

Abstract
Objective
Thiopurine S-methyltransferase *3A is degraded much more rapidly than is the “wild type” enzyme through a ubiquitin-proteasome dependent process. It also forms aggresomes, suggesting a possible dynamic balance between degradation and aggregation. We set out to identify genes encoding proteins participating in these processes.
Methods
Green fluorescent protein tagged TPMT*3A was expressed in a Saccharomyces cerevisiae gene deletion library, and flow-cytometry was used to screen for cells with high fluorescence intensity – indicating the loss of a gene essential for TPMT*3A degradation.
Results
Twenty-four yeast genes were identified in functional categories that included ubiquitin-dependent protein degradation, vesicle trafficking and vacuolar degradation. The presence of genes encoding proteins involved in vesicular transport and vacuolar degradation suggested a possible role in TPMT*3A degradation for autophagy – a process not previously identified as a pharmacogenomic mechanism. In support of that hypothesis, TPMT*3A aggregates increased dramatically in mutants for vacuolar protease and autophagy-related genes. Furthermore, TPMT*3A expression in human cells induced autophagy, and siRNA-mediated knockdown of ATG7, an autophagy-related human protein, enhanced TPMT*3A aggregation but not that of TPMT*3C or WT TPMT, indicating that autophagy contributes to TPMT*3A degradation in mammalian cells. We also demonstrated that UBE2G2, the human homologue of the E2 ubiquitin conjugating enzyme identified during the yeast genetic screen, was involved in TPMT*3A degradation in human cells.
Conclusion
These results indicate that autophagy should be considered among mechanisms responsible for the effects of pharmacogenetically significant polymorphisms that alter encoded amino acids.
Keywords: Thiopurine S-methyltransferase, TPMT, TPMT *3A, TPMT*3C, autophagy, protein misfolding, protein aggregation, protein degradation
Introduction
The thiopurine S-methyltransferase (TPMT) genetic polymorphism is a prototypic example of the clinical importance of pharmacogenomics [1–4] as well as a model system for the study of mechanisms responsible for the functional effects of nonsynonymous single nucleotide polymorphisms (SNPs) [5–7]. This polymorphism has such striking clinical significance [1, 2] that the United States Food and Drug Administration (FDA) highlighted TPMT as one of only two ‘valid biomarkers’ for pharmacogenomics in its original 2003 ‘Draft Guidance for Pharmacogenomic Data Submission’ [8], and the initial FDA public hearing on the inclusion of pharmacogenetic information in drug labeling dealt with TPMT and thiopurine drugs (http://www.fda.gov). TPMT is a cytosolic S-adenosyl-L-methionine (AdoMet)-dependent enzyme that catalyses the S-methylation of thiopurine drugs such as 6-mercaptopurine [9, 10], drugs that are used to treat acute lymphoblastic leukemia of childhood and inflammatory bowel disease [11]. However, these drugs, like many cytotoxic agents, have a narrow therapeutic index and can cause potentially life-threatening drug-related toxicity.
TPMT genetic polymorphisms are associated with large individual differences in tissue TPMT enzyme activity and protein levels [1, 12, 13]. Individuals with inherited decreases in TPMT activity – mainly as a result of the effects of the TPMT*3A allele (minor allele frequency in Caucasians of approximately 5%) [1] – are at greatly increased risk for severe life-threatening myelosuppression when treated with “standard” doses of thiopurine drugs [1, 14–17]. The TPMT*3A variant allele contains two nonsynonymous SNPs that result in Ala154Thr and Tyr240Cys alterations in the encoded amino acid sequence [18]. Levels of TPMT enzyme activity and protein are virtually undetectable in the tissues of subjects homozygous for TPMT*3A or in cultured mammalian cells transfected with this allele [1, 5–7, 12–15, 18, 19]. That is true because the TPMT*3A variant allozyme is degraded much more rapidly than is the WT enzyme [5–7]. This process of accelerated degradation involves molecular chaperones and ubiquitination of the variant allozymes, followed by proteasome-mediated degradation [5, 6]. TPMT*3C, with only the Tyr240Lys alteration in amino acid sequence, is the most common variant allele in East Asia and does not display the striking acceleration in degradation or the same tendency to aggregate that are observed with TPMT*3A [5, 20]. Recent studies in mammalian cells have also demonstrated that, when proteasome-mediated degradation is inhibited, TPMT*3A can aggregate with aggresome formation, almost certainly as a result of protein misfolding, [7]. For example, E. coli recombinant TPMT*3A eluted from a size-exclusion column entirely as aggregated protein, while the wild type (WT) enzyme and TPMT*3C displayed a lesser tendency to aggregate [7].
This series of observations suggests that a dynamic balance may exist for TPMT*3A among protein folding, proteasome-mediated degradation and aggregation. However, the identity of the proteins responsible for and participating in these cellular processes remains unclear. Knowledge of these mechanisms and the participating proteins has potential importance for pharmacogenomics, both because of their direct relevance to clinically significant variation in drug efficacy and toxicity [2] and because of their potential contribution to our mechanistic understanding of other important pharmacogenomic traits. Therefore, in the present study we have used Sacchoramyces cerevisiae as a genetic “model system” to identify 24 yeast genes that participate in TPMT*3A degradation/aggregation. Specifically, a yeast gene deletion library “screen” identified genes encoding proteins involved in both proteasome-mediated degradation and autophagy. We then used siRNA to “knockdown” in mammalian cells the expression in key genes in both pathways and demonstrated that results obtained with the yeast system also applied to mammalian cells.
Methods
Yeast strains, plasmids and media
We used four isogenic series of yeast strains in these studies: w303-1A (MATa can1–100 his3–11, 15 leu2–3, 112 trp1-1 ura3-1 ade2-1), YPH500 (MATα ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1), BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and BY4743 (MATa/MATα his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 lys2Δ0/LYS2 met15Δ0/MET15 ura3Δ0/ura3Δ0). The genotype of the tub4-1 strain is MATα ura3-52 lys2-801 ade2-101 trp1Δ63 his3Δ200 leu2Δ1 tub4-1 (also known as ESM208) [21]. The yeast enhanced GFP [22], human WT TPMT and TPMT*3A [7] cDNA ORFs were amplified using the PCR, and amplicons were cloned into a pRS426-based expression vector with the GAL1-10 promoter. GFP fusions were constructed in the same vectors by inserting the GFP ORF at the N-terminus of the WT TPMT or TPMT*3A ORFs to create pRS-GFPTPMT and pRS-GFPTPMT*3A plasmids, respectively. Construct sequences were verified by sequencing both strands. Yeast media and culture were prepared using standard procedures [23]. Cells transformed with TPMT constructs were grown in synthetic complete media lacking uracil (SC-Ura) and were switched to galactose medium (SC-Ura+Galactose) to allow the induction of expression. The cells were also treated with 20 μM MG132 (EMD Chemicals, Inc., San Diego, CA) and/or 69 μM benomyl (Sigma-Aldrich Corp., St. Louis, MO) to inhibit proteasomes or to disrupt the microtubule cytoskeleton, respectively [7, 24].
Yeast screening methods
The yeast homozygous diploid deletion pool, a collection of 4,667 yeast deletion strains, was obtained from Invitrogen (Carlsbad, CA). pRS-GFPTPMT*3A was used to transform the pooled deletion strains using the lithium acetate method [23]. Approximately 40,000 individual transformants were observed (~10-fold coverage) after plating onto SC-Ura plates to select for the plasmid. Transformants were cultured in SC-Ura+Galactose for 8 h at 30°C, and the cells were pelleted and resuspended in phosphate buffered saline (PBS). After sonication for 10 s, the cell suspension was added to a flow cytometer, and flow-cytometric sorting was used to collect cells with high fluorescence intensity. The high fluorescence cells were repooled and cultured once again in liquid medium. This process of selection for clones with high fluorescence intensity was repeated three times. At the end of that process, cells were plated to obtain individual colonies for further analysis.
Identification and testing of yeast gene deletion strains that displayed enhanced fluorescence intensity
Genomic DNA was isolated from colonies with enhanced fluorescence. The PCR was then used to amplify a 20-bp “barcode” sequence that uniquely identified each deletion strain, and the amplicons were sequenced. These sequences were then compared to those in the yeast gene deletion set database. One hundred and twenty eight individual colonies were sequenced. All strains identified in this fashion were streaked from the original frozen stocks of the yeast homozygous diploid gene deletion panel and were re-transformed with pRS-GFP-TPMT*3A. These re-transformed deletion strains were then tested using fluorescence microscopy to ensure that the enhanced fluorescence detected during the original flow cytometry study was not due to a second-site mutation.
RNA interference
Validated small interfering RNA (siRNA) directed against ATG7 as well as control siRNA were purchased from Qiagen (Valencia, CA). siRNA directed against UBE2G2 was purchased from Invitrogen (Carlsbad, CA). Cells cultured in 6-well plates were transfected with siRNAs at 50 nm final concentrations by using lipofectamine RNAiMAX (Invitrogen) and were incubated at 37°C for 48 h. Cells were then cotransfected with siRNAs at a 50 nm final concentration and with an HA-tagged TPMT*3A construct [7] by using X-tremeGENE siRNA Transfection Reagent (Roche Applied Science, Indianapolis, IN). The cells were incubated at 37°C for 48 h, and were then subjected to Western blot analysis and/or immunofluorescence microscopy.
Fluorescence microscopy
Yeast cells were grown overnight in SC-Ura+Glucose medium at 30°C. The cultures were then centrifuged at 10,000 g for 5 min. Pellets were resuspended in 3 mL SC-Ura+Galactose medium (A600 = 0.4), and were grown at 30°C for 8 h. The cells were then visualized using a Nikon Eclipse 80i microscope with a 60X oil immersion objective, and fluorescent images were acquired using a digital camera and SPOT software (Diagnostics Instruments, Inc., Sterling Heights, MI). To quantify aggregate formation in the cells, we randomly selected 3 fields, obtained photographs and counted all cells in each field – determining the percentage that displayed aggregate formation. Each field contained approximately 200 cells.
HEK293 cells were cotransfected with an HA-tagged TPMT*3A construct [7] and siRNA directed against ATG7, UBE2G2 or control siRNA, and the transfected cells were grown on coverslips for 48 h. The cells were fixed and permeabilized as described previously [7], and were stained for 30 min with an anti-hemagglutinin (HA) monoclonal antibody (Covance, Berkeley, CA), followed by incubation for 30 min with an anti-mouse antibody-fluorescein isothiocyanate (FITC) conjugate (Invitrogen). They were then visualized with a Zeiss LSM 510 Confocal Laser Scanning Microscope.
Western blot analysis
Yeast cellular extracts were prepared using the YeastBuster protein extraction reagent (EMD Chemicals, Inc., San Diego, CA). HEK293 cells were lysed on ice for 20 min with 1% Triton X-100 in Tris-buffered saline. Protein extracts from these lysates were subjected to SDS-PAGE using 10% or 15% gels, and Western blot analysis was performed. Antibodies against TPMT [13], ATG7 (Abgent, San Diego, CA), UBE2G2 (Abgent), and LC3 (Novus Biologicals, Littleton, CO) were used to perform the Western blots.
Data analysis
Group data were expressed as mean ± SEM and values were compared by the use of Student’s t test or by ANOVA using the “JMP” statistical package (Version 7.0, SAS Institute, Cary, NC). Genes identified as having an influence on TPMT*3A degradation and/or aggregation using the yeast deletion library were placed within functional categories on the basis of annotations provided by the Yeast Genome website (http:www.yeastgenome.org).
Results
TPMT*3A degradation and aggregation in yeast
To determine whether yeast might serve as a useful model system for the study of TPMT*3A degradation and aggregation, the human TPMT WT and TPMT*3A open reading frames (ORFs) were fused to the green fluorescent protein (GFP) ORF and were expressed in S. cerevisiae. It was reported previously that the human TPMT*3A variant allozyme was degraded much more rapidly than was the WT when expressed in yeast [19]. Consistent with those results, we observed dramatic differences in the levels of WT and TPMT*3A-GFP fusion protein, with only trace amounts of TPMT*3A-GFP detected by Western blot analysis 8 hours after expression (Fig. 1A). In addition, fluorescence from GFP-fusion protein containing WT TPMT was distributed diffusely throughout the yeast cytosol – as expected for a cytosolic protein (Fig. 1B). However, TPMT*3A-GFP fusion protein formed aggregates in a small percentage of yeast cells (<5%), with only very little background cytoplasmic fluorescence (Fig. 1B). Treatment of cells expressing the *3A-GFP variant fusion protein in the presence of the proteasome inhibitor MG132 resulted in a dramatic increase (to >40%) in the number of cells that displayed aggregates (Fig. 1B), very similar to observations made with regard to TPMT*3A aggresome formation in mammalian cells [7].
Figure 1.
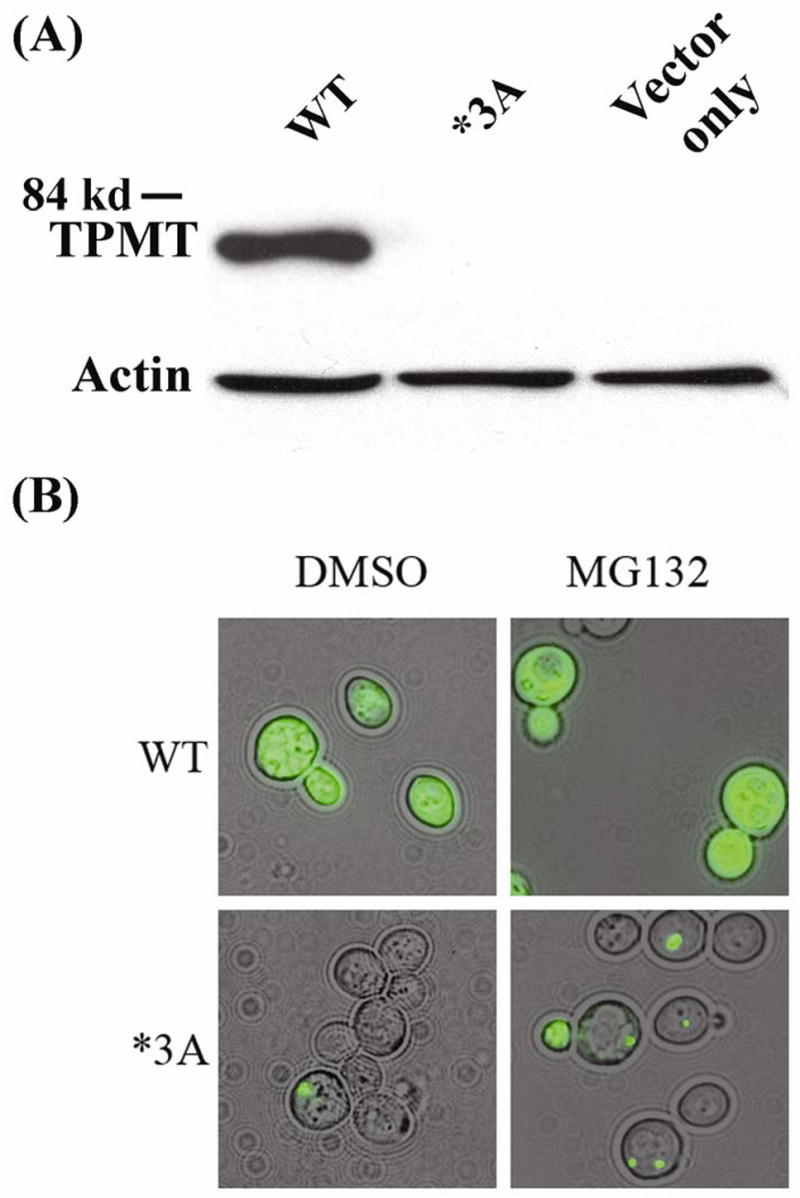
Human WT TPMT-GFP and TPMT*3A-GFP protein levels and aggregation in yeast. (A) S. cerevisiae strain W303-1A was transformed with human TPMT WT and TPMT*3A GFP fusion proteins or with an “empty” construct. Western blot analysis of cytosol from transformants was performed with anti-human TPMT antibody. Each lane was loaded with 0.25 μg of protein. The antibody was raised using a peptide present in both WT TPMT and TPMT*3A, i.e. neither of the *3A SNPs altered the amino acid sequence of the epitope used to generate the antibody. Identical results were obtained when anti-GFP was used to probe the membrane. (B) Epifluorescence microscopy of W303-1A S. cerevisiae transformed with human TPMT WT or *3A GFP fusion protein constructs. Transformants were grown for 8 h at 30°C in SC-Ura+Galactose.
Microtubule disruption and TPMT*3A aggregation in yeast
In mammalian cells, an intact microtubule cytoskeleton is required for aggresome formation and dynein, a microtubule motor, is responsible for the directed transport of misfolded proteins along microtubules to form aggresomes [25, 26]. To study the possible functional involvement of microtubules in TPMT*3A aggregation in yeast, cells expressing TPMT*3A-GFP were treated with the microtubule-depolymerizing drug benomyl, both in the presence and absence of MG132 (Fig. 2A). Virtually all benomyl-treated cells in which the TPMT*3A-GFP aggregates could be visualized exhibited numerous foci of fluorescence (Fig. 2A) as compared with cells treated with MG132 alone (see Figs. 1B and 2A), resembling TPMT*3A microaggregates that are observed in mammalian cells after microtubule disruption [7]. In contrast, only a small proportion of cells expressing TPMT*3A-GFP that were treated with DMSO or DMSO plus MG132 exhibited multiple fluorescent spots (Fig. 2A, bar graph).
Figure 2.

Microtubules, dynein and TPMT*3A-GFP aggregation. A total of 200 cells was counted for each experiment to obtain the data shown in the bar graphs. (A) W303-1A S. cerevisiae expressing TPMT*3A GFP were grown for 4 h in SC-Ura+Galactose, and were then treated with DMSO, with DMSO plus MG132, or DMSO plus the microtubule-depolarizing drug benomyl —with or without MG132. TPMT*3A-GFP aggregates were visualized with epifluorescence microscopy. (B) TUB4 wild type and a γ-tubulin temperature-sensitive (tub4-1) strain were transformed with TPMT*3A-GFP expression constructs and were then grown in SC-Ura+Galactose for 8 h at 23°C (the permissive temperature) or 37°C (the restrictive temperature). TPMT*3A-GFP aggregates were visualized using epifluorescence microscopy. (C) TPMT*3A-GFP was expressed in the parental BY4741 (WT) yeast strain and in dynein gene deletion strains (dyn1Δ, dyn2Δ, dyn3Δand pac11Δ. TPMT*3A-GFP aggregates were visualized with epifluorescence microscopy. Cells with multiple aggregates (≥2) and cells with single aggregates were counted, and the percentages of cells that exhibited multiple aggregates among cells that contained aggregates were calculated in each experiment.
The essential yeast gene TUB4 encodes γ–tubulin, a major component of the spindle pole body that is required for organizing and nucleating microtubules [27]. Temperature-sensitive (Ts) mutants of TUB4 have defects in microtubule organization and mitotic spindle formation [21]. In an attempt to confirm the results of the pharmacologic studies in which we used benomyl to disrupt microtubule function, we expressed TPMT*3A-GFP in TUB4 wild type and tub4-1 Ts mutant yeast strains at both the permissive (23°C) and restrictive (37°C) temperatures. Although, at the permissive temperature TUB4 and tub4-1 cells exhibited similar patterns of fluorescent TPMT*3A-GFP aggregates, a larger number of microaggregates was observed in tub4-1 cells at the restrictive temperature when compared with TUB4 WT cells (Fig. 2B) –supporting the results of the pharmacologic studies performed with benomyl.
Cytoplasmic dynein is a protein complex that functions as a microtubule motor by moving along microtubules in association with various cellular cargoes [28]. DYN1, DYN2, DYN3 and PAC11 encode different dynein subunits in yeast. Yeast dyn1Δ, dyn2Δ, dyn3Δ and pac11Δ mutants expressing TPMT*3A-GFP also exhibited numerous small foci of fluorescence in a large percentage of cells in comparison with the single spot of fluorescence seen in most WT yeast cells (Fig. 2C), suggesting defects in the mutant yeast cells in the transport of TPMT*3A-GFP along microtubules to form single large aggregates. These results suggested that not only the microtubule system, but also dynein was involved in TPMT*3A aggregate formation in yeast, once again paralleling results obtained in mammalian cells [7, 25].
Molecular chaperones and TPMT*3A-GFP aggregation in yeast
Molecular chaperones have profound effects on the degradation and aggregation of misfolded proteins [6, 29]. TPMT*3A in mammalian cells is associated with hsp70, hsp90 and the cochaperone hop, and hsp90 has been shown to be involved in targeting TPMT*3A for degradation and the addition of the hsp90 inhibitor geldanamycin accelerates the degradation of human WT TPMT [6]. To study the effects of molecular chaperones on TPMT*3A-GFP aggregation in yeast, TPMT*3A-GFP was expressed in hsc82Δ (an ortholog of human hsp90), ssa1Δ (an ortholog of human hsp70), ydj1Δ (an ortholog of human hsp40), sti1Δ (an ortholog of human hop) and hsp104Δ mutant strains. Deletion of these genes significantly increased TPMT*3A-GFP aggregates, quantitatively assessed on the basis of the frequency of inclusion bodies in the cells (Fig. 3A), suggesting that chaperone proteins also participate in TPMT*3A-GFP aggregation and/or clearance in yeast.
Figure 3.

Molecular chaperone, ATG and vacuolar (lysosomal) protease gene deletion and TPMT*3A-GFP aggregate formation in yeast. (A) TPMT*3A-GFP was expressed in the parental BY4741 (WT) yeast strain and molecular chaperone gene deletion strains (hsc82Δ, ssa1Δ, ydj1Δ sti1Δ and hsp104Δ. Cells were grown in SC-Ura+Galactose for 8 h at 30°C and the proportion of cells with aggregates was determined. A total of 200 cells were counted to obtain the bar graph data for each strain. Human orthologs for the deleted genes are listed in parentheses. (B) TPMT*3A-GFP was expressed in the parental WT yeast strain BY4741, in vacuolar (lysosomal) protease gene deletion strains (cps1Δ, ape3Δ) and in ATG gene deletion strains (atg3Δ, atg5Δ, atg7Δ atg8Δ atg10Δ and atg12Δ). Cells were grown for 8 h at 30°C in SC-Ura+Galactose and the proportion of cells with aggregates was determined. A total of 200 cells were counted for each strain to obtain the bar graph data. P values in both panels refer to comparisons with the WT yeast strain.
This series of experiments had demonstrated that TPMT*3A-GFP protein is rapidly degraded in yeast cells but that it is also capable of forming both large “aggresome”-like structures as well as microaggregates – similar to the situation observed in mammalian cells [7]. Furthermore, these processes – once again similar to the situation in mammalian cells [6, 7] – involve chaperone proteins, the microtubule system and dynein. Therefore, yeast appeared to be a useful model system for the study of these processes; a system for which a large number of high through-put molecular “tools” are available. We took advantage of one of those “tools” – a yeast gene deletion library – to explore the biology underlying the disposition of this pharmacogenetically important protein.
Yeast genome-wide screen for genes that participate in TPMT*3A degradation/aggregation
WT yeast cells expressing TPMT*3A-GFP exhibit very low levels of fluorescence intensity (see Fig. 1B), probably as a result of the rapid degradation of the fusion protein. We took advantage of that fact to screen a yeast gene deletion library for genes involved in the degradation of TPMT*3A-GFP. Specifically, a collection of 4,667 yeast strains that contained deletions of non-essential genes were transformed with the TPMT*3A-GFP expression construct, and flow-cytometry was used to screen for cells with high fluorescence intensity – indicating the loss of a gene essential for TPMT*3A degradation. Of the 4,667 mutants studied, we identified 24 that displayed enhanced fluorescence. Those mutant strains displayed dramatically enhanced TPMT*3A-GFP aggregation (see Supplemental Fig. S1 for examples). We also showed that enhanced TPMT*3A aggregation in these yeast mutants was not an artifact of fusion to GFP (Supplemental Fig. S2). The 24 mutant strains isolated by flow cytometry were then streaked from the original frozen stocks of the yeast gene deletion library and were confirmed by retransforming them with the TPMT*3A-GFP expression construct. The mutations in these strains involved genes that clustered within functionally-related categories, including ubiquitin-dependent protein degradation, microtubule formation, vesicle trafficking and vacuolar degradation (Table 1).
Table 1.
Mutant yeast strains that exhibited high fluorescence intensity and were selected by flow-cytometry analysis. The “Human Ortholog” column indicates yeast genes with known human orthologs.
| Strain and Pathway | Human Ortholog of Deleted Gene | Function of Deleted Gene |
|---|---|---|
| Ubiquitin- proteasome pathway | ||
| ubc7Δ | Yes | Ubiquitin conjugating enzyme |
| hul5Δ | Yes | Hect domain E3 ubiquitin-protein ligase |
| cue1Δ | No | Recruiting the ubiquitin-conjugating enzyme Ubc7p to the ER |
| ubx4Δ | No | UBX (ubiquitin regulatory X) domain-containing protein |
| ubp14Δ | Yes | Ubiquitin-specific protease |
| rpn10Δ | Yes | Non-ATPase base subunit of the 26S proteasome |
| Vesical trafficking | ||
| snc1Δ | Yes | v-SNARE involved in membrane fusion |
| syn8Δ | Yes | Endosomal SNARE related to mammalian syntaxin 8 |
| erv46Δ | Yes | Localized to COPII-coated vesicles; membrane fusion |
| prm9Δ | No | Involved in COPII binding |
| erp1Δ | Yes | Localized to COPII-coated vesicles |
| erp2Δ | Yes | Localized to COPII-coated vesicles |
| pex22Δ | No | Putative peroxisomal membrane protein |
| vps54Δ | No | Vacuolar Protein Sorting |
| snx4Δ | Yes | Sorting nexin |
| Chaperones | ||
| ssa1Δ | Yes | Member of heat shock protein 70 (hsp70) family |
| cne1Δ | Yes | Calnexin; integral membrane ER chaperone |
| Microtubule system | ||
| bud14Δ | No | Involved in bud-site selection; A regulator of dynein |
| ady3Δ | No | Interact with spindle pole body components |
| Vacuolar (lysosomal) protease | ||
| lap4Δ | Yes | Vacuolar aminopeptidase |
| Other | ||
| pca1Δ | Yes | Cadmium transporting P-type ATPase |
| Unknown | ||
| yml089cΔ | No | Unknown |
| yal045cΔ | No | Unknown |
| ydr026cΔ | Yes | Protein of unknown function that may interact with ribosomes |
Of the deleted genes identified during our selection process, more than one third encoded proteins involved in vesicular transport, vacuolar protein sorting and vacuolar import. Examples included vesicle membrane receptor proteins (SNARE) and components of the coatomer protein complex-II (COPII) (Table 1). These observations raised the possibility that, in addition to aggresome formation and degradation via a ubiquitin-proteasome-mediated process, autophagy might also play a role in TPMT*3A-GFP degradation. Several recent studies have suggested that the ubiquitination of defective proteins may either target those proteins to the proteasome or, when aggregation can occur, to the autophagy system [30, 31].
Autophagy is a highly conserved process in eukaryotes in which cytoplasm is sequestered into vesicles and is then delivered to vacuoles/lysosomes for breakdown [32]. ATG genes, such as ATG7 and ATG12, encode critical components required for the biogenesis of autophagic vesicles [33]. To determine whether autophagy might suppress TPMT*3A aggregate accumulation and, as a result, whether the mutation of genes important for autophagy might enhance aggregate accumulation, TPMT*3A-GFP was expressed in the yeast atg mutants atg3Δ, atg5Δ, atg7Δ, atg8Δ, atg10Δ and atg12Δ as well as the vacuolar (lysosomal) protease mutants cps1Δ and ape3Δ. The frequency of cells containing aggregated TPMT*3A-GFP was dramatically increased in these mutants (Fig. 3B), indicating that, in yeast, autophagy appeared to be involved in the degradation of TPMT*3A-GFP and/or the clearance of TPMT*3A-GFP aggregates.
Yeast genetic screen and mechanisms of TPMT*3A mammalian cell degradation
We next examined the relevance of selected genes identified using the yeast genetic screen, but now moving to studies performed with mammalian cells. The UBC7 gene in yeast encodes an E2 ubiquitin-conjugating enzyme [34], and expression of TPMT*3A-GFP in a ubc7Δ mutant strain resulted in dramatically enhanced aggregation (Supplemental Fig. S1). Therefore, we “knocked down” UBE2G2, the human ortholog for UBC7, in human embryonic kidney HEK293 cells (Fig. 4A), and observed enhanced formation of large TPMT*3A aggregates in these cells (Fig. 4B), suggesting that use of the yeast genetic screen was a relevant approach for identifying mammalian genes that participate in TPMT*3A degradation. Furthermore, when similar experiments were performed with TPMT*3C, a variant that, unlike TPMT*3A, fails to display either dramatically enhanced degradation or aggregate formation, we did not see evidence of enhanced aggregate formation after UBE2G2 knockdown (Fig. 4B). However, Western blot analysis did show increased levels of TPMT*3C protein levels after knockdown of this E2 conjugating enzyme (Fig. 4C).
Figure 4.

TPMT*3A degradation and aggregation in mammalian cells. (A) Immunoblot analysis of UBE2G2 in HEK293 cells transiently transfected with UBE2G2 siRNA. The bar graph shows quantification of the Western blot data (N = 3). (B) Loss of UBE2G2 resulted in increased TPMT*3A aggregation in HEK293 cells but not for WT TPMT or TPMT*3C. HEK293 cells expressing HA-tagged WT TPMT, TPMT*3A or TPMT*3C were transfected with control siRNA or siRNA directed against UBE2G2 and were then stained with anti-HA antibody. Large TPMT*3A aggregates were visualized with confocal epifluorescence microscopy. The bar graph data shows data for aggregate formation. A total of 200 cells were counted in each experiment (N = 3). (C) Immunoblot analysis of WT TPMT, TPMT*3C and TPMT*3A with or without siRNA knockdown of UBE2G2.
To test the hypothesis that autophagy might participate in TPMT*3A degradation in mammalian cells, we expressed TPMT*3A in HEK293 cells and examined the appearance of the phosphatidylethanolamine-conjugated form of LC3, the human homologue of yeast ATG8. During the formation of autophagosomes, LC3 is cleaved to generate a soluble form known as LC3-I [35]. LC3-I, in turn, is lipidated by mammalian ATG7 and ATG3 homologues to form LC3-II that localizes to autophagosomes and autolysomes [35]. Therefore, LC3-II quantity is a marker for autophagosome formation [35]. Cells expressing TPMT*3A accumulated more LC3-II than did cells expressing only GFP (Fig. 5A). We also treated the cells with 3-methyladenine, an inhibitor of autophagy, or with NH4Cl, an inhibitor of lysosomal protein degradation [36, 37]. The Western blots in Fig. 5B show that both treatments inhibited TPMT*3A degradation. We next examined the possible contribution of autophagy to TPMT*3A clearance in mammalian cells by siRNA knockdown of ATG7 (Fig. 5C), an essential component of the mammalian autophagy pathway. Inhibition of autophagy by transfecting cells with siRNA directed against ATG7 dramatically increased the number of microaggregates in HEK293 cells expressing TPMT*3A (Fig. 5D) – but not those expressing TPMT*3C – suggesting that autophagy may also play a role in the degradation and/or clearance of TPMT*3A aggregates in mammalian cells.
Figure 5.

Autophagy and TPMT WT, TPMT*3A and TPMT*3C degradation and aggregation. (A) Immunoblot analysis of LC3 in HEK293 cells expressing GFP or TPMT*3A. (B) HA-tagged WT TPMT, TPMT*3A and TPMT*3C were transiently expressed in HEK293 cells. The cells were treated with 10 mM 3-methyladenosine (an autophagy inhibitor), 50 mM NH4Cl (a lyosomal inhibitor) or water for 48 h. TPMT allozymes were detected with an anti-HA antibody. The blot on the left did not include TPMT*3A without inhibition of autophagy, while the blot on the right did. (C) Immunoblot analysis of ATG7 in HEK293 cells transiently transfected with ATG7-specific siRNA treatment. The bar graph shows quantification of the Western blot densities (N = 3). (D) Enhanced TPMT*3A aggregation in HEK293 cells after ATG7-specific siRNA treatment. HEK293 cells expressing HA-tagged WT TPMT, TPMT*3A or TPMT*3C were transfected with control siRNA or siRNA directed against ATG7 and were stained with anti-HA antibody. Aggregates were visualized with confocal epifluorescence microscopy. A total of 200 cells were counted in each experiment. The bar graph shows microaggregate formation for cells transfected with WT TPMT, TPMT*3A and TPMT*3C after ATG7 knockdown (N = 3).
Discussion
Our studies demonstrated dramatic differences in TPMT protein levels when WT and TPMT*3A-GFP fusion constructs were expressed in yeast, with only trace quantities of detectable TPMT*3A-GFP (Fig. 1A), consistent with previous reports [19]. We also observed that TPMT*3A-GFP aggregates were formed in yeast cells (Fig. 1B), aggregates with characteristics similar to those of TPMT*3A aggresomes in mammalian cells [6]. In addition, deletion of yeast molecular chaperone genes greatly enhanced TPMT*3A aggregation (Fig. 3A), paralleling results obtained with mammalian cells [7]. We also demonstrated that both pharmacological and gene deletion-induced disruption of the microtubule-dynein system resulted in a shift from single large aggregates to the formation of numerous microaggregates (Fig. 2) – also reflecting results obtained during our previous studies performed with mammalian cells [7]. We then used a yeast gene deletion panel to identify 24 candidate genes that might participate in TPMT*3A aggregation and/or degradation (Table 1). Those results demonstrated that the use of a yeast gene deletion panel can be a powerful tool for the identification of candidate genes encoding proteins that participate in pathways involved in the recognition of misfolded proteins as well as their trafficking and targeting for degradation and/or aggregation.
Nonsynonymous SNPs are a common type of gene sequence variation associated with the functional effects of pharmacologically relevant genes [3]. Altered protein level is a common explanation for the functional effect of this type of genetic polymorphism [3, 12], and, in many cases, accelerated protein degradation is the underlying mechanism [1, 5, 6]. It is known that the rapid degradation of TPMT*3A in mammalian cells involves ubiquitination of the variant allozyme, followed by proteasome-mediated degradation [5, 6, 19]. Our yeast gene deletion library studies resulted in the identification of genes encoding multiple components of this pathway, including E2 ubiquitin conjugating enzymes, E3 ubiquitin ligases and subunits of the proteasome. However, an unanticipated finding was the observation that efficient TPMT*3A clearance also requires autophagy.
Recent data from studies of neurodegenerative diseases associated with aggregation-prone proteins such as mutant huntingtin with polyglutamine-expansions or mutant forms of α-synuclein have shown that both proteasome and autophagy-dependent processes participate in the clearance of these proteins [38]. Those studies also demonstrated that dependence on the autophagy pathway for clearance is correlated with the tendency of these proteins to aggregate [38]. Furthermore, recent studies suggest that there is a dynamic balance between the ubiquitin-proteasome system and autophagy, and that HDAC6, a microtubule-associated deacetylase that interacts with polyubiquitinated proteins, is an essential mechanistic link in this interaction [39, 40]. We demonstrated previously that HDAC6 is involved in TPMT*3A aggresome formation in mammalian cells [7]. Therefore, autophagy might represent an important route for the clearance of TPMT*3A aggregates and/or aggregate precursors. It remains unclear whether autophagy degrades only soluble species and oligomers, or whether it can also directly clear larger aggregates. In the neurons of mice with neuronally restricted autophagy-gene knockouts, wild type cellular proteins that are not usually aggregate-formation prone will form inclusions, suggesting that aggregates can arise from the failure of autophagy to clear soluble proteins [30, 31]. Exactly how TPMT*3A is selected for targeting for autophagy-dependent degradation is currently unknown, but should be the subject for future studies.
Autophagy may be compromised by means other than defects in ATG proteins. Recent studies suggest a role for microtubules in autophagy [41–43]. HDAC6 and microtubules are required for the autophagic degradation of aggregated huntingtin [41], and both HDAC6 and microtubules are required for TPMT*3A aggresome formation [7]. Dynein dysfunction retards the clearance of aggregation-prone proteins like mutant huntingtin by reducing autophagosome-lysosome fusion [42]. As mentioned previously, HDAC6, dynein and microtubules are all involved in aggresome formation – including the formation of TPMT*3A aggresomes [7, 25, 26, 44]. Furthermore, it has been suggested that aggresome formation and autophagic sequestration could be cytoprotective pathways involved in the clearance of toxic aggregation-prone proteins such as those containing long polyglutamine repeats [43, 45]. In the present study, we observed that the disruption of microtubules or interference with the function of dynein in yeast decreased aggresome formation but increased the formation of small aggregates (Fig. 2), observations which could help to explain the impaired clearance of TPMT*3A aggregates through autophagy. The genes identified during our yeast genome-wide screen and the genes that we tested using gene deletion strains clustered into several functionally-related categories, including ubiquitin-proteasome-mediated protein degradation, molecular chaperones, the microtubule system, vesicle trafficking, vacuolar degradation and the biogenesis of autophagic vesicles (Table 1), suggesting that autophagy should be considered among mechanisms responsible for the effects of pharmacogenetically significant polymorphisms that alter encoded amino acids.
Finally, we also observed that TPMT*3C, the most common variant allozyme in East Asian populations [20], behaves differently from TPMT*3A, the most common variant allozyme in Caucasians [1]. Those observations were not surprising on the basis of a great deal of evidence which indicates that TPMT*3A is much more highly misfolded than TPMT*3C [6, 7], with numerous functional consequences, including those reported here. TPMT*3C also displays a difference in behavior from TPMT*3A in that it is rapidly degraded in mammalian cells but not in yeast [5–7], unlike TPMT*3A which is rapidly degraded in both. Whether that difference is due to the striking differences between these two variant allozymes in their degree of aggregation and misfolding is unclear, but it is clear that caution must be exercised in extrapolating functional observations from one species to another.
In summary, our observations support the conclusion that a dynamic balance appears to exist among protein folding, protein degradation and protein aggregation for TPMT*3A. That balance can be shifted either by mutations or by interventions that alter the cellular environment. For example, inhibition of proteasome or autophagy-mediated degradation results in a shift toward aggregate formation. Finally, our use of a yeast genetic screen demonstrates that this approach might be applied to study molecular mechanisms involved in the degradation of many unstable proteins – including other proteins of pharmacogenomic significance. This series of observations also has important implications for our understanding of cellular mechanisms in pharmacogenomics, mechanisms that contribute to inherited variation in drug response phenotypes.
Supplementary Material
Acknowledgments
We thank Elmar Schiebel for providing the S. cerevisiae strain ESM208.
Supported in part by National Institutes of Health (NIH) grants R01 GM28157, R01 GM35720 and U01 GM61388 (The Pharmacogenetics Research Network) (to R.M.W.), an Astella-ASPET Award (to L.W.), a Pancreatic SPORE Development Award (to L.W.) and by a PhRMA
Abbreviations
- TPMT
Thiopurine S-methyltransferase
- TPMT*3C
TPMT*3A
- SNP
single nucleotide polymorphism
- FDA
Food and Drug Administration
- S AdoMet
-adenosyl-L-methionine
- WT
wild type
- ORF
open reading frame
- GFP
green fluorescent protein
References
- 1.Wang L, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: insights, challenges and future directions. Oncogene. 2006;25:1629–38. doi: 10.1038/sj.onc.1209372. [DOI] [PubMed] [Google Scholar]
- 2.Weinshilboum R. Inheritance and drug response. N Engl J Med. 2003;348:529–37. doi: 10.1056/NEJMra020021. [DOI] [PubMed] [Google Scholar]
- 3.Weinshilboum R, Wang L. Pharmacogenetics: inherited variation in amino acid sequence and altered protein quantity. Clin Pharmacol Ther. 2004;75:253–8. doi: 10.1016/j.clpt.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Weinshilboum RM, Wang L. Pharmacogenetics and pharmacogenomics: development, science, and translation. Annu Rev Genomics Hum Genet. 2006;7:223–45. doi: 10.1146/annurev.genom.6.080604.162315. [DOI] [PubMed] [Google Scholar]
- 5.Tai HL, Fessing MY, Bonten EJ, Yanishevsky Y, d’Azzo A, Krynetski EY, et al. Enhanced proteasomal degradation of mutant human thiopurine S-methyltransferase (TPMT) in mammalian cells: mechanism for TPMT protein deficiency inherited by TPMT*2, TPMT*3A, TPMT*3B or TPMT*3C. Pharmacogenetics. 1999;9:641–50. doi: 10.1097/01213011-199910000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Wang L, Sullivan W, Toft D, Weinshilboum R. Thiopurine S-methyltransferase pharmacogenetics: chaperone protein association and allozyme degradation. Pharmacogenetics. 2003;13:555–64. doi: 10.1097/01.fpc.0000054124.14659.99. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Nguyen TV, McLaughlin RW, Sikkink LA, Ramirez-Alvarado M, Weinshilboum RM. Human thiopurine S-methyltransferase pharmacogenetics: variant allozyme misfolding and aggresome formation. Proc Natl Acad Sci U S A. 2005;102:9394–9. doi: 10.1073/pnas.0502352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.US Department of Health and Human Services Food and Drug Administration. US Department of Health and Human Services Food and Drug Administration. 2003.
- 9.Remy CN. Metabolism of thiopyrimidines and thiopurines. S-Methylation with S-adenosylmethionine transmethylase and catabolism in mammalian tissues. J Biol Chem. 1963;238:1078–84. [PubMed] [Google Scholar]
- 10.Woodson LC, Weinshilboum RM. Human kidney thiopurine methyltransferase. Purification and biochemical properties. Biochem Pharmacol. 1983;32:819–26. doi: 10.1016/0006-2952(83)90582-8. [DOI] [PubMed] [Google Scholar]
- 11.Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43:329–39. doi: 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- 12.Salavaggione OE, Wang L, Wiepert M, Yee VC, Weinshilboum RM. Thiopurine S-methyltransferase pharmacogenetics: variant allele functional and comparative genomics. Pharmacogenet Genomics. 2005;15:801–15. doi: 10.1097/01.fpc.0000174788.69991.6b. [DOI] [PubMed] [Google Scholar]
- 13.Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–62. [PMC free article] [PubMed] [Google Scholar]
- 14.Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM. Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J Pediatr. 1991;119:985–9. doi: 10.1016/s0022-3476(05)83063-x. [DOI] [PubMed] [Google Scholar]
- 15.Lennard L, Van Loon JA, Weinshilboum RM. Pharmacogenetics of acute azathioprine toxicity: relationship to thiopurine methyltransferase genetic polymorphism. Clin Pharmacol Ther. 1989;46:149–54. doi: 10.1038/clpt.1989.119. [DOI] [PubMed] [Google Scholar]
- 16.Schaeffeler E, Fischer C, Brockmeier D, Wernet D, Moerike K, Eichelbaum M, et al. Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics. 2004;14:407–17. doi: 10.1097/01.fpc.0000114745.08559.db. [DOI] [PubMed] [Google Scholar]
- 17.Schutz E, Gummert J, Mohr F, Oellerich M. Azathioprine-induced myelosuppression in thiopurine methyltransferase deficient heart transplant recipient. Lancet. 1993;341:436. doi: 10.1016/0140-6736(93)93028-y. [DOI] [PubMed] [Google Scholar]
- 18.Szumlanski C, Otterness D, Her C, Lee D, Brandriff B, Kelsell D, et al. Thiopurine methyltransferase pharmacogenetics: human gene cloning and characterization of a common polymorphism. DNA Cell Biol. 1996;15:17–30. doi: 10.1089/dna.1996.15.17. [DOI] [PubMed] [Google Scholar]
- 19.Tai HL, Krynetski EY, Schuetz EG, Yanishevski Y, Evans WE. Enhanced proteolysis of thiopurine S-methyltransferase (TPMT) encoded by mutant alleles in humans (TPMT*3A, TPMT*2): mechanisms for the genetic polymorphism of TPMT activity. Proc Natl Acad Sci U S A. 1997;94:6444–9. doi: 10.1073/pnas.94.12.6444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Collie-Duguid ES, Pritchard SC, Powrie RH, Sludden J, Collier DA, Li T, et al. The frequency and distribution of thiopurine methyltransferase alleles in Caucasian and Asian populations. Pharmacogenetics. 1999;9:37–42. doi: 10.1097/00008571-199902000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Spang A, Geissler S, Grein K, Schiebel E. gamma-Tubulin-like Tub4p of Saccharomyces cerevisiae is associated with the spindle pole body substructures that organize microtubules and is required for mitotic spindle formation. J Cell Biol. 1996;134:429–41. doi: 10.1083/jcb.134.2.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cormack BP, Bertram G, Egerton M, Gow NA, Falkow S, Brown AJ. Yeast-enhanced green fluorescent protein (yEGFP)a reporter of gene expression in Candida albicans. Microbiology. 1997;143(Pt 2):303–311. doi: 10.1099/00221287-143-2-303. [DOI] [PubMed] [Google Scholar]
- 23.Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. Methods Enzymol. 1991;194:3–933. [PubMed] [Google Scholar]
- 24.Muchowski PJ, Ning K, D’Souza-Schorey C, Fields S. Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proc Natl Acad Sci U S A. 2002;99:727–32. doi: 10.1073/pnas.022628699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnston JA, Illing ME, Kopito RR. Cytoplasmic dynein/dynactin mediates the assembly of aggresomes. Cell Motil Cytoskeleton. 2002;53:26–38. doi: 10.1002/cm.10057. [DOI] [PubMed] [Google Scholar]
- 26.Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143:1883–98. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sobel SG, Snyder M. A highly divergent gamma-tubulin gene is essential for cell growth and proper microtubule organization in Saccharomyces cerevisiae. J Cell Biol. 1995;131:1775–88. doi: 10.1083/jcb.131.6.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamamoto A, Hiraoka Y. Cytoplasmic dynein in fungi: insights from nuclear migration. J Cell Sci. 2003;116:4501–12. doi: 10.1242/jcs.00835. [DOI] [PubMed] [Google Scholar]
- 29.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–51. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 30.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 31.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 32.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(Suppl 2):1542–52. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–6. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 34.Jungmann J, Reins HA, Schobert C, Jentsch S. Resistance to cadmium mediated by ubiquitin-dependent proteolysis. Nature. 1993;361:369–71. doi: 10.1038/361369a0. [DOI] [PubMed] [Google Scholar]
- 35.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Field MC, Moran P, Li W, Keller GA, Caras IW. Retention and degradation of proteins containing an uncleaved glycosylphosphatidylinositol signal. J Biol Chem. 1994;269:10830–7. [PubMed] [Google Scholar]
- 37.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 39.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 40.Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy. 2007;3:643–5. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- 41.Iwata A, Riley BE, Johnston JA, Kopito RR. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280:40282–92. doi: 10.1074/jbc.M508786200. [DOI] [PubMed] [Google Scholar]
- 42.Ravikumar B, Acevedo-Arozena A, Imarisio S, Berger Z, Vacher C, O’Kane CJ, et al. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat Genet. 2005;37:771–6. doi: 10.1038/ng1591. [DOI] [PubMed] [Google Scholar]
- 43.Webb JL, Ravikumar B, Rubinsztein DC. Microtubule disruption inhibits autophagosome-lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol. 2004;36:2541–50. doi: 10.1016/j.biocel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 44.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 45.Taylor JP, Tanaka F, Robitschek J, Sandoval CM, Taye A, Markovic-Plese S, et al. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum Mol Genet. 2003;12:749–57. doi: 10.1093/hmg/ddg074. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.