

Abstract
Chagas disease, caused by infection with the protozoan parasite Trypanosoma cruzi, is a major public health problem in Central and South America. The pathogenesis of Chagas disease is complex and the natural course of infection is not completely understood. The recent development of bioluminescence imaging technology has facilitated studies of a number of infectious and non-infectious diseases. We developed luminescent T. cruzi to facilitate similar studies of Chagas disease pathogenesis. Luminescent T. cruzi trypomastigotes and amastigotes were imaged in infections of rat myoblast cultures, which demonstrated a clear correlation of photon emission signal strength to the number of parasites used. This was also observed in mice infected with different numbers of luminescent parasites, where a stringent correlation of photon emission to parasite number was observed early at the site of inoculation, followed by dissemination of parasites to different sites over the course of a 25 day infection. Whole animal imaging from ventral, dorsal and lateral perspectives provided clear evidence of parasite dissemination. The tissue distribution of T. cruzi was further determined by imaging heart, spleen, skeletal muscle, lungs, kidneys, liver and intestines ex vivo. These results illustrate the natural dissemination of T. cruzi during infection and unveil a new tool for studying a number of aspects of Chagas disease, including rapid in vitro screening of potential therapeutic agents, roles of parasite and host factors in the outcome of infection, and analysis of differential tissue tropism in various parasite-host strain combinations.
Keywords: Trypanosoma cruzi, Luciferase, Bioluminescent imaging, Chagas disease
1. Introduction
Trypanosoma cruzi, the causative agent of Chagas disease, is an intracellular, eukaryotic parasite of the family Trypanosomatidae. Endemic to vast regions of Central and South America, Chagas disease remains the leading form of infectious heart disease worldwide (Kirchhoff et al., 2004). While previous reports from the World Health Organization estimated that 16–18 million people are infected with Chagas disease (Moncayo, 1999, 2003), a more recent analysis indicates that this number has been reduced to nearly 8 million, due to progress in the control of vectorial transmission in Latin American countries (World Health Organization, www.who.int/mediacentre/news/releases/2007/pr36/en/index.html). However, despite the optimistic nature of this statistic, the report also states that cases are now being identified outside the typical endemic regions due to increasing incidences of blood transmission (Kirchhoff et al., 2006) and organ transplantation (Nowicki et al., 2006). Chagas disease can occur in an acute phase, typically characterized by high parasitism, fever and lymphadenopathy, but more commonly progresses to a chronic phase where cardiac alterations or gastrointestinal disorders are observed.
Although the tissue tropism can vary among parasite strains (Melo and Brener, 1978), it is generally thought that, while capable of invading virtually any cell in the body (Lenzi et al., 1996), T. cruzi preferentially targets neuronal and muscle cell-types (Lenzi et al., 1996) and its associated pathogenicities have typically been found to correspond to parasitosis of the myocardium (Ben Younes-Chennoufi et al., 1988; Jones et al., 1993; Bellotti et al., 1996; Anez et al., 1999; Zhang and Tarleton, 1999) or digestive tract (Vago et al., 1996). These organs are often the focus of Chagas disease research, since these are the well-characterized disease manifestations. While other studies have identified trypanosomes in liver, spleen and lung tissue (Melo and Brener, 1978) and, more recently, bone and cartilage (Morocoima et al., 2006), these distributions generally follow the use of immunosuppressive therapy (Calabrese et al., 1992; Calabrese, 1999; Taniwaki et al., 2005) to facilitate robust parasite proliferation and expansion. Regardless of the mode of infection or treatment regimen, the sacrifice of animals has typically been required to obtain information on dissemination of parasites and detection of parasites in specific tissues following infection. Furthermore, quantification of whole animal and organ-specific parasite burden has been both cumbersome and inconsistent, incorporating such techniques as PCR amplification or in situ hybridization of parasite-specific genes from tissue (Lane et al., 1997, 2003; Zhang and Tarleton, 1999; James et al., 2002), parasite antigen-specific immunofluoresence (Ben Younes-Chennoufi et al., 1988; Chandler and Watts, 1988; Taniwaki et al., 2007) and the counting of either nests of parasite amastigotes in tissue sections or free-swimming trypomastigotes in the blood (Nunes et al., 1990; Mortatti et al., 1992; Russo et al., 1996; Pinto et al., 1999). While these approaches have certainly been adequate for a variety of studies of experimental Chagas disease pathogenesis, they are also accompanied by significant limitations.
During the past several years, bioluminescence imaging (BLI) techniques have overcome these limitations in the analysis of many disease processes, including various models of cancer tumorigenesis (Edinger et al., 2002; Vooijs et al., 2002; Shachaf et al., 2004; Lyons et al., 2006; Wendt et al., 2007) and infections caused by bacteria, viruses, fungi and parasites, nicely summarized in a recent review (Hutchens and Luker, 2007). The incorporation of in vivo BLI has not only provided a means by which to evaluate the spatiotemporal progression of disease in real-time, but has brought about the opportunity to observe potentially biologically relevant interactions of host and pathogen, in the case of infectious disease, that may have otherwise gone unnoticed. In general, BLI detects light resulting from the reaction of luciferase enzymes with a specific substrate. This is made possible by utilizing either bacterial luciferase genes capable of encoding both enzyme and substrate that are typically transferred to other species of bacteria, or by using luciferase enzymes from higher organisms such as the firefly or sea pansy (Hutchens and Luker, 2007). While the use of bacterial genes precludes the use of exogenous substrate to initiate the luciferase enzymatic reaction, employing genes from the firefly and other such organisms requires that luciferin substrate be provided to produce detectable light.
In this study, we sought to develop bioluminescent T. cruzi for in vivo BLI analysis of infection using our well-established model of experimental Chagas disease. To do this, we engineered the Brazil strain of T. cruzi to express firefly luciferase using standard transfection methods. Following infection of animals with different numbers of luminescent T. cruzi trypomastigotes, we observed clear qualitative and quantitative differences in parasite burden up to 2 weeks p.i., after which time a similar burden was achieved and maintained throughout the remainder of the acute infection. In addition to in vivo imaging of parasite infection in A/J mice, we were able to detect luminescence in all three life cycle stages of T. cruzi by different methods. Finally, we illustrate the ability to detect luminescence in several harvested organs in a terminal, ex vivo analysis.
2. Materials and methods
2.1. Parasites
The Brazil, heart-derived strain of T. cruzi (Hyland et al., 2007) was used for the experiments described here. Epimastigotes, used for transfection and specific in vitro assays, were maintained in supplemented liver digest neutralized tryptose medium (LDNT) as described previously (Kirchhoff et al., 1984). Epimastigote transfectant cultures, consisting of differentiated metacyclic trypomastigotes, were used to infect monolayers of H9C2 rat myoblasts (American Type Culture Collection, Manassas, VA) from which trypomastigotes could be continually passaged and isolated. These trypomastigotes were used for all animal infections described.
2.2. Generation of bioluminescent Trypanosoma cruzi
For stable integration of the firefly luciferase gene into the tubulin locus, we used plasmid pBS:THT-x-T (the generous gift of Wesley Van Voorhis, University of Washington). In this pBluescript (Stratagene, La Jolla, CA)-based plasmid, the HygTK gene is flanked by β-tubulin untranslated/intergenic regions (UTR/IR) and the gene of interest is flanked by α-tubulin intergenic regions (Weston et al., 1999). The MluI site in the β-α UTR/IR was converted to a unique SalI site for plasmid linearization prior to transfection. The firefly luciferase gene was amplified by PCR from the pGL3 basic vector (Promega, Madison, WI) with forward primer 5′-GGATCCATGGAAGACGCCAAAAACATAAAG-3′ and reverse primer 5′-TCTAGATTACACGGCGATCTTTCC-3′ which included the BamHI and XbaI restriction sites (underlined), respectively. The resulting amplicon was ligated into the pCR-blunt vector (Invitrogen, Carlsbad, CA) and was selected for kanamycin resistance. The luciferase coding sequence was then liberated by digestion with BamH1 and Xba1 and the purified insert was directionally cloned into pBS:THT-x-T using these sites. The resulting plasmid, pBS:THT-Luc-T was linearized with SalI and 10 μg of DNA was transfected into T. cruzi by electroporation with a Gene Pulser (Bio-Rad, Hercules, CA) using the following conditions: 500 μF, 450 V, in a 0.2 cm cuvette. Briefly, mid-log stage (~2 × 107/ml) epimastigotes of T. cruzi Brazil heart strain (Hyland et al., 2007) were harvested by centrifugation, washed twice in PBS supplemented with 1 mg/L glucose and resuspended at a final concentration of 1 × 108 cells in 0.4 mL of electroporation buffer (PBS with 0.5 mM MgCl2 and 0.1 mM CaCl2). Following electroporation, the cells were placed on ice for 15 min and were transferred to flasks containing 5 mL of LDNT. Forty-eight hours post transfection, cells were selected for resistance to hygromycin (Boehringer–Mannheim, Mannheim, Germany) at 0.75 mg/ml. Drug-resistant parasites were analyzed for luciferase activity 6 weeks following transfection.
2.3. In vitro bioluminescent imaging
To confirm the expression of luciferase in antibiotic resistant transfectants, T. cruzi epimastigotes were serially diluted from 1 × 106 to 500 parasites in Dulbecco’s PBS (GibcoBRL, Grand Island, NY) into a black 96-well plate (Costar, Acton, MA). A 50 μL cell suspension was mixed with 50 μL of Steady Glo reagent (Promega, Madison, WI), according to the manufacturer’s instructions. After 5 min, the plate was read using a SpectraMax Gemini XS (Molecular Devices, Sunnyvale, CA) and analyzed with SoftmaxPro 4.8 software (Molecular Devices). Wild-type (untransfected) Brazil heart T. cruzi was included as a negative control. For further analysis of luminescence in trypomastigote and amastigotes life stages, active, in vitro rat myoblast infections, using a variety of parasite-to-myoblast ratios (as described in Fig. 1) were also analyzed for bioluminescence. Infections were given 5 days to become established in a clear 6-well plate (BD Biosciences, San Jose, CA), luciferin was added to 150 μg/ml, and plates were imaged using the Xenogen IVIS system (see below for a description of the IVIS imaging system and analysis software) after a 5 min incubation.
Fig. 1.

Generation and analysis of luminescent Trypanosoma cruzi. (A) Construct and cloning strategy used to generate luminescent T. cruzi. pBS:THT-x-T (obtained from Wesley Van Voorhis, University of Washington), a plasmid with a pBluescript backbone and a hygromycin-resistance gene, was modified by insertion of the firefly luciferase gene. After linearization with SalI, the plasmid was transfected into T. cruzi epimastigotes and the Hyg and Luc genes integrated into the tubulin locus by homologous recombination. (B) Serial dilutions of antibiotic-resistant, T. cruzi epimastigote transfectants and wild-type epimastigotes were examined for luciferase activity. A black, 96-well plate containing 50 μL suspensions of parasites, mixed with 50 μL of Steady Glo reagent was read by a SpectraMax Gemini XS Microplate Spectrofluorometer and analyzed with SoftmaxPro 4.8 software. (C) Luminescent trypomastigotes and amastigotes were examined for luciferase activity in active, in vitro rat myoblast infections using parasite:myoblast ratios of 40:1, 20:1, 10:1, 5:1, 2:1 or uninfected (NI). Five days p.i., D-luciferin was added to each well and the 6-well plate was imaged with the Xenogen IVIS system after a 5-min incubation (see Materials and methods for detailed description). In the pseudocolor image the luciferase activity or photon intensity ranges from the lowest intensity (blue) to highest intensity (red). Abbreviations: RLU, relative light units; min, minimum.
2.4. Experimental animals and T. cruzi infections
Four to 6 week old male A/J mice (Jackson Laboratories, Bar Harbor, ME) were housed under specific pathogen-free conditions. Mice were infected by i.p. injection of either 1 × 106, 1 × 105, or our standard quantity of 1 × 104 Brazil heart strain, luminescent T. cruzi trypomastigotes derived from infection of tissue culture H9C2 rat myoblasts. Wild-type Brazil heart (untransfected) T. cruzi and uninfected controls that had received an i.p. injection of PBS were included for in vivo analysis. The use and care of mice were conducted in accordance with the guidelines of the Center for Comparative Medicine at Northwestern University.
2.5. In vivo and ex vivo bioluminescent imaging
Prior to bioluminescent imaging, mice were anesthetized with 1.5% isofluorane. After anesthesia was achieved, 150 mg/kg body weight of substrate luciferin potassium salt dissolved in PBS and filtered through a 0.22 μm filter (Molecular Therapeutics, Ann Arbor, MI) was administered by a single i.p. injection. Mice were placed into the camera chamber, where a controlled flow of 1.5% isofluorane in air was administered through a nose cone via a gas anesthesia system designed to work in conjunction with the bioluminescent imaging system (IVIS 100; Xenogen, Alameda, CA). This imaging system consists of a cooled charge-coupled device camera mounted on a light-tight specimen chamber, a camera controller, and a Windows computer system. In order to allow adequate dissemination of the luciferin substrate (Contag et al., 1995), mice were maintained for 10 min after injection of the substrate. Mice were imaged in dorsal, ventral and left lateral positions by capturing a grayscale body image overlaid by a pseudocolor image representing the spatial distribution of the detected photons. Images were collected with 0.5 to 2 min integration times depending on signal intensity. For the analysis of parasites in specific organs, mice were administered the luciferin substrate, as described, maintained for 5 min and sacrificed for organ harvest. Data acquisition and analysis were performed by using the LivingImage software (Xenogen) where luminescence could be quantified as the sum of all detected photon counts per second within a chosen region of interest.
3. Results
3.1. Evaluation of Trypanosoma cruzi bioluminescence
To examine the live, in vivo dissemination of T. cruzi in a non-invasive manner, we engineered bioluminescent epimastigotes by integrating the firefly luciferase coding sequence into the tubulin locus. The luciferase coding sequence from the pGL3 basic vector was directionally subcloned into the pBS:THT-x-T tubulin integration vector (Weston et al., 1999), and integrated into the intergenic regions of parasite α- and β-tubulin after electroporation (Fig. 1A). Integration of the luciferase gene into the tubulin locus was confirmed by luciferase Southern blot hybridization of genomic DNA digested with SacII (Weston et al., 1999) as well as chromosomes prepared by pulsed-field gradient gel electrophoresis (data not shown). Eight weeks post transfection, an antibiotic-resistant epimastigote polyclonal population displayed notable luminescence measured by a standard plate-reading apparatus, indicating successful integration and expression of the luciferase gene (Fig. 1B). Further analysis of this luminescent population was conducted by allowing the epimastigote population to differentiate into metacyclic trypomastigotes. Once metacyclogenesis occurred, trypomastigotes were used to infect myoblasts at variable ratios from which both infectious trypomastigotes and intracellular, replicating amastigotes could be imaged for luminescence. The imaging of this in vitro infection revealed significant bioluminescence with an expected correlation of signal strength to parasite number (Fig. 1C).
3.2. In vivo characterization of luciferase T. cruzi
Once the myoblast infections were established and propagated by serial passage, it was not possible to determine with accuracy the number of parasites responsible for generating the luminescent signals shown in Fig. 1C. However, based on a comparison of the number of trypomastigotes used to initiate the in vitro infection and the numbers of epimastigotes used for the experiment of Fig. 1B, it appeared that the IVIS instrument was far superior to the standard plate-reading apparatus in luminescence sensitivity. For this reason, and to determine the minimal inoculum required to follow the course of infection in our mouse model of Chagas disease, we infected mice with 106, 105 or 104 (our normal inoculum) luminescent trypomastigotes by i.p. injection. Imaging of these mice as soon as 1 h p.i. revealed the capability of the IVIS system to detect all amounts used, with clear differences in signal strength (Fig. 2A). The magnitude of light emission was noticeably higher in the in vivo infection than in the in vitro infection when comparing the maximum values indicated on the scales. Interestingly, while infection with 104 luminescent trypomastigotes produced a signal clearly above that of wild-type parasites in vivo (not shown), nearly 2 × 105 epimastigotes were required to generate a luminescent signal above that of wild-type cells (Fig. 1B). The dissemination of parasites was monitored in infections initiated with the different inocula over 25 days, showing the highest parasite burden at 10 days p.i. when the highest number of trypomastigotes was used, but then a reduced and more dispersed burden was similar among the three animals by 3 weeks p.i. (Fig. 2A). When quantified, the signal intensity in all infections indicates a peak of parasite burden at 10 days p.i. when using either 1 × 106 or 1 × 105 parasites, with a slight lag in peak signal (14 days p.i.) when using 1 × 104 parasites (Fig. 2B). Additionally, at 2 weeks p.i., the original number of parasites used for infection became irrelevant as photon emission, or luminescence, was indistinguishable among the groups (Fig. 2B) and parasitosis appears to become more organ-specific at later time-points (Fig. 2A). It is noteworthy that the 10-fold differences of parasite quantity used for the initial infection correlated with 10-fold differences in luminescence signal intensity observed 1 h p.i. (Fig. 2B).
Fig. 2.


Luminescent T. cruzi imaged at various times p.i. with an IVIS imaging system. (A) Trypomastigotes were isolated from in vitro myoblast infections and different amounts, as indicated, were injected i.p. into A/J mice. Mice were imaged ventrally starting 1 h after infection and monitored the days p.i. as shown. For all images shown, the color scale ranges from blue (just above background with a minimum set to 15,000 photons/sec/cm2/sr) to red (maximum of 1 × 106 photons/sec/cm2/sr). (B) The course of whole-body parasite burden expressed in terms of the photonic signal resulting from infection of A/J mice with either 106, 105 or 104 luminescent or 106 wild-type T. cruzi trypomastigotes. The total light emission from the entire mouse body was measured and data points were generated from the analysis of at least two mice per infection condition. Day 0 p.i. corresponds to measurements acquired 1 h p.i. (see A). The signal shown for wild-type infection corresponds to background noise of the IVIS instrument. Abbreviation: min, minimum.
3.3. Course of intraperitoneal infection observed in the A/J-Brazil strain-strain model of experimental Chagas disease
Since our experimental model of acute Chagas disease is normally initiated with the i.p. injection of 104 Brazil strain trypomastigotes, we conducted a thorough analysis of parasite dissemination from three perspectives. As expected, the majority of luminescence observed early in the infection was in the lower left quadrant, where the parasites were administered. This was consistently observed from ventral, dorsal and left lateral viewpoints (Fig. 3). As the infection progressed, the overall intensity of luminescence decreased and specific regions retained parasites, indicative of possible occupancy in specific organs. For instance, at both 21 and 25 days p.i., it appeared that parasites were persistent in regions corresponding to lung or heart, as might be expected (Fig. 3, ventral). The persistence of luminescence emitted from the lower portion of the mouse abdomen suggested a general maintenance of parasite proliferation in this i.p. region or specific habitation of parasites in the gastrointestinal tract. To further examine the parasite burden in an organ-specific manner, several organs were harvested from animals 25 days p.i. and imaged for luminescence. These animals were provided D-luciferin substrate, allowed time for adequate systemic dissemination and sacrificed for organ harvest. As shown in Fig. 4, the majority of T. cruzi was found in the gastrointestinal system, particularly the large intestine (LI), with a notable presence in the small intestine (SI), lungs (L) and kidneys (K). Smaller amounts of luminescence were also observed in the heart (H) and skeletal muscle (Sk) while either a completely absent or barely detectable luminescent signal was seen in spleen (Sp) and liver (Lv). Whole blood (B) revealed a low level signal.
Fig. 3.

Course of parasite dissemination in a mouse model of experimental Chagas heart disease. A/J mice were injected i.p. with 1 × 104 luminescent Trypanosoma cruzi trypomastigotes and imaged either ventrally, dorsally or laterally over the course of infection prior to death typically observed by 30 days p.i.. For all images shown, the color scale ranges from blue (just above background with a minimum set to 50,000 photons/sec/cm2/sr) to red (maximum of 1 × 106 photons/sec/cm2/sr). The minimum for this scale was adjusted to avoid signal saturation during the peak of signal intensity. The ventral, dorsal and lateral perspectives for each timepoint were taken from the same animal and all images are representative of at least two animals. Abbreviation: min, minimum.
Fig. 4.
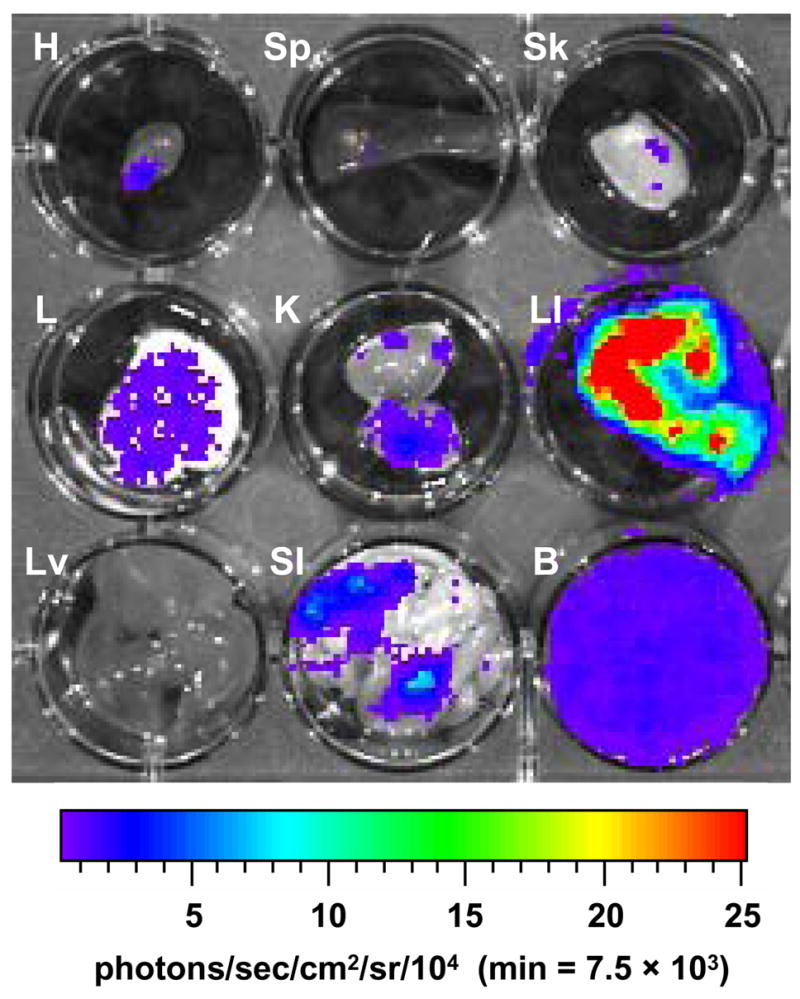
Detection of luminescent T. cruzi in the internal organs of infected A/J mice. Mice were infected with 104 trypomastigotes and injected with D-luciferin substrate (as described in Materials and methods) prior to sacrifice and organ dissection. Twenty-five days p.i. luminescence was analyzed in heart (H), spleen (Sp), skeletal muscle (Sk), lung (L), kidney (K), large intestine (LI), liver (Lv), small intestine (SI) and whole blood (B). For all images shown, the color scale ranges from blue (just above background with a minimum set to 7,500 photons/sec/cm2/sr) to red (maximum of 2.5 × 105 photons/sec/cm2/sr). The minimum and maximum for this scale was adjusted to enhance signal detection while avoiding saturation and is consistent for all organs imaged. Abbreviation: min, minimum.
4. Discussion
To make bioluminescent imaging of T. cruzi possible, a method in which the firefly luciferase enzyme could be stably expressed within the parasite was required. The pBS:THT-x-T plasmid, having been previously employed for the expression of an FL-160-GFP fusion in T. cruzi (Weston et al., 1999), was chosen for its ease of modification, possession of an antibiotic resistance gene for positive selection, and design for integration into an essential region of the trypanosome genome (tubulin locus) to ensure its maintained expression. This transfection strategy produced transgenic parasites that could be used for animal infections without the need for continuous drug selection, unlike those using episomal vectors. The administration of hygromycin following transfection produced a population of epimastigotes that readily display luminescent activity using a standard plate-reading luminometer (Fig. 1B). Although the modest sensitivity of this instrument precludes assessment of luciferase activity at the single-cell level, there is potential for such a tool in the screening of potential parasiticidal compounds. This high-throughput, efficient method has been effective for the investigation of anti-Leishmania compounds (Ashutosh et al., 2005; Lang et al., 2005) and could similarly be applied to luminescent T. cruzi.
We employed the IVIS instrument, typically used for whole animal imaging, to assess bioluminescence of trypomastigotes and amastigotes from a live, in vitro infection of rat cardiomyocytes. This method provides a more complete picture of luminescence by providing a visualization of light and the ability to quantify emitted photons in a specific region of interest. In addition to the utility of luminescent trypanosomes for drug screening, studies of host and parasite factors involved in susceptibility and resistance to infection could be accomplished using this technology. For instance, specific deletion or knockdown of parasite or host cell components thought to be required for invasion could be quickly analyzed by determining whether infections can be established and maintained in culture before moving to an animal model.
The ability to image T. cruzi with IVIS instrumentation on a whole animal level will also enable investigators to conduct important studies of virulence from both host and parasite perspectives. We initially had to determine the threshold of detection as it relates to our standard mouse infection regimen. We previously established an experimental model of Chagas disease in which A/J mice are infected with 104 trypomastigotes and analyzed in the acute phase (21 days p.i.) at which time severe inflammation, fibrosis and parasitosis of the heart is typically observed. To assess the sensitivity of the IVIS instrument for imaging our luciferase parasites we found that, while luminescence detection was minimal 1 h p.i., the proliferation and dissemination over a 25 day period was sufficient to produce a maintained signal throughout the course of infection (Fig. 2A). Interestingly, when infecting with either 10-or 100-fold more trypomastigotes, an initial correlation of luminescence to parasite number was observed (Fig. 2B, 1 h p.i.), followed by a gradual equilibration of parasite burden by 14 days p.i.. One possible explanation for this could be a combination of enhanced immunity against T. cruzi coupled with an impaired capacity for immune evasion typically associated with the parasite. A number of reports have described different ways in which T. cruzi is capable of evading host immune responses during the acute phase of infection, allowing the persistence to gradually contribute to chronic pathology (Kierszenbaum, 1981; Garcia et al., 1997; Brodskyn et al., 2002; Kotner and Tarleton, 2007). With the extreme nature of infecting with 105 or 106 trypomastigotes, an inoculum far surpassing any used for a variety of T. cruzi-based experimental animal systems, the ability of the parasite to avoid the robust adaptive immune response mounted could be reduced. By 10 days p.i., we observed a peak in parasite burden in these animals followed by a decrease which approaches that seen in animals infected with our normal quantity of parasites. The timing of this event likely corresponds to the peak of the adaptive immune response, which appears to control the infection once the parasite burden reaches a certain point. Interestingly, animals infected with the lowest number of parasites show increased parasite burden until 14 days p.i., at which point the animals appear to have the same burden as those receiving higher inocula. In all cases, once the adaptive immune response presumably initiated full control of parasitism, we observed a sharp decline in burden by 21 days p.i., at which point parasites appeared to have taken up residence in specific organs, rather than being dispersed throughout the animal. As the animals progress to 25 days p.i., the luminescent signal plateaued, suggestive of a scenario in which host immunity has controlled the infection and parasite persistence has been achieved. In this particular experimental disease model, using the Brazil parasite strain with A/J mice, animals succumb to disease by 30 days p.i., prohibiting the examination of luminescence into the chronic phase of disease. The development of luminescent T. cruzi in other parasite strains and performing infections in different strains of mice will facilitate the investigation of parasite dissemination in other, long-term chronic animal models of Chagas disease.
Further analysis of our standard experimental model, using 104 luminescent trypanosomes, provided a clearer picture of parasite dissemination by imaging from three perspectives (Fig. 3; ventral, dorsal and lateral). The site of injection, in the lower left quadrant, displayed the most prominent photon emission early after infection, suggesting that parasites immediately invade and initiate replication in surrounding tissue. The infection spread over the next 2 weeks, at which point the peak of parasite load was observed, followed by the sharp decrease in luminescent signal. By 3 weeks p.i., the intensity in the lower abdominal region was decreased but maintained, and luminescence was observed in areas of the thoracic region from the ventral perspective, suggestive of parasitosis of heart or lung. In addition, the dorsal view suggested potential parasitism of either the spleen or kidney at both 21 and 25 days p.i.. While the signal observed in the abdominal region diminished over time, it remained strong enough to potentially mask the parasite burden associated with nearby tissue (e.g. skeletal muscle, liver).
In order to overcome this issue, we analyzed specific organs from infected mice 25 days p.i.. After allowing adequate dissemination of the injected luciferin substrate, mice were sacrificed and organs (heart, skeletal muscle, spleen, lungs, kidneys, liver, blood, large and small intestine) were imaged using IVIS (Fig. 4). As anticipated, due to the typical anatomical sites associated with Chagas disease, we observed luminescent signals from heart and skeletal muscle as well as gastrointestinal organs. Substantial photon emission was observed in both lung and kidney tissue while nearly a complete absence of signal was observed in spleen and liver. This was surprising, given the significant roles of these organs in the reticuloendothelial system. While the blood appears to have high parasitemia based on visual appearance, the overall signal is actually quite low with respect to the scale of intensity (Fig. 4; B). This low-level parasitemia is in agreement with previous findings at the acute timepoint used for analysis. While others have reported parasite distribution to lung, spleen and liver in cases when animals have been immunosuppressed (Calabrese et al., 1992) and even parasitism of bone and cartilage (Morocoima et al., 2006), it has become increasingly clear from a number of studies that the genetics of the parasite and host play a defining role in the tissue distribution of T. cruzi (Melo and Brener, 1978; Ben Younes-Chennoufi et al., 1988; McCabe et al., 1989; Andrade et al., 2002; Franco et al., 2003; Marinho et al., 2004). Although the pathogenesis of Chagas disease is multivariate, the persistence of T. cruzi has been suggested as playing a fundamental role in disease progression (Anez et al., 1999; Zhang and Tarleton, 1999; Tarleton, 2001). Despite the continued debate and variability of the mechanism of pathogenesis, the development of luminescent T. cruzi provides the ability to quickly screen organs and tissue samples for the presence of parasites. Further correlation of whole body and harvested organ luminescence should permit the analysis of tissue-specific parasitization in a non-invasive manner using this powerful technology. In infections in which small numbers of microorganisms persist at extremely low levels, methods of detection proven to be far more sensitive (e.g., PCR) will continue to be essential for specific analyses. The observation of low-level parasitemia indicated by ex vivo luminescence of blood, in conjunction with the apparent absence of parasites in blood-filtering organs, illustrates the limitations of luminescence detection. This limitation does not come unexpectedly, since the detection of a minimal presence of trypanosomes has also been challenging with a number of other methods (see Introduction). Although luminescent trypanosomes do not overcome all the obstacles encountered in this field of research, they do provide a powerful solution to a number of experimental challenges that previously required invasive, more costly, analyses.
During the past several years several groups have employed IVIS technology to a variety of parasitic infections (Heussler and Doerig, 2006; Hutchens and Luker, 2007). Recent applications to Toxoplasma gondii virulence have determined the importance of IFN-γ and Toll-like receptor signaling to parasite dissemination to the CNS following immunosuppression (Dellacasa-Lindberg et al., 2007) or to overall host resistance (Hitziger et al., 2005), respectively. These studies were made possible using transgenic, knockout mice coupled with multiple strains of luminescent Toxoplasma. Others have used multiple strains to determine differences in replication capacity over time and to examine the reactivation of parasites during a chronic infection (Saeij et al., 2005). Dissemination patterns resulting from different routes of Toxoplasma infection have also been analyzed (Boyle et al., 2007). In addition to in vitro drug screening, bioluminescent Leishmania amazonensis has also been used in vivo and ex vivo to examine the response to various therapeutics on both living mice and extracted, parasitized organs (Lang et al., 2005). Results of our study confirm the applicability of IVIS technology for the study of Chagas disease pathogenesis. In addition to the experiments conducted in other parasitic disease models, we will now able to address questions pertaining to the relevance of parasite burden to the magnitude of organ-specific autoimmunity (Leon et al., 2001; Hyland et al., 2007), conduct rapid screening of new potential parasiticidal drugs and test strategies for vaccine development against this parasite.
Acknowledgments
We are grateful to Dr. Wesley Van Voorhis for the generous gift of the pBS-THT-x-T plasmid which was easily modified for use in transfection, and to Dr. Dixon Kaufman and Courtney Larson for the use of and expert technical assistance on the IVIS 100 BLI instrument and software. This work was supported by NIH grant HL075822 (to DME) and predoctoral fellowship from the American Heart Association (to KVH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrade LO, Machado CRS, Chiari E, Pena SDJ, Macedo AM. Trypanosoma cruzi: role of host genetic background in the differential tissue distribution of parasite clonal populations. Exp Parasitol. 2002;100:269–275. doi: 10.1016/s0014-4894(02)00024-3. [DOI] [PubMed] [Google Scholar]
- Anez N, Carrasco H, Parada H, Crisante G, Rojas A, Fuenmayor C, Gonzalez N, Percoco G, Borges R, Guevara P, Ramirez JL. Myocardial parasite persistence in chronic chagasic patients. Am J Trop Med Hyg. 1999;60:726–732. doi: 10.4269/ajtmh.1999.60.726. [DOI] [PubMed] [Google Scholar]
- Ashutosh Gupta, Ramesh S, Sundar S, Goyal N. Use of Leishmania donovani field isolates expressing the luciferase reporter gene in in vitro drug screening. Antimicrob Agents Chemother. 2005;49:3776–3783. doi: 10.1128/AAC.49.9.3776-3783.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellotti G, Bocchi EA, de Moraes AV, Higuchi ML, Barbero-Marcial M, Sosa E, Esteves-Filho A, Kalil R, Weiss R, Jatene A, Pileggi F. In vivo detection of Trypanosoma cruzi antigens in hearts of patients with chronic Chagas’ heart disease. Am Heart J. 1996;131:301–307. doi: 10.1016/s0002-8703(96)90358-0. [DOI] [PubMed] [Google Scholar]
- Ben Younes-Chennoufi A, Hontebeyrie-Joskowicz M, Tricottet V, Eisen H, Reynes M, Said G. Persistence of Trypanosoma cruzi antigens in the inflammatory lesions of chronically infected mice. Trans R Soc Trop Med Hyg. 1988;82:77–83. doi: 10.1016/0035-9203(88)90269-6. [DOI] [PubMed] [Google Scholar]
- Boyle JP, Saeij JP, Boothroyd JC. Toxoplasma gondii: inconsistent dissemination patterns following oral infection in mice. Exp Parasitol. 2007;116:302–305. doi: 10.1016/j.exppara.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Brodskyn C, Patricio J, Oliveira R, Lobo L, Arnholdt A, Mendonca-Previato L, Barral A, Barral-Netto M. Glycoinositolphospholipids from Trypanosoma cruzi interfere with macrophages and dendritic cell responses. Infect Immun. 2002;70:3736–3743. doi: 10.1128/IAI.70.7.3736-3743.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese KS, Bauer PG, Lagrange PH, Goncalves da Costa SC. Trypanosoma cruzi infection in immunosuppressed mice. Immunol Lett. 1992;31:91–96. doi: 10.1016/0165-2478(92)90016-h. [DOI] [PubMed] [Google Scholar]
- Calabrese KS. Immunosuppressive drugs as a tool to explore immunopathology in experimental Chagas disease. Mem Inst Oswaldo Cruz. 1999;94(Suppl 1):273–276. doi: 10.1590/s0074-02761999000700046. [DOI] [PubMed] [Google Scholar]
- Chandler FW, Watts JC. Immunofluorescence as an adjunct to the histopathologic diagnosis of Chagas’ disease. J Clin Microbiol. 1988;26:567–569. doi: 10.1128/jcm.26.3.567-569.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contag CH, Contag PR, Mullins JI, Spilman SD, Stevenson DK, Benaron DA. Photonic detection of bacterial pathogens in living hosts. Mol Microbiol. 1995;18:593–603. doi: 10.1111/j.1365-2958.1995.mmi_18040593.x. [DOI] [PubMed] [Google Scholar]
- Dellacasa-Lindberg I, Hitziger N, Barragan A. Localized recrudescence of Toxoplasma infections in the central nervous system of immunocompromised mice assessed by in vivo bioluminescence imaging. Microbes Infect. 2007;9:1291–1298. doi: 10.1016/j.micinf.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Edinger M, Cao YA, Hornig YS, Jenkins DE, Verneris MR, Bachmann MH, Negrin RS, Contag CH. Advancing animal models of neoplasia through in vivo bioluminescence imaging. Eur J Cancer. 2002;38:2128–2136. doi: 10.1016/s0959-8049(02)00410-0. [DOI] [PubMed] [Google Scholar]
- Franco DJ, Vago AR, Chiari E, Meira FC, Galvao LM, Machado CR. Trypanosoma cruzi: mixture of two populations can modify virulence and tissue tropism in rat. Exp Parasitol. 2003;104:54–61. doi: 10.1016/s0014-4894(03)00119-x. [DOI] [PubMed] [Google Scholar]
- Garcia IE, Lima MR, Marinho CR, Kipnis TL, Furtado GC, Alvarez JM. Role of membrane-bound IgM in Trypanosoma cruzi evasion from immune clearance. J Parasitol. 1997;83:230–233. [PubMed] [Google Scholar]
- Heussler V, Doerig C. In vivo imaging enters parasitology. Trends Parasitol. 2006;22:192–195. doi: 10.1016/j.pt.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Hitziger N, Dellacasa I, Albiger B, Barragan A. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell Microbiol. 2005;7:837–848. doi: 10.1111/j.1462-5822.2005.00517.x. [DOI] [PubMed] [Google Scholar]
- Hutchens M, Luker GD. Applications of bioluminescence imaging to the study of infectious diseases. Cell Microbiol. 2007;9:2315–2322. doi: 10.1111/j.1462-5822.2007.00995.x. [DOI] [PubMed] [Google Scholar]
- Hyland KV, Leon JS, Daniels MD, Giafis N, Woods LM, Bahk TJ, Wang K, Engman DM. Modulation of autoimmunity by treatment of an infectious disease. Infect Immun. 2007;75:3641–3650. doi: 10.1128/IAI.00423-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James MJ, Yabsley MJ, Pung OJ, Grijalva MJ. Amplification of Trypanosoma cruzi-specific DNA sequences in formalin-fixed raccoon tissues using polymerase chain reaction. J Parasitol. 2002;88:989–993. doi: 10.1645/0022-3395(2002)088[0989:AOTCSD]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Jones EM, Colley DG, Tostes S, Lopes ER, Vnencak-Jones CL, McCurley TL. Amplification of a Trypanosoma cruzi DNA sequence from inflammatory lesions in human chagasic cardiomyopathy. Am J Trop Med Hyg. 1993;48:348–357. doi: 10.4269/ajtmh.1993.48.348. [DOI] [PubMed] [Google Scholar]
- Kierszenbaum F. On evasion of Trypanosoma cruzi from the host immune response. Lymphoproliferative responses to trypanosomal antigens during acute and chronic experimental Chagas’ disease. Immunology. 1981;44:641–648. [PMC free article] [PubMed] [Google Scholar]
- Kirchhoff LV, Hieny S, Shiver GM, Snary D, Sher A. Cryptic epitope explains the failure of a monoclonal antibody to bind to certain isolates of Trypanosoma cruzi. J Immunol. 1984;133:2731–2735. [PubMed] [Google Scholar]
- Kirchhoff LV, Weiss LM, Wittner M, Tanowitz HB. Parasitic diseases of the heart. Front Biosci. 2004;9:706–723. doi: 10.2741/1255. [DOI] [PubMed] [Google Scholar]
- Kirchhoff LV, Paredes P, Lomeli-Guerrero A, Paredes-Espinoza M, Ron-Guerrero CS, Delgado-Mejia M, Pena-Munoz JG. Transfusion-associated Chagas disease (American trypanosomiasis) in Mexico: implications for transfusion medicine in the United States. Transfusion. 2006;46:298–304. doi: 10.1111/j.1537-2995.2006.00715.x. [DOI] [PubMed] [Google Scholar]
- Kotner J, Tarleton R. Endogenous CD4(+) CD25(+) regulatory T cells have a limited role in the control of Trypanosoma cruzi infection in mice. Infect Immun. 2007;75:861–869. doi: 10.1128/IAI.01500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane JE, Olivares-Villagomez D, Vnencak-Jones CL, McCurley TL, Carter CE. Detection of Trypanosoma cruzi with the polymerase chain reaction and in situ hybridization in infected murine cardiac tissue. Am J Trop Med Hyg. 1997;56:588–595. doi: 10.4269/ajtmh.1997.56.588. [DOI] [PubMed] [Google Scholar]
- Lane JE, Ribeiro-Rodrigues R, Olivares-Villagomez D, Vnencak-Jones CL, McCurley TL, Carter CE. Detection of Trypanosoma cruzi DNA within murine cardiac tissue sections by in situ polymerase chain reaction. Mem Inst Oswaldo Cruz. 2003;98:373–376. doi: 10.1590/s0074-02762003000300013. [DOI] [PubMed] [Google Scholar]
- Lang T, Goyard S, Lebastard M, Milon G. Bioluminescent Leishmania expressing luciferase for rapid and high throughput screening of drugs acting on amastigote-harbouring macrophages and for quantitative real-time monitoring of parasitism features in living mice. Cell Microbiol. 2005;7:383–392. doi: 10.1111/j.1462-5822.2004.00468.x. [DOI] [PubMed] [Google Scholar]
- Lenzi HL, Oliveira DN, Lima MT, Gattass CR. Trypanosoma cruzi: paninfectivity of CL strain during murine acute infection. Exp Parasitol. 1996;84:16–27. doi: 10.1006/expr.1996.0086. [DOI] [PubMed] [Google Scholar]
- Leon JS, Godsel LM, Wang K, Engman DM. Cardiac myosin autoimmunity in acute Chagas heart disease. Infect Immun. 2001;69:5643–5649. doi: 10.1128/IAI.69.9.5643-5649.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons SK, Lim E, Clermont AO, Dusich J, Zhu L, Campbell KD, Coffee RJ, Grass DS, Hunter J, Purchio T, Jenkins D. Noninvasive bioluminescence imaging of normal and spontaneously transformed prostate tissue in mice. Cancer Res. 2006;66:4701–4707. doi: 10.1158/0008-5472.CAN-05-3598. [DOI] [PubMed] [Google Scholar]
- Marinho CR, Bucci DZ, Dagli ML, Bastos KR, Grisotto MG, Sardinha LR, Baptista CR, Goncalves CP, Lima MR, Alvarez JM. Pathology affects different organs in two mouse strains chronically infected by a Trypanosoma cruzi clone: a model for genetic studies of Chagas’ disease. Infect Immun. 2004;72:2350–2357. doi: 10.1128/IAI.72.4.2350-2357.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe RE, Meagher S, Mullins B. Trypanosoma cruzi: explant organ cultures from mice with chronic Chagas’ disease. Exp Parasitol. 1989;68:462–469. doi: 10.1016/0014-4894(89)90131-8. [DOI] [PubMed] [Google Scholar]
- Melo RC, Brener Z. Tissue tropism of different Trypanosoma cruzi strains. J Parasitol. 1978;64:475–482. [PubMed] [Google Scholar]
- Moncayo A. Progress towards interruption of transmission of Chagas disease. Mem Inst Oswaldo Cruz. 1999;94(Suppl 1):401–404. doi: 10.1590/s0074-02761999000700079. [DOI] [PubMed] [Google Scholar]
- Moncayo A. Chagas disease: current epidemiological trends after the interruption of vectorial and transfusional transmission in the Southern Cone countries. Mem Inst Oswaldo Cruz. 2003;98:577–591. doi: 10.1590/s0074-02762003000500001. [DOI] [PubMed] [Google Scholar]
- Morocoima A, Rodriguez M, Herrera L, Urdaneta-Morales S. Trypanosoma cruzi: experimental parasitism of bone and cartilage. Parasitol Res. 2006;99:663–668. doi: 10.1007/s00436-006-0211-2. [DOI] [PubMed] [Google Scholar]
- Mortatti RC, Fonseca LS, Coelho J, Oliveira A, Moreno M. Follow-up of patent and subpatent parasitemias and development of muscular lesions in mice inoculated with very small numbers of Trypanosoma cruzi. Exp Parasitol. 1992;75:233–239. doi: 10.1016/0014-4894(92)90183-b. [DOI] [PubMed] [Google Scholar]
- Nowicki MJ, Chinchilla C, Corado L, Matsuoka L, Selby R, Steurer F, Mone T, Mendez R, Aswad S. Prevalence of antibodies to Trypanosoma cruzi among solid organ donors in Southern California: a population at risk. Transplantation. 2006;81:477–479. doi: 10.1097/01.tp.0000195778.08411.b8. [DOI] [PubMed] [Google Scholar]
- Nunes MP, Coutinho SG, Louis JA, Souza WJ. Trypanosoma cruzi: quantification in tissues of experimentally infected mice by limiting dilution analysis. Exp Parasitol. 1990;70:186–192. doi: 10.1016/0014-4894(90)90099-x. [DOI] [PubMed] [Google Scholar]
- Pinto PL, Takami R, Nunes EV, Guilherme CS, Oliveira OC, Jr, Gama-Rodrigues J, Okumura M. Life cycle of Trypanosoma cruzi (Y strain) in mice. Rev Hosp Clin Fac Med Sao Paulo. 1999;54:141–146. doi: 10.1590/s0041-87811999000500002. [DOI] [PubMed] [Google Scholar]
- Russo M, Starobinas N, Marcondes MC, Minoprio P, Honteyberie-Joskowicz M. The influence of T cell subsets on Trypanosoma cruzi multiplication in different organs. Immunol Lett. 1996;49:163–168. doi: 10.1016/0165-2478(96)02498-4. [DOI] [PubMed] [Google Scholar]
- Saeij JP, Boyle JP, Grigg ME, Arrizabalaga G, Boothroyd JC. Bioluminescence imaging of Toxoplasma gondii infection in living mice reveals dramatic differences between strains. Infect Immun. 2005;73:695–702. doi: 10.1128/IAI.73.2.695-702.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, Yang Q, Bishop JM, Contag CH, Felsher DW. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- Taniwaki NN, Andreoli WK, Calabrese KS, da Silva S, Mortara RA. Disruption of myofibrillar proteins in cardiac muscle of Calomys callosus chronically infected with Trypanosoma cruzi and treated with immunosuppressive agent. Parasitol Res. 2005;97:323–331. doi: 10.1007/s00436-005-1429-0. [DOI] [PubMed] [Google Scholar]
- Taniwaki NN, da Silva CV, da Silva S, Mortara RA. Distribution of Trypanosoma cruzi stage-specific epitopes in cardiac muscle of Calomys callosus, BALB/c mice, and cultured cells infected with different infective forms. Acta Trop. 2007;103:14–25. doi: 10.1016/j.actatropica.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Tarleton RL. Parasite persistence in the aetiology of Chagas disease. Int J Parasitol. 2001;31:550–554. doi: 10.1016/s0020-7519(01)00158-8. [DOI] [PubMed] [Google Scholar]
- Vago AR, Macedo AM, Adad SJ, Reis DD, Correa-Oliveira R. PCR detection of Trypanosoma cruzi DNA in oesophageal tissues of patients with chronic digestive Chagas’ disease. Lancet. 1996;348:891–892. doi: 10.1016/S0140-6736(05)64761-7. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Jonkers J, Lyons S, Berns A. Noninvasive imaging of spontaneous retinoblastoma pathway-dependent tumors in mice. Cancer Res. 2002;62:1862–1867. [PubMed] [Google Scholar]
- Wendt MK, Cooper AN, Dwinell MB. Epigenetic silencing of CXCL12 increases the metastatic potential of mammary carcinoma cells. Oncogene. 2008;27:1461–1471. doi: 10.1038/sj.onc.1210751. [DOI] [PubMed] [Google Scholar]
- Weston D, La Flamme AC, Van Voorhis WC. Expression of Trypanosoma cruzi surface antigen FL-160 is controlled by elements in the 3′ untranslated, the 3′ intergenic, and the coding regions. Mol Biochem Parasitol. 1999;102:53–66. doi: 10.1016/s0166-6851(99)00079-1. [DOI] [PubMed] [Google Scholar]
- Zhang L, Tarleton RL. Parasite persistence correlates with disease severity and localization in chronic Chagas’ disease. J Infect Dis. 1999;180:480–486. doi: 10.1086/314889. [DOI] [PubMed] [Google Scholar]