

Abstract
Cellular membranes obtained from the 1321N1 and A172 astrocytoma cell lines were immobilized on a chromatographic phase to create cellular membrane affinity chromatography (CMAC) columns, CMAC(1321N1) and CMAC(A172). The columns were characterized using frontal affinity chromatography with [3H]-epibatidine as the marker ligand and epibatidine, nicotine, and methyllycaconitine as the displacers. The results indicated that the columns contained homomeric α7 nicotinic acetylcholine receptors (α7 nAChR) and heteromeric nicotinic acetylcholine receptors (αxβy nAChRs), which was confirmed by the addition of subtype-specific inhibitors, κ-bungarotoxin (α7 nAChR) and K-bungarotoxin (αxβy nAChR) to the mobile phase. The presence of two additional ligand-gated ion channels (LGICs), γ-aminobutyric acid (GABAA) and N-methyl-d-aspartic acid (NMDA), was established using frontal affinity chromatography with flunitrazepam and diazepam (GABAA receptor) and MK-801 and NMDA (NMDA receptor). The presence of the four LGICs was confirmed using confocal microscopy and flow cytometry. The results indicate that the CMAC(1321N1) and CMAC(A172) columns contain four independently functioning LGICs, that the columns can be used to characterize binding affinities of small molecules to each of the receptors, and that the CMAC approach can be used to probe the expression of endogenous membrane receptors.
Gliomas are a family of malignant tumors found in the adult central nervous system, which originate from three different glial elements.1 One of these elements, astrocytic tumors, includes astrocytomas, anaplasic astrocytomas, and glioblastoma multiforma.1 Although the genetics and cellular biology of malignant gliomas have been extensively studied, cf. refs 1 and 2, current treatment options are limited and the patient's prognosis is poor.
A possible therapeutic target for the treatment of malignant gliomas is the homomeric α7 nicotinic acetylcholine receptor (α7 nAChR). This receptor has been associated with the growth and metastasis of malignant neuroendocrine neoplasms such as small cell lung carcinomas,3-5 and the α7 nAChR antagonist κ-cobratoxin has been shown to reduce tumor growth in nonsmall cell lung cancer tumors engrafted in nude mice.6 While functional α7 nAChRs have been identified in cultured rat astrocytes7 and in autopsy samples from patients with Alzheimer's disease,8,9 to our knowledge, α7 nAChRs have not been identified or studied in astrocytomas.
One approach to the study of α7 nAChR expression in astrocytoma cell lines is cellular membrane affinity chromatography (CMAC) employing columns containing membrane fragments from the target cells. We have recently reported the development of CMAC(α7 nAChR) columns obtained using transfected cell lines that stably express α7 nAChRs and demonstrated that these columns could be used to study agonist-receptor binding.10 We have also recently demonstrated that CMAC columns could be created using membrane fragments from a 1321N1 astrocytoma cell line stably transfected with cDNA for the purinergic P2Y1 receptor and used to study interactions with the expressed receptor.11 Thus, it appeared likely that CMAC columns created using astrocytoma cell lines could be used in the characterization of α7 nAChR endogenously expressed in these cells. The results from the chromatographic studies using CMAC columns created from the 1321N1 and A172 astrocytoma cell lines, CMAC(1321N1) and CMAC(A172) columns, are reported below.
The data from these studies indicated that the 1321N1 and A172 cell lines express functional α7 nAChRs. The chromatographic data was supported by confocal microscopy and flow cytometry. This represents the first report of CMAC columns containing an endogenously expressed human neuronal nAChR. The results from the chromatography studies suggested that the cell lines also expressed heteromeric nAChRs, (αxβy nAChRs), which was confirmed using subtype selective inhibitors. The data indicate that CMAC(1321N1) and CMAC(A172) columns can be used for the direct online determination of the relative affinities of ligands to homomeric and heteromeric nAChRs, which represents an improvement over the current approach which uses multiple competitive membrane binding studies.12
nAChRs are members of the ligand-gated ion channel (LGIC) superfamily that also includes the γ-aminobutyric acid (GABAA) and N-methyl-d-aspartic acid (NMDA) receptors.13 The presence of functional GABAA receptors in human-derived astrocytomas and glioblastoma multiformas has been reported, while the presence of NMDA receptors in these tumors has been suggested but not proven.2 In the current studies, the presence of functional NMDA and GABAA receptors on the CMAC(1321N1) and CMAC(A172) columns was confirmed using competitive displacement chromatography, confocal microscopy, and flow cytometry.
The results of this study demonstrate that the CMAC columns contain four functional LGICs that are endogenously expressed in the 1321N1 and A172 cell lines and that these receptors independently bind known ligands. The data suggest that the CMAC columns can be used to identify and characterize ligands which bind to one or more of the LGICs as well as study the expression of these receptors.
MATERIALS AND METHODS
Materials
Aprotinin, 2-mercaptoethanol, benzamidine, leupeptin, tosyl-amido-2-phenylethyl-chloromethyl ketone (TPCK), phenylmethanesulfonyl fluoride (PMSF), cholate, glycerol, HEPES buffer, TRIS buffer, ethylenediaminetetraacetic acid (EDTA) ammonium acetate, NaCl, MgCl2, CaCl2, ethylene glycol-bis (2-aminoethylether) N,N,N′,N′ tetraacetic acid (EGTA), epibatidine (EB), methyllycaconitine (MLA), nicotine (NIC), flunitrazepam (FTZ), diazepam (DAZ), MK-801, N-methyl-d-aspartic acid (NMDA), α-bungarotoxin (α-BTx), fluorescent isothiocyanate-labeled α-bungarotoxin (FITC-α-BTx), l-glutamate, glycine, paraformaldehyde, and sodium acetate were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). HPLC grade ammonium acetate and 0.1 M ammonium hydroxide solution were purchased from Fisher Scientific (Pittsburgh, PA). κ-Bungarotoxin (κ-BTx) was obtained from Biotoxins (St. Cloud, FL). [3H]-EB (9.1 mCi/mol), [3H]-FTZ (87 mCi/mol), and [3H]-MK-801 (20 mCi/mol) were purchased from Amersham Life Science Products (Boston, MA). IAM liquid chromatographic stationary phase (IAM-PC, 12 μm, 300 Å) was purchased from Regis Chemical Co. (Morton Grove, IL).
Cell Lines
The human 1321N1 astrocytoma cell line was obtained from European Collection of Cell Cultures (Sigma-Aldrich). The A172 astocytoma cell line was obtained from American Tissue Culture Collection (ATCC, Manassas, VA). The cells were seeded in either T-150 culture flasks (Fisher Scientific) or glass bottom wells (Mat Tek Corp, Ashland, MA) with Dulbecco's modified Eagle's medium (DMEM) containing 4 mM glutamine and 4.5 g/L glucose (Mediatech, Inc., Herndon, VA) supplemented with 10% fetal bovine serum (Fetal Clone III, Hyclone, Logan, UT), penicillin (100 U/mL, Mediatech), streptomycin (100 μg/mL, Mediatech) and maintained at 37 °C in a humidified atmosphere containing 5% CO2. In T-150 flasks, cells were subcultured or harvested for experiments at ∼90% confluence and the medium was replaced at 3−4 day intervals. In glass bottom wells, the cells were seeded at 2 × 105 cells/well and used at ∼50% confluence.
Preparation of CMAC(1321N1) and CMAC(A172) Columns
Cellular membrane fragments from the 1321N1 and A172 cell lines were immobilized on the IAM stationary phase as previously described.10,11 In brief, 107 cells were homogenized for 3 × 30 s at the setting of 11 on a model PT-2100 homogenizer (Kinematica, Luzern, Switzerland) in 10 mL of homogenization buffer. As previously demonstrated, the composition of the buffer is dependent upon the type of cell and the target protein.10 When 1321N1 cells were utilized, the solubilization buffer was composed of HEPES buffer [20 mM, pH 8.0] containing 500 mM NaCl, 5 mM 2-mercaptoethanol, 100 μM benzamidine, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 50 μg/mL TPCK, and 100 μg/mL PMSF. When A172 cells were utilized, the solubilization buffer was composed of HEPES buffer [20 mM, pH 7.5] containing 150 mM NaCl, 1.5 mM MgCl2, 1 mM CaCl2, 1 mM EGTA, 1 μg/mL aprotinin, 10 μg/mL leupeptin, and 1 mM PMSF. The resulting homogenate was centrifuged at 700g for 5 min, the pellet was discarded, and the supernatant was centrifuged at 100 000g for 30 min at 4 °C. The resulting pellet was suspended in 10 mL of solubilization buffer supplemented with 2% (w/v) cholate and 10% glycerol. The resulting mixture was rotated at 150 rpm using an orbital shaker for 18 h at 4 °C and centrifuged at 100 000g for 25 min. The supernatant was collected and added to 200 mg of IAM particles, and the resulting mixture was rotated at room temperature for 1 h at 150 rpm and then dialyzed for 1 day using HEPES buffer [20 mM, pH 8.0] containing 500 mM NaCl and 1 mM EDTA (1321N1 cells) or HEPES buffer [20 mM, pH 7.5] containing 150 mM NaCl and 1 mM EGTA (A172 cells). The suspension was centrifuged for 3 min at 4 °C at 700g, and the pellet was washed with ammonium acetate [10 mM, pH 7.4], centrifuged, and packed into an HR 5/2 column (Amersham Pharmacia Biotech, Uppsala, Sweden) to yield a 150 × 5-mm (i.d.) chromatographic bed.
Chromatographic Studies
The CMAC columns were placed in a chromatographic system consisting of a LC-10AD isocratic HPLC pump (Shimadzu, Columbia, MD), a 50-mL sample super-loop (Amersham Pharmacia Biotech), and an IN/US systemβ-ram model 3 online scintillation detector (IN/US, Tampa, FL) with a dwell time of 2 s and running Laura Lite 3 software. The mobile phases used in the study were ammonium acetate [10 mM, pH 7.4] (nAChR studies); ammonium acetate [10 mM, pH 7.4] containing 5 nM α-BTx or 1 nM κ-BTx (homomeric and heteromeric nAChR studies, respectively); Tris [50 mM, pH 7.4] (GABAA); Tris [50 mM, pH 7.4] containing 1 μM l-glutamate and 1 μM glycine (NMDA). The mobile phase was delivered at 0.2 mL/min at room temperature.
The competitive displacement studies were conducted using frontal chromatographic techniques as previously described.14,15 For nAChR studies, the marker ligand was [3H]-EB (60 pM) and the displacers were EB (7.5−150 pM), MLA (1−40 pM) and NIC (10−200 nM). For GABAA receptor studies, the marker ligand was [3H]-FTZ (25 pM) and the displacers were FTZ (0.5−5 nM) and DAZ (0.1−10 nM). For NMDA receptor studies, the marker ligand was [3H]-MK-801 (50 pM) and the displacers were NMDA (0.2−5 nM) and MK-801 (0.5−200 nM).
Data Analysis
The dissociation constants for the marker (KdM) and displacer ligands (Kd) were calculated using a previously described approach.14,15 In brief, the dissociation constants of the displacer ligand (Kd), as well as the number of the active binding sites of the immobilized LGIC receptor (P) can be calculated using eq 1
| (1) |
where [D] is the concentration of displacer ligand, V is the retention volume of the displacer ligand, and Vmin is the retention volume of the displacer ligand when the specific interaction is completely suppressed. From the plot of [D](V - Vmin) versus [D], the dissociation constant values (Kd), for the displacer ligand can be obtained. The data was analyzed by nonlinear regression with a sigmoidal response curve using Prism 4 software (Graph Pad Software Inc., San Diego, CA) running on a personal computer.
Confocal Microscopy Studies
Confocal images were taken with an LSM410 confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY) using an excitation wavelength of 488 nm from an argon laser and a 515−540 nm emission filter. Histograms comparing average intensity versus frequency of the confocal images were constructed using Zeiss LSM 510 Meta software. The α7 nAChR and GABA studies were conducted by slight modifications of previously described procedures,10 see Supporting Information for details.
The NMDA studies were carried out using a previously described indirect method,16 which had been modified for use in this study. In brief, cultured wells were washed and permeabilized in 0.2% Triton X-100 in PBS for 5 min, washed twice with 1 mL of PBS, and 1 mL of 10% normal horse serum (NHS) containing 0.05% sodium azide was added and the solution left at room temperature for 1 h. The wells were emptied, and 100 μL of NHS containing a 1:50 dilution of mouse anti-NMDA R1 (Invitrogen, Carlsbad, CA) was added; the samples were incubated at room temperature for 15 min and washed three times with 1 mL of PBS (5 min between washings), and the wells were emptied. A 100 μL aliquot of 10% NHS containing 0.05% sodium azide was added and, where indicated, a 1:200 dilution of horse fluorescin conjugated antimouse IgG (Vector Laboratories, Burlingame, CA) was added, and the samples were incubated in the dark for 15 min at room temperature and washed three times with 1 mL of PBS, and 1 mL of PBS was added followed by immediate fluorescent confocal examination.
Flow Cytometry
The cell suspensions were analyzed for forward and side scatter measurements with a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA) using an excitation wavelength of 488 nm from an argon laser and a 530 nm bandpass filter emission collection lens. From each sample, 104 cells were analyzed and gated to exclude debris.
The α7 nAChR was investigated as follows: Culture flasks were washed once with 1X PBS, and the cells were detached using a scraper. The suspension was centrifuged for 5 min at 170g, the supernatant removed, and the cellular pellet suspended in 10 mL of 1X PBS. A 3 × 106 cells aliquot was transferred to a new tube and centrifuged at 170g for 5 min; the supernatant was removed, and the pellet was suspended in 1 mL of 1X PBS containing 1% paraformaldehyde (fixation buffer) and left at room temperature for 20 min. The mixture was centrifuged at 170g for 5 min, the supernatant removed, the cells washed with 1 mL of 1X PBS and centrifuged at 170g for 5 min. The wash was repeated, and the cell pellet was suspended in 600 μL of 1X PBS, and 198 μL aliquots (∼1−106 cells per sample) were placed in 2 mL eppendorf tubes. Samples were either preincubated by adding 1 mM EB (2 μL of 100 mM stock solution) or 2 μL of 1X PBS to yield a final volume of 200 μL per sample. Cells were manually mixed and incubated on ice for 30 min, then stained with 1.2 μL of FITC-α-BTx [1.2 μg/mL in 1X PBS], manually mixed, and incubated on ice for 30 min.
The GABAA and NMDA receptors were studied in similar manner, and the procedures are presented in the Supporting Information.
RESULTS
Frontal Chromatography Studies
Cellular membrane fragments obtained from the 1321N1 and A172 astrocytoma cell lines were used to create the CMAC(1321N1) and CMAC(A172) columns used in this study, and as in previous studies, cf. ref 10, no attempt was made to isolate or purify the LGIC receptors. Instead, the immobilized receptors were identified and characterized using frontal displacement chromatography.
The initial chromatographic experiments examined the effect of serial mobile phase concentrations of the displacer ligands EB, NIC, and MLA on the chromatographic retention of the nAChR marker ligand [3H]-EB. All of the displacers produced concentration-dependent decreases in [3H]-EB retention, cf. Figure S-1 in the Supporting Information, and the data was used to calculate the affinities, Kd values, of the displacers for the immobilized nAChR, Table 1.
Table 1.
Chromatographically Determined Kd Values (Kd(Exp)) for the nAChR Ligands Epibatidine, Methyllycaconitine, and Nicotine, the GABAA Receptor Ligands, Flunitrazepam and Diazepam, and the NMDA Receptor Ligands, MK-801 and N-Methyl-D-Aspartic Acid, Determined on the CMAC(1321N1) and CMAC(A172) Columnsa
| column | ligand | Kd (Exp) MP [nM] | Kd (Lit) [nM] | Kd (Exp) MP + α-BTx [nM] | Kd(Exp) MP + κ-BTx [nM] |
|---|---|---|---|---|---|
| CMAC(1321N1) | |||||
| epibatidine | 0.25 | 0.64−0.82 (α7)b 0.005−0.16 (αxβy)c | 0.0086 | 0.467 | |
| methyllycaconitine | 7.3 | 2.2d | NA | 2.0 | |
| nicotine | 154 | 5600−7600 (α7)b 0.40−440 (αxβy)c | 82.4 | 4,570 | |
| flunitrazepam | 1.05 | 1.3e | |||
| diazepam | 3.21 | 1.0e | |||
| N -methyl-D-aspartic acid | 1.67 | 0.6−1.2e | |||
| MK801 | 9.02 | 10.0f | |||
| CMAC(A172) | |||||
| epibatidine | 0.012 | 0.64−0.82 (α7)b 0.005−0.16 (αxβy)c | 0.033 | 0.141 | |
| methyllycaconitine | 4.2 | 2.2d | NA | 5.3 | |
| nicotine | 104.8 | 5600−7600 (α7)b 0.40−440 (αxβy)c | 50.1 | 12,600 | |
| flunitrazepam | 0.81 | 1.3e | |||
| diazepam | 0.78 | 1.0e | |||
| N-methyl-D-aspartic acid | 0.62 | 0.6−1.2e | |||
| MK801 | 11.7 | 10.0f | |||
MP represents the mobile phase used in the experiments; MP + α-BTx indicates that 5 nM α-bungarotoxin had been added to the mobile phase; MP + κ-BTx indicates that 1 nM κ-bungarotoxin had been added to the mobile phase; NA represents no affect on the retention of the marker ligand.
Reference 10.
Reference 18.
Reference 17.
Reference 21.
Reference 22.
On the CMAC(1321N1) column, the calculated Kd value for MLA, a specific ligand for the α7 nAChR, was 7.3 (±7.0) nM, which was consistent with the previously reported value of 2.2 nM.17 The Kd values obtained for EB, 0.25 (±0.20) nM, was about 2-fold lower than previously reported affinities obtained on CMAC columns containing stably expressed α7 nAChR10 and suggested that the binding to the CMAC(1321N1) column was stronger than the CMAC(α7 nAChR) columns. The Kd value obtained for NIC, 155 (±45) nM, was ∼30-fold stronger than the previously reported Kd values for NIC binding to the CMAC(α7 nAChR)10 and was more consistent with the values reported for the binding of NIC to αxβy nAChRs, 0.40−440 nM,18 Table 1. The data suggest that the CMAC(1321N1) column may contain functional αxβy nAChRs in addition to the α7 nAChR. The observed standard errors were larger than those usually obtained using the CMAC approach; however, as the marker ligand epibatidine binds to both subtypes, this can be expected.
On the CMAC(A172) column, the observed Kd value for MLA, 4.2 (±0.4) nM, was also consistent with the previously reported affinity for the α7 nAChR, Table 1. However, the calculated Kd values for EB, 0.012 (±0.007) nM, and NIC, 105 (±48) nM, were >50-fold stronger than previously reported values for the α7 nAChR and were consistent with those reported for binding to αxβy nAChRs.18 These results suggested that the CMAC(A172) column may also contain αxβy nAChRs in addition to the α7 nAChR.
The possibility that the CMAC(1321N1) and CMAC(A172) columns contained both αxβy nAChRs and α7 nAChRs was investigated using selective inhibitors of homomeric or heteromeric nAChR subtypes. In one series of studies, the α7 nAChR-selective inhibitor α-BTx19 was added to the mobile phase, and the second series of studies utilized κ-BTx, a selective antagonist of heteromeric nAChRs.20
The addition of α-BTx to the mobile phase reduced the retention of [3H]-EB on the CMAC(1321N1) column (Figure 1a, curves A and B) and CMAC(A172) column (Figure 1b, curves A and B), indicating that the antagonist had blocked specific binding to α7 nAChRs contained within these columns but that other specific binding sites were still functional. The addition of increasing concentrations of the α7 nAChR-specific ligand MLA did not affect the retention of EB on either column, which indicates that the α-BTx had inhibited α7 nAChR binding. The addition of increasing concentrations of EB and NIC to the mobile phase did produce competitive displacements of [3H]-EB and allowed for the calculation of the respective Kd values, Table 1. Under these experimental conditions, the calculated Kd values for EB on the CMAC(1321N1) column was reduced from 0.25 to 0.0086 (±0.00048) nM, while there was no significant change in the Kd value obtained on the CMAC(A172) column, 0.033 (±0.0046) nM, Table 1. The same trend was observed when NIC was the displacing ligand where the Kd dropped by ∼50% on both columns, to 82.4 (±1.1) and 50.1(±16.7) nM, Table 1. The observed affinities are consistent with previously reported data obtained using heteromeric nAChRs, Table 1, and indicate that both the CMAC(1321N1) and CMAC-(A172) columns contained αxβy nAChRs which were unaffected by the α-BTx.
Figure 1.

The chromatographic traces obtained for 60 pM [3H]-EB on the CMAC(1321N1) column (panel A) and CMAC(A172) column (panel B) where A is the trace obtained using a mobile phase composed of ammonium acetate [10 mM, pH 7.4]; B is the trace obtained after the addition of 5 nM α-BTx to the mobile phase; and C is the trace obtained after the addition of 1 nM κ-BTx to the mobile phase.
The addition of κ-BTx to the mobile phase reduced the retention of [3H]-EB on the CMAC(1321N1) column (Figure 1a, curves A and C) and CMAC(A172) column (Figure 1b, curves A and C) indicating that the antagonist had blocked specific binding to αxβy nAChRs contained within these columns and that other specific binding remained active on the columns. The addition of increasing concentrations of MLA produced the expected reduction in [3H]-EB retention on both columns and the calculated Kd values, 2.0 (±0.8) and 5.3 (±0.5) nM, were consistent with the affinity of MLA for the α7 nAChR, Table 1. The results indicated that functional α7 nAChRs were present on both columns. This was confirmed through the calculation of the Kd values for EB and NIC. The presence of κ-BTx in the mobile phase reduced the affinity of EB on the CMAC(1321N1) and CMAC(A172) columns to 0.467 (±0.162) and 0.141 (±0.016) nM, respectively, and the affinity for NIC was decreased by 4 570 (±830) and 12 600 (±5 240) nM, respectively, Table 1. The observed affinities are consistent with previously reported data for binding to the α7 nAChR. The results indicate that the addition of κ-BTx had reduced or eliminated binding to αxβy nAChRs contained within the CMAC(1321N1) and CMAC(A172) columns and the observed specific binding interactions occurred at unaffected α7 nAChRs.
The presence of multiple nAChR subtypes on the CMAC-(1321N1) and CMAC(A172) columns was also demonstrated by the biphasic chromatographic traces obtained using a 7.5 pM concentration of [3H]-EB, parts a and b of Figure 2, respectively. In frontal affinity chromatography, a biphasic curve suggests the presence of two distinct binding interactions with the marker ligand, which have significant differences in affinity for the marker. In this study, curve A is consistent with the binding of EB to the lower affinity α7 nAChR and curve B with the binding of EB to αxβy nAChRs contained within the CMAC columns.
Figure 2.
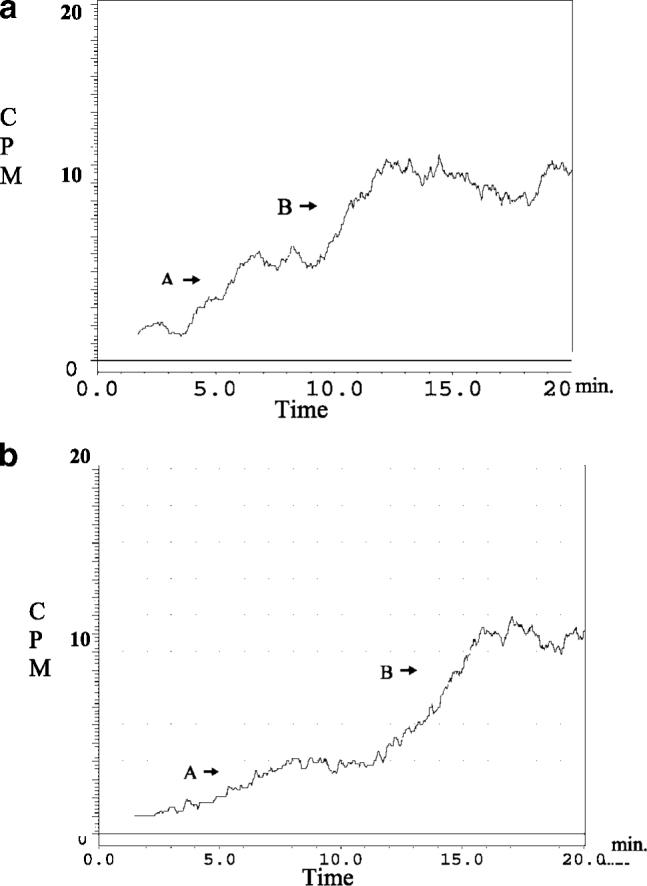
The chromatographic traces obtained for 7.5 pM [3H]-EB on the CMAC(1321N1) column (panel a) and CMAC(A172) column (panel b) present a biphasic curve where A represents the binding to the homomeric nAChR receptor and B represents the binding to the heteromeric nAChR.
When the GABAA receptor agonist [3H]-FTZ was used as the marker ligand, the expected frontal curves were observed on the CMAC(1321N1) and CMAC(A172) columns indicating that the compound was specifically bound to the immobilized membranes. Increasing concentrations of FTZ and DAZ produced concentration dependent decreases in the frontal curves, and the data were used to calculate the apparent Kd values for the displacers, Table 1. The calculated Kd values for FTZ on the CMAC(1321N1), 1.05 (±0.06) nM, and CMAC(A172) column, 0.81 (±0.56) nM, were consistent with previously reported affinities obtained using membrane binding studies and frontal affinity chromatography.21 Similar results were obtained when DAZ was used as the displacer, and although a 4-fold difference in the Kd values for diazepam was observed, 3.21 (±0.23) nM (CMAC(1321N1)) versus 0.78 (±0.09) nM (CMAC(A172)), both results were consistent with the previously reported values, 1.3 and 1.0 nM.21
When the NMDA receptor antagonist [3H]-MK-801 was used as the marker ligand, the expected frontal curves were observed on the CMAC(1321N1) and CMAC(A172) columns, which indicated that the compound was specifically bound to the immobilized membranes. Increasing concentrations of NMDA and MK-801 produced concentration dependent decreases in the frontal curves, and the data were used to calculate the apparent Kd values for the displacers, Table 1. The calculated Kd values for NMDA, 1.67 (±0.60) and 0.62 (±0.16) nM, and MK-801, 9.02 (±4.06) and 11.7 (±6.9) nM, on the CMAC(1321N1) and CMAC(A172) columns, respectively, were consistent with previously reported affinities obtained using membrane binding and frontal affinity chromatography studies.21,22
In order to determine if the chromatographic results reflected cross-selectivity between the immobilized nAChR, GABAA, and NMDA receptors, competitive displacement studies were conducted using the specific receptor markers and 5 nM concentrations of these markers as displacers. When [3H]-EB was the marker, the addition of FTZ or MK-801 had no effect on EB retention while EB produced a significant reduction in the breakthrough volume, Figure S-2A in the Supporting Information. In a similar manner, the addition of nAChR and NMDA ligands had no effect on the retention of the GABAA marker, Figure S-2B in the Supporting Information, and the addition of nAChR and GABAA ligands had no effect on the retention of the NMDA marker, Figure S-2C in the Supporting Information. The data indicate that for these ligands and markers the three LGICs function independently of each other.
Confocal Microscopy and Flow Cytometric Analysis
The use of FITC-α-BTx and fluorescent confocal microscopy to measure the expression of functional α7 nAChRs on the cellular surface of macrophages has been previously described,23 and we have recently described its use with immobilized cellular membrane fragments from cell lines expressing the α7 nAChR.10 In this study, the same approach was used with intact 1321N1 and A172 cell lines, and the images indicated that FITC-α-BTx was bound to the 1321N1 test cells and that the measured emissions corresponding to the samples stained with FITC-α-BTx were significantly reduced when the cells were pretreated with EB, Figure S-3 in the Supporting Information. The results demonstrate that the 1321N1 and A172 cell lines express functional α7 nAChR.
With the use of the same approach, the GABAA receptors on the 1321N1 cell lines were examined using fluorescent tagged muscimol (FL-MUS) using a previously described protocol.24 The addition of 100 μM FL-MUS stained the test cells while the extent of staining was significantly reduced by pretreatment with 10 mM MUS, Figure 3A-C. The same results were obtained when A172 cells were studied indicating that both cell lines expressed the GABAA receptor.
Figure 3.

The confocal microscopy images obtained with 1321N1 astrocytoma cell lines where A, control; B, after incubation with 100 μM fluorescent tagged muscimol (fl-MUS, a GABAA receptor marker); C, cells pretreated with 10 mM cold MUS prior to incubation with 100 μM fl-MUS; D, control; E, after incubation with mouse anti-NMDA R1; F, after incubation with mouse anti-NMDA R1 followed by fluorescein conjugated antimouse IgG. Confocal images were taken with an LSM410 confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY) using an excitation wavelength of 488 nm from an argon laser and a 515−540 nm emission filter.
As previously described, a direct staining approach can not be used to study NMDA receptors.16 Therefore, the 1321N1 and A172 cell lines were studied using an indirect technique involving the initial labeling with mouse anti-NMDA followed by visualization with secondary fluorescein antimouse IgG. The data obtained using the 1321N1 cell line, Figure 3D-F and the A172 cell line indicated that both cell lines expressed NMDA receptors.
In order to measure the relative quantities of α7 nAChRs in the 1321N1 and A172 cell lines, flow cytometry analyses were conducted following staining with FITC-α-BTx. When 1321N1 cells were labeled with FITC-α-BTx, 92% of the fluorescence was observed in the M2 region and pretreatment of the samples with 1 mM EB reduced this to 44%, Figure 4A. Similarly, when the A172 cells were labeled with FITC-α-BTx, 58% of the fluorescence was observed in the M2 region and pretreatment with 1 mM EB reduced this to 25%, Figure 4B. The data indicates that the expression of functional α7 nAChRs is greater in the 1321N1 cell line than in the A172 cell line, and this result is consistent with the results from the chromatographic studies.
Figure 4.
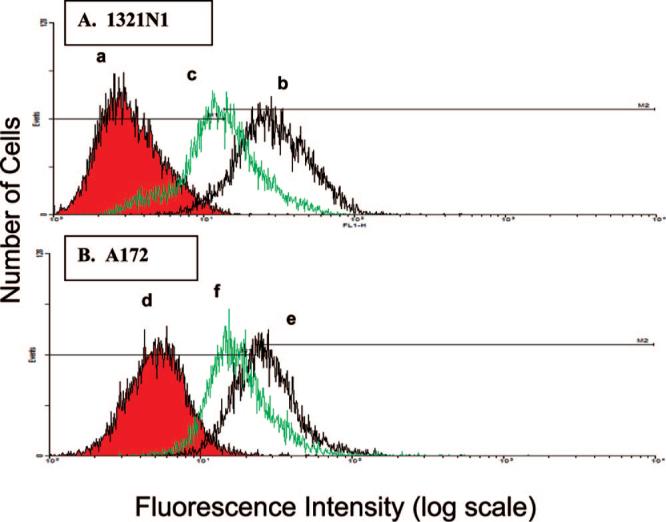
Results from flow cytometry studies utilizing the 1321N1 (panels a-c) and A172 (panels e and f) astrocytoma cell lines where panels a and d are untreated cells; panels b and e are cells treated with FITC-αBTx; and panels c and f are preincubation with EB followed by treatment with FITC-RBTx. See text for experimental details.
Flow cytometry studies were also undertaken with the fluorescent markers for the GABAA and NMDA receptors. The results are consistent with the data from the chromatographic and confocal studies and support the conclusion that the 1321N1 and A172 cell lines express these receptors (Figures S-4 and S-5 in the Supporting Information, respectively).
DISCUSSION
The results of this study indicate that the CMAC(1321N1) and CMAC(A172) columns contain the α7 nAChR, heteromeric nAChRs, GABAA, and NMDA receptors. The data also indicates that the four LGICs retained their ability to selectively and independently bind competitive agonists and antagonists with affinities which are consistent with previously reported values, Table 1. The presence of multiple LGICs on a CMAC column is consistent with an earlier column prepared using solubilized rat forebrain tissue.21 In the earlier study, active nAChR, GABAA, and NMDA receptors were identified, but only the heteromeric nAChR and GABAA were characterized.
While the presence of nAChRs in the tissue-derived CMAC column was confirmed, it was not determined which heteromeric subtypes of the receptors were present. This was not investigated based upon previous studies which had indicated that rat forebrain primarily contains the heteromeric α4β2 nAChR,25 and the CMAC column-determined Kd values for EB and NIC were consistent with the reported affinities for the α4β2 nAChR.21 However, recent data suggests that the rat forebrain may also contain α7 nAChRs, cf. ref 26, and the tissue-derived CMAC column may have indeed contained homomeric and heteromeric nAChRs.
The data from this study establishes the presence of homomeric and heteromeric nAChRs on the CMAC(1321N1) and CMAC(A172) columns. Although the detection of the endogenous expression of the α7 nAChR in 1321N1 and A172 astrocytoma cell lines is a new finding, it is consistent with previous studies that have identified this receptor in astrocytes.7-9 The presence of heteromeric nAChR subtypes in these astrocytomas has not been previously reported nor have they been identified in astrocytes, as previous immunohistochemistry studies on tissue samples obtained from patients with Alzheimer's disease did not indicate the presence of heteromeric subunits in the astrocytes present in these samples.8,9 The difference between the composition of nAChRs in tissue-derived astrocytes and the astrocytoma cell lines may simply be a reflection of different genetic expressions in closely related but not identical cell types. It may also reflect the difficulty of identifying and characterizing nAChR receptor subtypes using a standard biological approach. This problem was addressed in a recent review of nAChRs in the rodent brain in which the authors observed that the issues associated with the identification and characterization of native nAChR subtypes is “complicated by the large number of nAChR subtypes, the expression of multiple subtypes within a given tissue or cell type, and the lack of subtype-specific pharmacological tools”.27
In this study, the CMAC approach was able to detect and characterize the expressed homomeric and heteromeric nAChRs in a direct and relatively simple manner. It is a clear improvement over a recently proposed approach that involves a flow scheme based upon EB binding affinities and multiple competitive binding studies.12 In addition, it appears that the CMAC method was able to qualitatively determine the relative expression of these receptors in the two cell lines. This is based upon the following data contained in Table 1: (1) the experimentally determined Kd value for EB was 20-fold lower on the CMAC(A172) column relative to the CMAC(1321N1) column; (2) the addition of α-BTx to the mobile phase had a greater effect on the Kd value determined on the CMAC(1321N1) column; (3) the addition of κ-BTx to the mobile phase had a greater effect on the Kd value determined on the CMAC(A172) column. These results suggest that there is a greater expression of the α7 nAChR relative to heteromeric nAChRs in the 1321N1 cell line as compared to the A172 cell line. The qualitative homomeric/heteromeric nAChR relationships derived from the chromatographic results were confirmed by the data from the flow cytometry studies, Figure 4.
The presence of GABAA receptors in the astrocytoma-derived CMAC columns was an expected result as previous reports have identified this receptor in human-derived astrocytoma cell lines.2 However, the identification and characterization of NMDA receptors on the CMAC(1231N1) and CMAC(A172) columns was an unexpected result. The NMDA receptor is a member of a LGIC subfamily, the ionotropic glutamate receptors, which also includes the α-amino-3-hydroxy-5-methyl-4-isoxazole proprionic acid (AMPA) and kainite receptors.2 The AMPA receptor, but not the NMDA receptor, has been identified in tissue samples and isolated astrocytes obtained from the brains of patients with epilepsy.2 In this study we were unable to establish the presence of the AMPA receptor in the 1321N1 and A172 cell lines using RT-PCR and Western blot techniques (data not shown). Thus, to our knowledge, the results of this study represent the initial report of the identification and characterization of the NMDA receptor and not the AMPA receptor in astrocytoma cell lines.
CONCLUSIONS
Previous studies in the development and characterization of CMAC columns have utilized stably transfected cell lines, cf. refs 10 and 11. The results of this study demonstrate that CMAC columns can be produced using membrane fragments from native cell lines and that the expression of the LGICs characterized was sufficient to create viable CMAC columns. The use of nontransfected cells to create a chromatographic column containing an immobilized transmembrane protein has been previously reported by Lundahl et al.28 In the previous study, membranes from red blood cells were entrapped in proteoliposomes, and the resulting column was used to study interactions with the GLUT1 glucose transporter. The data from this study confirm Lundahl's earlier observations and demonstrate that the CMAC approach can be employed to characterize multiple, native transmembrane proteins and can be envisioned as a method to map cellular membrane surfaces. The use of CMAC columns to identify multiple drug transporters and G-protein coupled receptors in the membranes of astrocytoma cell lines is currently under study, and the results will be reported elsewhere.
The results from this study also indicate that the co-immobilized nAChRs, GABAA, and NMDA receptors bound receptor-specific marker ligands in an independent manner. This suggests that cell-based CMAC columns can be used to not only pattern the nAChRs contained within these cells but also develop a “chromatographic” array to study multiple interactions between test compounds and tissue-expressed receptors, transporters, and ion channels. This possibility is currently under study, and the results will be reported elsewhere.
ACKNOWLEDGMENT
This work was supported by funds from the Intramural Research Program of the National Institute on Aging, NIH. The authors would like to thank Magdalena Juhaszova from the Laboratory of Cardiovascular Sciences, National Institute on Aging/NIH and Christa M. Morris, Francis J. Chrest, Robert Wersto, and Brittany Frank from the Research Resources Branch, National Institute on Aging/NIH for their assistance in this project.
Footnotes
SUPPORTING INFORMATION AVAILABLE
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Shapiro JR. Curr. Neuro. Neurosci. Rep. 2001;1:217–224. doi: 10.1007/s11910-001-0021-y. [DOI] [PubMed] [Google Scholar]
- 2.Verkhratsky A, Steinhauser C. Brain Res. Rev. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- 3.Codignola A, Tarroni P, Cattaneo MG, Vicentini LM, Clementi F, Sher E. FEBS Lett. 1994;342:286–290. doi: 10.1016/0014-5793(94)80518-0. [DOI] [PubMed] [Google Scholar]
- 4.Quik M, Chan J, Patrick J. Brain Res. 1994;655:161–167. doi: 10.1016/0006-8993(94)91610-1. [DOI] [PubMed] [Google Scholar]
- 5.Sciamanna MA, Griesmann GE, Williams CL, Lennon VA. J. Neurochem. 1997;69:2302–2311. doi: 10.1046/j.1471-4159.1997.69062302.x. [DOI] [PubMed] [Google Scholar]
- 6.Grozio A, Paleari L, Catassi A, Servent D, Cilli M, Piccardi F, Paganuzzi M, Cesario A, Granone P, Mourier G, Russo P. Int. J. Cancer. 2008;122:1911–1915. doi: 10.1002/ijc.23298. [DOI] [PubMed] [Google Scholar]
- 7.Sharma G, Vijayaraghavan S. Proc. Natl. Acad. Sci. U.S.A. 2001;98:4148–4153. doi: 10.1073/pnas.071540198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teaktong T, Graham A, Court J, Perry R, Jaros E, Johnson M, Hall R, Perry E. Glia. 2003;41:207–211. doi: 10.1002/glia.10132. [DOI] [PubMed] [Google Scholar]
- 9.Yu WF, Guan ZZ, Bogdanovic N, Nordberg A. Exp. Neurol. 2005;192:215–225. doi: 10.1016/j.expneurol.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 10.Moaddel R, Oliveira RV, Kimura T, Hyppolite P, Juhaszova M, Xiao Y, Kellar KJ, Bernier M, Wainer IW. Anal. Chem. 2008;80:48–54. doi: 10.1021/ac701943b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moaddel R, Calleri E, Massolini G, Frazier CR, Wainer IW. Anal. Biochem. 2007;364:216–218. doi: 10.1016/j.ab.2007.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, Marks MJ. Biochem. Pharmacol. 2007;74:1235–1246. doi: 10.1016/j.bcp.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arias HR. Neurochem. Int. 2000;36:595–645. doi: 10.1016/s0197-0186(99)00154-0. [DOI] [PubMed] [Google Scholar]
- 14.Moaddel R, Wainer IW. Anal. Chim. Acta. 2006;564:97–105. doi: 10.1016/j.aca.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 15.Moaddel R, Jozwiak K, Wainer IW. Med. Res. Rev. 2007;27:713–753. doi: 10.1002/med.20091. [DOI] [PubMed] [Google Scholar]
- 16.Weiss SW, Albers DS, Iadarola MJ, Dawson TM, Dawson VL, Standaert DG. J. Neuro. Sci. 1998;18:1725–1734. doi: 10.1523/JNEUROSCI.18-05-01725.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whiteaker P, Davies ARL, Marks MJ, Blagrough IS, Potter BVL, Wolstenholme AJ, Collins AC, Wonnacott S. Eur. J. Neurosci. 1999;11:2689–2696. doi: 10.1046/j.1460-9568.1999.00685.x. [DOI] [PubMed] [Google Scholar]
- 18.Moaddel R, Jozwiak K, Whittington K, Wainer IW. Anal. Chem. 2005;77:895–901. doi: 10.1021/ac048826x. [DOI] [PubMed] [Google Scholar]
- 19.Chen D, Pattrick JW. J. Biol. Chem. 1997;272:24024–24029. doi: 10.1074/jbc.272.38.24024. [DOI] [PubMed] [Google Scholar]
- 20.Grant GA, Luetje CW, Summers R, Xu XL. Biochemistry. 1998;37:12166–12171. doi: 10.1021/bi981227y. [DOI] [PubMed] [Google Scholar]
- 21.Moaddel R, Cloix J-F, Ertem G, Wainer IW. Pharm. Res. 2002;19:104–107. doi: 10.1023/a:1013619802766. [DOI] [PubMed] [Google Scholar]
- 22.Kalb RG, Lidow MS, Halsted MJ, Hockfield S. Proc. Natl. Acad. Sci. U.S.A. 1992;89:8502–8506. doi: 10.1073/pnas.89.18.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susaria S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 24.Borodinsky LN, Spitzer NC. Proc. Natl. Acad. Sci. U.S.A. 2007;104:335–340. doi: 10.1073/pnas.0607450104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flores CM, Rogers SW, Pabreza LA, Wolfe BA, Kellar KJ. Mol. Pharmacol. 1992;41:31–37. [PubMed] [Google Scholar]
- 26.Briggs CA, Schrimpf MR, Anderson DJ, Gubbins EJ, Gronlien JH, Hakerud M, Ween H, Thorin-Hagene K, Malysz J, Li J, Bunnelle WH, Gopalakrishnan M, Meyer MD. Br. J. Pharmacol. 2008;153:1054–1061. doi: 10.1038/sj.bjp.0707649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gotti C, Moretti M, Gaimarri A, Zanardi A, Clementi F, Zoli M. Biochem. Pharmacol. 2007;74:1102–1111. doi: 10.1016/j.bcp.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 28.Brekkan E, Lundqvist A, Lundahl P. Biochemistry. 1996;35:12141–12145. doi: 10.1021/bi9603231. [DOI] [PubMed] [Google Scholar]