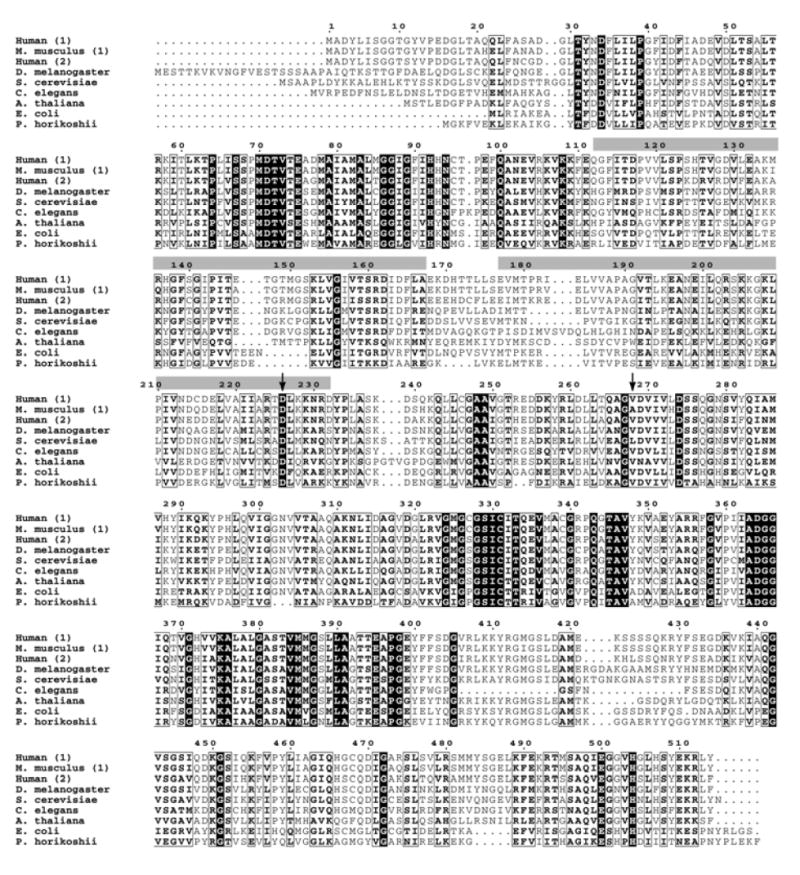
IMPDH sequence comparisons across species.
6. CONCLUSIONS
Thus similar approaches were used to identify the RP1 and RP10 (IMPDH1) genes; mutations in both of these genes cause a substantial fraction of cases of autosomal dominant retinitis pigmentosa; and the clinical phenotypes are similar. At a functional level, though, the two gene products are strikingly different. The RP1 protein has very little sequence similarity to other genes, is limited to rod and cone photoreceptors, and plays an unknown role in the biology of the interconnecting cilium. In contrast, IMPDH1 is ubiquitously expressed, is a member of a highly-conserved family of enzymes, and has well-characterized biochemical properties. Efforts to identify retinal disease genes are undertaken to help patients affected with these disorders, but characterization of these genes often reveals new biological processes.
Acknowledgments
Supported by grants from the Foundation Fighting Blindness and the George Gund Foundation, the William Stamps Farish Fund, the M.D. Anderson Foundation, the John S. Dunn Foundation, and Alfred W. Lasher III; and by grants EY05235 (D.G.B), EY07142 (S.P.D.), and EY14170 (S.P.D.) from the National Eye Institute-National Institutes of Health.
References
- Berson EL, Grimsby JL, Adams SM, McGee TL, Sweklo E, Pierce EA, Sandberg MA, Dryja TP. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1) Invest Ophthalmol Vis Sci. 2002;42:2217–2224. [PubMed] [Google Scholar]
- Blackshaw S, Fraioli RE, Furukawa T, Cepko CL. Comprehensive analysis of photoreceptor gene expression and the identification of candidate retinal disease genes. Cell. 2001;107:579–589. doi: 10.1016/s0092-8674(01)00574-8. [DOI] [PubMed] [Google Scholar]
- Blanton SH, Heckenlively JR, Cottingham AW, Friedman J, Sadler LA, Wagner W, Friedman LH, Daiger SD. Linkage mapping of autosomal dominant retinitis pigmentosa (RP1) to the pericentric region of human chromosome 8. Genomics. 1991;11:857–869. doi: 10.1016/0888-7543(91)90008-3. [DOI] [PubMed] [Google Scholar]
- Bowne SJ, Daiger SP, Hims MM, Sohocki MM, Malone KA, McKie AB, Heckenlively JR, Birch DG, Inglehearn CF, Bhattacharya SS, Bird A, Sullivan LS. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1999;11:2121–2128. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowne SJ, Sullivan LS, Blanton SH, Cepko CL, Blackshaw S, Birch DG, Hughbanks-Wheaton D, Heckenlively JR, Daiger SP. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet. 2002;11:559–568. doi: 10.1093/hmg/11.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich K, Jacobi FK, Tippmann S, Schmid R, Zrenner E, Wissinger B, Apfelstedt-Sylla E. A novel mutation of the RP1 gene (Lys778ter) associated with autosomal dominant retinitis pigmentosa. Brit J Ophthalmol. 2002;86:328–332. doi: 10.1136/bjo.86.3.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field LL, Heckenlively JR, Sparks RS, Garcia CA, Farson C, Zedalis D, Sparkes MC, Crist M, Tideman S, Spence MA. Linkage analysis of five pedigrees affected with typical autosomal dominant retinitis pigmentosa. J Med Genet. 1982;19:266–270. doi: 10.1136/jmg.19.4.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Cheon K, Nusinowitz S, Liu Q, Bei D, Atkins K, Azimi A, Daiger SP, Farber DB, Heckenlively JR, Pierce EA, Sullivan LS, Zuo J. Progressive photoreceptor degeneration, outer segment dysplasia and rhodopsin mis-localization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc Nat Acad Sci USA. 2002;99:5698–5703. doi: 10.1073/pnas.042122399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu JJ, Spychala J, Mitchell BS. Regulation of the human inosine monophosphate dehydrogenase type I gene. Utilization of alternative promoters. J Biol Chem. 1997;272:4458–466. doi: 10.1074/jbc.272.7.4458. [DOI] [PubMed] [Google Scholar]
- Guillonneau X, Piriev NI, Danciger M, Kozak CA, Cideciyan AV, Jacobson SG, Farber DB. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RP1 locus. Hum Mol Genet. 1999;8:1541–1546. doi: 10.1093/hmg/8.8.1541. [DOI] [PubMed] [Google Scholar]
- Inglehearn CF, McHale JC, Keen TJ, Skirton H, Lunt PW. A New family linked to the RP1 dominant retinitis pigmentosa locus on chromosome 8q. J Med Genet. 1999;36:646–648. [PMC free article] [PubMed] [Google Scholar]
- Jordan SA, Farrar GJ, Kenna P, Humphries MM, Sheils DM, Kumar-Singh R, Sharp EM, Ayuso C, Benitez J, Humphries P. Localization of an autosomal dominant retinitis pigmentosa gene to chromosome 7q. Nat Genet. 1993;4:54–58. doi: 10.1038/ng0593-54. [DOI] [PubMed] [Google Scholar]
- Kennan A, Aherne A, Palfi A, Humphries M, McKee A, Stitt A, Simpson DAC, Demtroder K, Orntoft T, Ayuso C, Kenna PF, Farrar GJ, Humphries P. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho-/- mice. Hum Mol Genet. 2002;11:547–557. doi: 10.1093/hmg/11.5.547. [DOI] [PubMed] [Google Scholar]
- Kerr KM, Hedstrom L. The roles of conserved carboxylate residues in IMP dehydrogenase and identification of a transition state analog. Biochem. 1997;36:13365–13373. doi: 10.1021/bi9714161. [DOI] [PubMed] [Google Scholar]
- Liu Q, Zhou J, Daiger SP, Farber DB, Heckenlively JR, Smith JE, Sullivan LS, Zuo J, Milam AH, Pierce EA. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest Ophthalmol Vis Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- McGuire RE, Gannon AM, Sadler-Sullivan LA, Rodriguez JA, Daiger SP. Evidence for a major gene (RP10) for autosomal dominant retinitis pigmentosa on chromosome 7q: linkage mapping in a second, unrelated family. Hum Genet. 1995;95:71–74. doi: 10.1007/BF00225078. [DOI] [PubMed] [Google Scholar]
- McGuire RE, Jordan SA, Braden VV, Bouffard GG, Humphries P, Green ED, Daiger SP. Mapping the RP10 locus for autosomal dominant retinitis pigmentosa on 7q: refined genetic positioning and localization within a well-defined YAC contig. Genome Res. 1996;6:255–266. doi: 10.1101/gr.6.4.255. [DOI] [PubMed] [Google Scholar]
- Millán JM, Martínez F, Vilela C, Beneyto M, Prieto F, Nájera C. An autosomal dominant retinitis pigmentosa family with close linkage to D7S480 on 7q. Hum Genet. 1995;96:216–218. doi: 10.1007/BF00207382. [DOI] [PubMed] [Google Scholar]
- Mohamed Z, Bell C, Hammer HM, Converse CA, Esakowitz L, Haites NE. Linkage of a medium sized Scottish autosomal dominant retinitis pigmentosa family to chromosome 7q. J Med Genet. 1996;33:714–715. doi: 10.1136/jmg.33.8.714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne A, Vithana E, Khaliq S, Hameed A, Deller J, Abu-Safieh L, Kermani S, Leroy BP, Mehdi SQ, Moore AT, Bird AC, Bhattacharya SS. RP1 protein truncating mutations predominate at the RP1 adRP locus. Invest Ophthalmol Vis Sci. 2000;41:4069–4073. [PubMed] [Google Scholar]
- Pierce EA, Quinn T, Meehan T, McGee TL, Berson EL, Dryja TP. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- Sullivan LS, Heckenlively JR, Bowne SJ, Zuo J, Hide WA, Gal A, Denton M, Inglehearn CF, Blanton SH, Daiger SP. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet. 1999;22:255–259. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S-Y, Denton M, Sullivan LS, Daiger SP, Gal A. Genetic mapping of RP1 on 8q11-q21 in an Australian family with autosomal dominant retinitis pigmentosa reduces the critical region to 4 cM between D8S601 and D8S285. Hum Genet. 1996;98:741–743. doi: 10.1007/s004390050296. [DOI] [PubMed] [Google Scholar]