

Photoreactivation is the reversal of the harmful effects of far-UV radiation (200–300 nm) on organisms, such as growth delay, mutation, cell death, and cancer, by concomitant or subsequent exposure of the organism to near-UV/blue light (300–500 nm). The two major lesions induced in DNA by UV light are cyclobutane pyrimidine dimers (Pyr<>Pyr2 or CPD), which constitute ∼80–90% of the photoproducts, and pyrimidine-pyrimidone (6-4) photoproducts (Pyr[6-4]Pyr), which account for the 10–20% of the UV lesions. Photoreactivation results from the repair of these lesions in situ by flavoproteins called photoreactivating enzymes (photolyase) that use a blue-light photon as a co-substrate. Photolyases that repair these two photoproducts are evolutionarily related but functionally distinct. Enzymes that repair CPDs are referred to as CPD photolyase, and enzymes that repair (6-4) photoproducts are called (6-4) photolyase. For historical reasons and as a matter of common practice, the term “photolyase” without further qualification means CPD photolyase, and it will be used as such in this review, which celebrates the 50th anniversary of the discovery of photolyase.
Historical Perspective
Photoreactivation was discovered by Kelner, who found that the lethal effect of UV radiation on Streptomyces griseus could be reversed if, following UV radiation, the irradiated bacterial culture was exposed to visible light (1). Resurrection of the UV light-killed cells by light attracted the interest of many physicists in part because the reversal of the effect of high energy UV light by lower energy blue light was counterintuitive and seemed to run counter to the laws of physics. One such physicist was Claud S. Rupert, who eventually discovered photolyase (2). He and his colleagues used the DNA transformation assay to understand the molecular basis of photoreactivation. Extensive screening had revealed that photoreactivation was not universally distributed in the biological world. Of note, it was known that Escherichia coli possessed photoreactivation, but Haemophilus influenzae, which is a naturally transformable species, did not. Rupert did the following experiment (Fig. 1). He irradiated DNA isolated from a streptomycin-resistant H. influenzae strain with a UV dose that reduced the transformation efficiency by 30-fold. Then, he mixed the irradiated DNA with cell-free extract made from either H. influenzae or E. coli, and the mixtures were incubated either in dark or under light and used to transform a streptomycin-sensitive H. influenzae strain. He found that incubating the damaged DNA with the H. influenzae extract either in dark or under light did not improve its transformation efficiency. In contrast, whereas incubating the damaged DNA with E. coli extract in the dark did not affect its transforming capacity, light increased its transforming efficiency by 10-fold. Rupert and colleagues concluded that E. coli contained a light-activated enzyme that repaired the UV light-induced DNA damage and named it photoreactivating enzyme (2), which later came to be known as photolyase.
FIGURE 1.

Key experiments in photolyase enzymology. A, historical experiment that led to the discovery of photolyase by Rupert et al. (2). This page from C. S. Rupert's notebook is the record of the experiment done on June 16, 1956 and shows repair of UV damage to H. influenzae DNA (TP = transforming principle) by E. coli extract in the presence of blue light. The table shows the results of a Haemophilus transformation assay with UV light-irradiated DNA. The titers of transformants are listed in the last two columns. Row A, unirradiated DNA; row B, irradiated DNA; row D, irradiated DNA mixed with E. coli extract and exposed to blue light; row E, same as in row D but kept in the dark; rows E–I, results from control experiments (2). B, flash photolysis. An E. coli strain dependent exclusively on photolyase for Pyr<>Pyr repair was irradiated with increasing UV doses and either kept in the dark (•) or exposed to a camera flash (○) before plating. Cells exposed to a flash after 1.6 J/m2 have approximately the same survival as cells irradiated with 0.4 J/m2 and kept in dark. Because 1 J/m2 produces 65 Pyr<>Pyr in the E. coli chromosome, this strain must have at least 65 × (1.6–0.4) = 78 photolyase molecules/cell (4).
Rupert continued to study the repair reaction in some detail using cell-free extracts or partially purified enzyme from E. coli and budding yeast and developed Scheme 1.
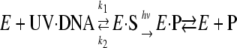 |
SCHEME 1 |
Thus, he concluded that the reaction proceeds by the classical Michaelis-Menten scheme with the notable exception that catalysis is absolutely dependent on light. Finally, Rupert introduced the then nascent flash photolysis technology to the field of photolyase (Fig. 1) and using this technology determined some of the fundamental enzymatic parameters both in vitro and in vivo (3, 4). Shortly after the discovery of photolyase, Pyr<>Pyr and (6-4) photoproducts were identified as the two major UV light-induced lesions in DNA. Photolyases from E. coli and budding yeast studied by Rupert and others repair only Pyr<>Pyr (5, 6). Recombinant DNA technology considerably accelerated the pace of characterization of these enzymes (4–7).
However, it was only in 1993 that a photolyase that repairs the (6-4) photoproduct was also discovered (8). Similarly, in 1993, it was found that an Arabidopsis thaliana protein with high sequence homology to photolyase had no repair activity but functioned as a blue-light receptor (cryptochrome) for plant growth and development (9). Finally, in 1996, cryptochrome was discovered in humans (10) and mice, and in 1998, it was shown to regulate the circadian clock in these and other animals by light-dependent and light-independent mechanisms (11–13). In this review, E. coli photolyase, which is the best characterized photolyase to date, will be discussed, and when necessary, it will be compared with some other photolyases that have been studied in some detail. Then, the structures and functions of (6-4) photolyase and cryptochrome will be reviewed briefly before addressing the issue of suitability of photolyase for in vivo enzymology.
Structure of Photolyase
Primary Structure—The photolyase/cryptochrome proteins, which are ∼400–600 amino acids in length, constitute a large and ancient flavoprotein family (14). Although sequence analysis is often sufficient to designate a new member of the family as a photolyase, a (6-4) photolyase, or a cryptochrome, frequently, the designation can be made only by functional testing. The three functional members of the family are not universally distributed. E. coli has only photolyase; marsupials have photolyase and cryptochrome; Drosophila possesses all three; placental mammals have only cryptochrome; and Bacillus subtilis and Caenorhabditis elegans have none.
Cofactors and Chromophores—All photolyases are known or presumed to contain FAD as the catalytic cofactor (5, 6). In addition, they contain a “second chromophore” that is not essential for activity but increases the efficiency of repair under limiting light conditions (15–17). The second chromophore, which functions as a light-harvesting photoantenna, is 5,10-methenyltetrahydrofolate (MTHF) in the majority of photolyases analyzed to date. In some rare species that can synthesize 5-deazaflavin, such as Anacystis nidulans, the second chromophore is 8-hydroxy-5-deazariboflavin (8-HDF). Finally, in some thermophilic bacteria, FMN and FAD have recently been identified as the second chromophores (18, 19). The E. coli photolyase in its native state contains FAD in the two-electron reduced and deprotonated FADH– form (λmax = 360 nm, ε360 = 5,000 m–1 cm–1), and hence, the yellow color of the enzyme is dominated by MTHF (λmax = 385 nm, ε385 = 25,000 m–1 cm–1). However, during purification under aerobic conditions, FADH– is oxidized to the rather stable FADH· blue neutral radical, and the enzyme exhibits dark blue color. Excessive handling of the enzyme causes further oxidation to oxidized FAD, and the enzyme acquires a bright yellow color (5, 6).
Crystal Structure of Photolyase—Crystal structures of several photolyases and of the cryptochrome from A. thaliana have been determined. All have the same basic architecture even when the sequence identity between members is as low as 25% and the enzymes have different second chromophores as in the case of E. coli photolyase, which contains MTHF, and A. nidulans photolyase, which contains 8-HDF (20, 21). Hence, as a representative of the entire family, the structure of the E. coli photolyase will be discussed (20). The enzyme is essentially globular in shape (Fig. 2) and is made up of two well defined domains, an N-terminal α/β-domain (residues 1–131) and a C-terminal α-helical domain (residues 204–472). The two domains are connected to one another with a long interdomain loop (residues 132–203) that wraps around the α/β-domain (Fig. 2A). The MTHF photoantenna is located in a shallow cleft between the two domains. In contrast to MTHF, the FAD cofactor is deeply buried within the α-helical domain and is held tightly in place by interactions with 14 amino acids. The photolyase/cryptochrome FAD has the unusual conformation of the adenine ring being stacked on top of the isoalloxazine ring. This conformation is thought to be important for regulating the rates of electron transfer to substrate and the subsequent charge recombination. Surface potential representation of the enzyme reveals a positively charged groove that runs the length of the enzyme. In the middle of this groove, a hole of the proper dimensions and polarity to accommodate a Pyr<>Pyr leads to the FAD located in the bottom of the hole (Fig. 2B), providing strong evidence that the dimer is pulled into the hole to be repaired and then released.
FIGURE 2.

Structure of photolyase. A, ribbon diagram representation. B, surface potential representation. The dashed box marks the hole leading to FAD. Positively (blue) and negatively (red) charged residues are highlighted.
Finally, the crystal structure revealed another feature of the enzyme of functional significance. Photochemical studies have shown that the second chromophore contributes to repair by fluorescence resonance energy transfer (FRET) to the catalytic factor FADH– (5, 6). According to Förster's FRET formula, the efficiency of transfer is inversely proportional to the distance between the donor and acceptor and to the angle between the transition dipole moments of the donor and acceptor. Energy transfer is favorable when the interchromophore distance is short and the transition dipole moments of the donor and acceptor have the same orientation and direction. The interchromophore distance is 16.8 Å in E. coli photolyase (20) and 17.5 Å in A. nidulans photolyase (21). Despite the more favorable distance between the chromophores in E. coli photolyase, the energy transfer efficiency from MTHF to FADH– is only ∼70% (22) compared with the energy transfer efficiency of nearly 100% in the A. nidulans enzyme (23). The crystal structures of these two enzymes provided an explanation to this seemingly paradoxical phenomenon: in E. coli photolyase, the transition dipole moments of the donor and acceptor are oriented at nearly 90° with one another (20). In contrast, the angle between the transition dipole moments of the two chromophores in A. nidulans photolyase is only 35.6 Å (21), more than sufficient to compensate for the longer interchromophore distance and achieve maximum efficiency.
Reaction Mechanism
Photolyase carries out catalysis according to Michaelis-Menten kinetics and the “bisubstrate ordered sequential mechanism”: the enzyme binds Pyr<>Pyr independent of light to form an E·S complex, which must then absorb a photon (second substrate) to initiate catalysis. Although the bisubstrate ordered sequential mechanism is quite common in enzymology, photolyase differs from all other such enzymes in that the second substrate is a photon and not a molecule.
DNA Binding—Both solution and crystallographic studies have shown that a T<>T bends the duplex by 30° toward the major groove and unwinds it by 9° (5, 6). These features of the DNA backbone are recognized by photolyase, which makes a moderately stable complex through ionic interactions between the positively charged groove on the surface of the enzyme and the first phosphate 5′ and the three phosphates 3′ to the T<>T on the damaged strand and some weaker interactions with the backbone of the complementary strand across from the dimer (24). These interactions further weaken the duplex in the immediate vicinity of the T<>T, leading to “flipping out” of the T<>T into the active-site cavity in the middle of the DNA binding groove (20) to make a high stability complex. Within this complex, the T<>Tis within van der Waals contact with FADH– such that high efficiency electron transfer can occur during catalysis. As is the case with other DNA-acting enzymes that employ base flipping in the course of substrate recognition, photolyase binds to a T<>Tin single-stranded DNA with higher affinity than to a T<>Tina duplex because it is energetically more favorable to flip out a dimer from single-stranded DNA than from a duplex (6).
The co-crystal structure of the enzyme-product complex of A. nidulans photolyase with a decamer duplex containing a centrally located T<>T analog has supported the binding model and has provided a more detailed view of the enzyme-substrate contacts (25). In addition to single- and double-stranded DNAs, photolyase binds and repairs short oligonucleotides containing T<>T. In fact, even a simple thymine base dimer binds photolyase with considerable affinity and is repaired efficiently (26). It is noteworthy that a class of photolyases with exquisite specificity to Pyr<>Pyr in single-stranded DNA was recently discovered (27, 28).
Catalysis—DNA repair by photolyase is through a radical
mechanism that involves light-initiated
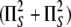 cycloconversion of the cyclobutane ring joining the two pyrimidines. The
overall quantum yield of repair (φ = number of Pyr<>Pyr repaired per
number of photons absorbed) is 0.7 for E. coli photolyase and 0.9 for
A. nidulans photolyase
(5,
6). Catalysis encompasses
several microscopic steps (Fig.
3), all of which have been directly observed in real time by
ultrafast (femtosecond-picosecond) absorption and fluorescence spectroscopy
(29,
30). First, a 300–500 nm
photon is absorbed by the second chromophore (MTHF or 8-HDF; ∼1 fs).
Second, the excitation energy is transferred from the photoantenna to
FADH– by FRET (τ = 160 ps for E. coli photolyase
and τ = 50 ps for A. nidulans photolyase)
(22,
28). Third,
1(FADH–)* transfers an electron to Pyr<>Pyr
(τ = 170 ps) to generate the
cycloconversion of the cyclobutane ring joining the two pyrimidines. The
overall quantum yield of repair (φ = number of Pyr<>Pyr repaired per
number of photons absorbed) is 0.7 for E. coli photolyase and 0.9 for
A. nidulans photolyase
(5,
6). Catalysis encompasses
several microscopic steps (Fig.
3), all of which have been directly observed in real time by
ultrafast (femtosecond-picosecond) absorption and fluorescence spectroscopy
(29,
30). First, a 300–500 nm
photon is absorbed by the second chromophore (MTHF or 8-HDF; ∼1 fs).
Second, the excitation energy is transferred from the photoantenna to
FADH– by FRET (τ = 160 ps for E. coli photolyase
and τ = 50 ps for A. nidulans photolyase)
(22,
28). Third,
1(FADH–)* transfers an electron to Pyr<>Pyr
(τ = 170 ps) to generate the
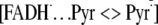 charge transfer complex. Finally, the cyclobutane ring is split, and
FADH· is restored to the catalytically active form by back-electron
transfer (τ = 560 ps). Thus, the entire catalysis reaction takes ∼1 ns
for both types of photolyases. The quantum yield of repair by
1(FADH–)* is close to unity for both enzymes
(5,
6). However, the overall
quantum yields of repair of the enzymes are dictated by the quantum yield of
FRET from the second chromophore to FADH–, which is
95–99% in A. nidulans photolyase and only ∼70% in the
E. coli enzyme (5,
6).
charge transfer complex. Finally, the cyclobutane ring is split, and
FADH· is restored to the catalytically active form by back-electron
transfer (τ = 560 ps). Thus, the entire catalysis reaction takes ∼1 ns
for both types of photolyases. The quantum yield of repair by
1(FADH–)* is close to unity for both enzymes
(5,
6). However, the overall
quantum yields of repair of the enzymes are dictated by the quantum yield of
FRET from the second chromophore to FADH–, which is
95–99% in A. nidulans photolyase and only ∼70% in the
E. coli enzyme (5,
6).
FIGURE 3.

UV photoproducts and reaction mechanisms of photolyases. A, structures of the two major UV photoproducts. PL, photolyase. B, reaction mechanisms of photolyases. Left, E. coli CPD photolyase; right, D. melanogaster (6-4) photolyase. For clarity, the two critical His residues essential for catalysis by stabilizing the putative oxetane intermediate by the general acid-base mechanism (33, 36) are not shown.
Finally, with respect to catalysis by photolyase, two points should be noted. First, even though it is commonly stated that photolyase uses light energy to repair DNA, the reaction catalyzed, strictly speaking, is not a photochemical reaction because the blue light that initiates catalysis does not have enough energy to split the cyclobutane ring. Rather, the enzyme uses blue light as a co-substrate to increase the reduction potential of FADH–, enabling it to reduce the Pyr<>Pyr and hence lower the free energy barrier for “chemical splitting” of the cyclobutane ring. Second, although the flavin cofactor of photolyase is involved in a redox reaction during catalysis, this is a cyclic redox reaction in which the net charge of the flavin and of the substrate/product remains unchanged at the end of the catalytic cycle. In this regard, photolyase is not alone as several other flavoenzymes, such as N-methylglutamate synthase and chorismate synthase, also catalyze reactions with no net redox change (31).
(6-4) Photolyase
In the (6-4) photoproduct, C-6 of the 5′-base is joined to C-4 of the 3′-base, and the –OH (or –NH2) group at C-4 of the 3′-base is transferred to C-5 of the 5′-base (Fig. 3A). In contrast to Pyr<>Pyr, in which breaking of the UV light-induced bonds restores the bases to their canonical forms, the breaking of the C-5–OH and C-6–C-4 bonds of the (6-4) photoproducts would not repair DNA but in fact would generate two damaged bases. Thus, formally an enzyme that repairs the (6-4) photoproduct must catalyze both bond breakage (lyase) and group transfer (transferase) reactions. Surprisingly, the (6-4) photolyase, which exhibits ∼30% sequence identity to the classical Pyr<>Pyr photolyase of E. coli (32), accomplishes this difficult task, albeit with a rather low quantum yield.
Structure—The structures of Drosophila melanogaster (6-4) photolyase alone and in complex with substrate/product have been solved (33). The general architecture of the enzyme is quite similar to that of E. coli photolyase with the important exception of having 8-HDF as the second chromophore.
Function—Biochemical and crystallographic data reveal that (6-4) photolyase has a reaction mechanism quite similar to that of Pyr<>Pyr photolyase (33–36). The positively charged groove on the enzyme surface binds mainly through ionic bonds to the damaged strand and makes some weaker contacts with the complementary strand. The strong ionic interaction with the severely distorted DNA backbone facilitates the 180° flip-out of the (6-4) photoproduct into the active-site cavity (33, 35).
As in the case of photolyase, the flavin in (6-4) photolyase is in the form
of FADH–, and it is presumed to generate the
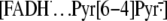 charge transfer complex by single-electron transfer from
1(FADH–)* that is generated by FRET from the
second chromophore or by direct light absorption
(5,
6). Because of the apparent
implausibility that the
charge transfer complex by single-electron transfer from
1(FADH–)* that is generated by FRET from the
second chromophore or by direct light absorption
(5,
6). Because of the apparent
implausibility that the
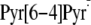 intermediate would undergo both C–C bond breakage and –OH group
transfer reactions in a concerted manner, it was proposed
(35) that the enzyme converts
the (6-4) photoproduct either thermally before light absorption or
photochemically following excitation to the four-membered oxetane
intermediate, so the concerted breaks of the C-4–C-6 and C-5–OH
bonds would generate the two canonical pyrimidines
(Fig. 3B). Although
some preliminary data seemed to support thermal formation of the oxetane
intermediate (35), the recent
crystallographic data strongly suggest that the oxetane intermediate may be a
very short-lived transition state intermediate that forms photochemically
(33).
intermediate would undergo both C–C bond breakage and –OH group
transfer reactions in a concerted manner, it was proposed
(35) that the enzyme converts
the (6-4) photoproduct either thermally before light absorption or
photochemically following excitation to the four-membered oxetane
intermediate, so the concerted breaks of the C-4–C-6 and C-5–OH
bonds would generate the two canonical pyrimidines
(Fig. 3B). Although
some preliminary data seemed to support thermal formation of the oxetane
intermediate (35), the recent
crystallographic data strongly suggest that the oxetane intermediate may be a
very short-lived transition state intermediate that forms photochemically
(33).
Cryptochrome
Cryptochrome was originally defined as a pigment of unknown (cryptic)
nature that mediates blue-light responses in plants. Currently, any protein
with sequence homology to photolyase but with no repair function is called
cryptochrome and is presumed to be a photosensory pigment
(37–41).
In A. thaliana, it has been shown conclusively that AtCRY1 and AtCRY2
function as blue-light photoreceptors
(9). The structure of AtCRY1
has been solved. It is very similar to that of photolyase except it lacks the
positively charged DNA binding groove
(42) and has an unstructured
C-terminal extension beyond the photolyase homology region. In
Drosophila, there is compelling evidence that DmCRY is a blue-light
photoreceptor that sets the circadian clock
(13). In mice, genetic
analysis has revealed that cryptochromes CRY1 and CRY2 are core clock proteins
that are essential for the functioning of the circadian clock in a
light-independent manner (12).
Although there are some genetic data implicating mouse CRYs in circadian
photoreception, this issue remains to be settled
(14,
38). Interestingly, some
insects, such as the monarch butterfly, possess both Drosophila-like
(called Type 1) and human-like (called Type 2) CRYs, and some insects, such as
the honeybee, possess only a human-like (Type 2) CRY that participates in the
circadian clock but apparently not in photoreception
(43). Furthermore, it has
recently been shown that DmCRY may function as a light-activated
magnetoreceptor (44). The
mechanisms of photoreception and magnetoreception by CRYs are not understood
at present. Even the redox state of FAD in plant and insect CRYs is a matter
of considerable debate
(45–47),
although the majority of action spectrum studies favor photolyase-like
FADH– or
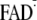 states
(44,
48).
states
(44,
48).
Photolyase and in Vivo Enzymology
A major goal in biochemistry is to obtain information on behaviors of enzymes that would reflect their in vivo functions. In enzymological terms, it is desirable to know the on- and off-rates, the Michaelis constants, and the kcat of an enzyme in vivo. For the vast majority of enzymes, this is not possible even with the currently available single-molecule and high resolution visualization technologies. In contrast, with photolyase this goal, was accomplished 40 years ago (3). This was possible because of three factors. First, the absolute dependence of catalysis by photolyase on light makes it possible to analyze the binding and catalysis steps independently. Second, the level of substrate in the cell can be controlled easily by simply changing the UV dose administered to the cell culture. Finally, a single light flash of millisecond-to-femtosecond duration is sufficient to convert all of the substrate bound to the enzyme into product in less than a nanosecond. Even though in recent years technically very advanced ultrafast flash systems have been used to investigate the progression of the photolyase enzyme-substrate along the reaction coordinates, a simple photographic flash unit is sufficient for most purposes. Indeed, camera flash photolysis was used in vivo 40 years ago to calculate some basic enzymological parameters such as the concentration of photolyase in the cell, the on- and off-rates and the association equilibrium constant of the enzyme, and the precise value of the photolytic cross-section and therefore the approximate value of the quantum yield of repair (3). Remarkably, when the E. coli photolyase was eventually purified and characterized, the values obtained with the defined enzyme-substrate system (49, 50) were, within experimental error, identical to those obtained in vivo (Table 1). Naturally, the two-chromophore system, the extinction coefficient, and other structural and mechanistic aspects of photolyase could not be addressed by the in vivo flash photolysis technology. Nevertheless, photolyase still occupies a unique position in biochemistry because of its easy accessibility to in vivo enzymology. In that regard, it is interesting to note that some recent commentaries on the history of enzymology and the future of the field stated that “...we do not know the real rates of target location for any in vivo system” (51) and “...we believe that a crucial next step will be to go beyond the milieu of dilute aqueous solutions and individual purified enzymes that has defined enzymology for the past 100 years... In vivo enzymology is the logical next step...” (italics added) (52). For photolyase, the future is now, or more accurately, it has been with us for 40 years!
TABLE 1.
Reaction constants for E. coli photolyase obtained in vivo and with purified enzyme in vitro
Reflections
The discovery of photolyase 50 years ago opened the era of DNA repair enzymology. The light dependence of catalysis has made it possible to chart the progression of the chemical reaction along reaction coordinates in real time at picosecond resolution. The same property of the enzyme has allowed detailed in vivo enzymology, which is considered a major goal of biochemical research in the future. Finally, the characterization of photolyase played a significant role in the discovery of cryptochrome in plants and the discovery of the closely related but functionally distinct cryptochromes in animals. These discoveries have led to mechanistic insight into blue-light responses in plants, the circadian clock and sleep homeostasis in humans, and magnetoreception in migratory animals. Who would have thought that an experiment done 50 years ago with H. influenzae DNA and E. coli cell-free extract would have major impact on research topics ranging from crop yield in plants, to sleep-wake cycle regulation in humans, and to global positioning systems in butterflies and birds?
Supplementary Material
Author's Choice—Final version full access.
I dedicate this paper to my Ph.D. advisor, Professor Claud S. Rupert, on the occasion of his 90th birthday and the 50th anniversary of his discovery of photolyase.
This work was supported, in whole or in part, by National Institutes of Health Grant GM31082. This minireview will be reprinted in the 2008 Minireview Compendium, which will be available in January, 2009.
Footnotes
The abbreviations used are: Pyr, pyrimidine; CPD, cyclobutane pyrimidine dimer; MTHF, 5,10-methenyltetrahydrofolate; 8-HDF, 8-hydroxy-5-deazariboflavin; FRET, fluorescence resonance energy transfer.
References
- 1.Kelner, A. (1949) Proc. Natl. Acad. Sci. U. S. A. 35 73–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rupert, C. S., Goodgal, S. H., and Herriott, R. M. (1958) J. Gen. Physiol. 41 451–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harm, W., Harm, H., and Rupert, C. S. (1968) Mutat. Res. 6 371–385 [DOI] [PubMed] [Google Scholar]
- 4.Sancar, A., and Rupert, C. S. (1978) Gene (Amst.) 4 295–308 [DOI] [PubMed] [Google Scholar]
- 5.Sancar, A. (1994) Biochemistry 33 2–9 [DOI] [PubMed] [Google Scholar]
- 6.Sancar, A. (2003) Chem. Rev. 103 2203–2237 [DOI] [PubMed] [Google Scholar]
- 7.Sancar, A., Smith, F. W., and Sancar, G. B. (1984) J. Biol. Chem. 259 6028–6032 [PubMed] [Google Scholar]
- 8.Todo, T., Takemori, H., Ryo, H., Ihara, M., Matsunaga, T., Nikaido, O., Sato, K., and Nomura, T. (1993) Nature 361 371–374 [DOI] [PubMed] [Google Scholar]
- 9.Ahmad, M., and Cashmore, A. R. (1993) Nature 366 162–166 [DOI] [PubMed] [Google Scholar]
- 10.Hsu, D. S., Zhao, X., Zhao, S., Kazantsev, A., Wang, R. P., Todo, T., Wei, Y. F., and Sancar, A. (1996) Biochemistry 35 13871–13877 [DOI] [PubMed] [Google Scholar]
- 11.Miyamoto, A., and Sancar, A. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 6097–6102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thresher, R. J., Vitaterna, M. H., Miyamoto, Y., Kazantsev, A., Hsu, D. S., Petit, C., Selby, C. P., Dawut, L., Smithies, O., Takahashi, J. S., and Sancar, A. (1998) Science 282 1490–1494 [DOI] [PubMed] [Google Scholar]
- 13.Stanewsky, R., Kaneko, M., Emery, P., Beretta, B., Wager-Smith, K., Kay, S. A., Rosbash, M., and Hall, J. C. (1988) Cell 95 681–692 [DOI] [PubMed] [Google Scholar]
- 14.Partch, C. L., and Sancar, A. (2005) Photochem. Photobiol. 81 1291–1304 [DOI] [PubMed] [Google Scholar]
- 15.Jorns, M. S., Sancar, G. B., and Sancar, A. (1984) Biochemistry 23 2673–2679 [DOI] [PubMed] [Google Scholar]
- 16.Johnson, J. L., Hamm-Alvarez, S., Payne, G., Sancar, G. B., Rajagopalan, K. V., and Sancar, A. (1988) Proc. Natl. Acad. Sci. U. S. A. 85 2046–2050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eker, A. P. M., Hessels, J. K. C., and van de Velde, J. (1988) Biochemistry 27 1758–1765 [Google Scholar]
- 18.Ueda, T., Kato, A., Kuramitsu, S., Terasawa, H., and Shimada, I. (2005) J. Biol. Chem. 280 36237–36243 [DOI] [PubMed] [Google Scholar]
- 19.Fujihashi, M., Numoto, N., Kobayashi, Y., Misushima, A., Tsujimura, M., Nakamura, A., Kawarabayasi, Y., and Miki, K. (2007) J. Mol. Biol. 365 903–910 [DOI] [PubMed] [Google Scholar]
- 20.Park, H. W., Kim, S. T., Sancar A., and Deisenhofer, J. (1995) Science 268 1866–1872 [DOI] [PubMed] [Google Scholar]
- 21.Tamada, T., Kitadokoro, K., Higuchi, Y., Inaka, K., Yasui, A., de Ruiter, P. E., Eker, A. P. M., and Miki, K. (1997) Nat. Struct. Biol. 4 887–891 [DOI] [PubMed] [Google Scholar]
- 22.Payne, G., and Sancar, A. (1990) Biochemistry 29 7715–7727 [DOI] [PubMed] [Google Scholar]
- 23.Kim, S. T., Heelis, P. F., and Sancar, A. (1992) Biochemistry 31 11244–11248 [DOI] [PubMed] [Google Scholar]
- 24.Husain, I., Sancar, G. B., Holbrook, S. R., and Sancar, A. (1987) J. Biol. Chem. 262 13188–13197 [PubMed] [Google Scholar]
- 25.Mees, A., Klar, T., Gnau, P., Hennecke, U., Eker, A. P., Carell, T., and Essen, L. O. (2004) Science 306 1789–1793 [DOI] [PubMed] [Google Scholar]
- 26.Kim, S. T., and Sancar, A. (1991) Biochemistry 30 8623–8630 [DOI] [PubMed] [Google Scholar]
- 27.Selby, C. P., and Sancar, A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 17696–17700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang, Y., Baxter, R., Smith, B. S., Partch, C. L., Colbert, C. L., and Deisenhofer, J. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 17701–17706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saxena, C., Sancar, A., and Zhong, D. (2004) J. Phys. Chem. B 108 18026–18033 [Google Scholar]
- 30.Kao, Y. T., Saxena, C., Wang, L., Sancar, A., and Zhong, D. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 14724–14728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bornemann, S. (2002) Nat. Prod. Rep. 19 761–772 [DOI] [PubMed] [Google Scholar]
- 32.Todo, T., Ryo, H., Yamamoto, K., Toh, H., Inui, T., Ayaki, H., Nomura, T., and Ikenaga, M. (1996) Science 272 109–112 [DOI] [PubMed] [Google Scholar]
- 33.Maul, M. J., Barends, T. R. M., Glas, A. F., Cryle, M. J., Domaratcheva, T., Schneider, S., Schlichting, I., and Carell, T. (2008) Angew. Chem., in press [DOI] [PubMed]
- 34.Kim, S. T., Malhotra, K., Smith, C. A., Taylor, J. S., and Sancar, A. (1994) J. Biol. Chem. 269 8535–8540 [PubMed] [Google Scholar]
- 35.Zhao, X., Liu, J., Hsu, D. S., Zhao, S., Taylor, J. S., and Sancar, A. (1997) J. Biol. Chem. 272 32580–32590 [DOI] [PubMed] [Google Scholar]
- 36.Hitomi, K., Nakamura, H., Kim, S. T., Mizukoshi, T., Ishikawa, T., Iwai, S., and Todo, T. (2001) J. Biol. Chem. 276 10103–10109 [DOI] [PubMed] [Google Scholar]
- 37.Cashmore, A. R. (2003) Cell 114 537–543 [PubMed] [Google Scholar]
- 38.Sancar, A. (2004) J. Biol. Chem. 279 34079–34082 [DOI] [PubMed] [Google Scholar]
- 39.Lin, C., and Todo, T. (2005) Genome Biol. 6 220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Losi, A. (2007) Photochem. Photobiol. 83 1283–1300 [DOI] [PubMed] [Google Scholar]
- 41.Öztürk, N., Song, S.-H., Özgür, S., Selby, C. P., Morrison, L., Partch, C., Zhong, D., and Sancar, A. (2007) Cold Spring Harbor Symp. Quant. Biol. 72 119–131 [DOI] [PubMed] [Google Scholar]
- 42.Brautigam, C. A., Smith, B. S., Ma, Z., Palnitkar, M., Tomchick, D. R., Machius, M., and Deisenhofer, J. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 12142–12147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yuan, Q., Matterville, D., Briscoe, A. D., and Reppert, S. M. (2007) Mol. Biol. Evol. 24 948–955 [DOI] [PubMed] [Google Scholar]
- 44.Gegear, R. J., Casselman, A., Waddell, S., and Reppert, S. M. (2008) Nature 454 1014–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song, S.-H., Öztürk, N., Denaro, T. R., Arat, N.Ö., Kao, Y. T., Zhu, H., Zhong, D., Reppert, S. M., and Sancar, A. (2007) J. Biol. Chem. 282 17608–17612 [DOI] [PubMed] [Google Scholar]
- 46.Öztürk, N., Song, S.-H., Selby, C. P., and Sancar, A. (2008) J. Biol. Chem. 283 3256–3263 [DOI] [PubMed] [Google Scholar]
- 47.Hoang, N., Schleicher, E., Kacprzak, S., Bouly, J. P., Picot, M., Wu, W., Berndt, A., Wolf, E., Bittl, R., and Ahmad, M. (2008) PLoS Biol. 6 e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Vickle-Chavez, S. J., and Van Gelder, R. N. (2007) J. Biol. Chem. 282 10561–10566 [DOI] [PubMed] [Google Scholar]
- 49.Husain, I., and Sancar, A. (1987) Nucleic Acids Res. 5 1109–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kavakli, I. H., and Sancar, A. (2004) Biochemistry 43 15103–15110 [DOI] [PubMed] [Google Scholar]
- 51.Widom, J. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 16909–16910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ringe, D., and Petsko, G. A. (2008) Science 320 1428–1429 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.