

Abstract
Nitric oxide and nitrovasodilators induce vascular smooth muscle cell relaxation in part by cGMP-dependent protein kinase I (PKG-Iα)-mediated activation of myosin phosphatase (MLCP). Mechanistically it has been proposed that protein-protein interactions between the N-terminal leucine zipper (LZ) domain of PKG-Iα ((PKG-Iα1-59) and the LZ and/or coiled coil (CC) domain of the myosin binding subunit (MBS) of MLCP are localized in the C terminus of MBS. Although recent studies have supported these interactions, the critical amino acids responsible for these interactions have not been identified. Here we present structural and biophysical data identifying that the LZ domain of PKG-Iα1-59 interacts with a well defined 42-residue CC motif (MBSct42) within the C terminus of MBS. Using glutathione S-transferase pulldown experiments, chemical cross-linking, size exclusion chromatography, circular dichroism, and isothermal titration calorimetry we identified a weak dimer-dimer interaction between PKG-Iα1-59 and this C-terminal CC domain of MBS. The Kd of this non-covalent complex is 178.0 ± 1.5 μm. Furthermore our 1H-15N heteronuclear single quantum correlation NMR data illustrate that this interaction is mediated by several PKG-Iα residues that are on the a, d, e, and g hydrophobic and electrostatic interface of the C-terminal heptad layers 2, 4, and 5 of PKG-Iα. Taken together these data support a role for the LZ domain of PKG-Iα and the CC domain of MBS in this requisite contractile complex.
Vascular smooth muscle cell tone in blood vessels is clearly coupled to the phosphorylation state of the myosin light chain. This process is tightly regulated by the rates of myosin light chain phosphorylation through the myosin light chain kinase and subsequent dephosphorylation via myosin phosphatase (MLCP)2 (1-6). MLCP is a heterotrimer comprised of a 130-kDa myosin binding subunit (MBS), a 37-kDa protein phosphatase inhibitor catalytic subunit, and a 20-kDa subunit with no ascribed function (1-5). The importance of cGMP-dependent PKG-Iα in vascular smooth muscle tone is supported by extensive studies demonstrating that PKG-Iα regulates pathways of NO-dependent vascular relaxation that involve the MBS of MLCP (6). Protein-protein interactions between PKG-Iα and MLCP are believed to be mediated by the N-terminal leucine zipper (LZ) domain of 59 residues in PKG-Iα (PKG-Iα1-59) and C-terminal 180 residues of MBS (MBSct180) in MLCP (1).
In previous studies by Atkinson et al. (7), they described the dimer properties of PKG-Iα (residues 1-39) and determined the structure of the PKG-Iα monomer within this leucine-zipper dimer. We and others have shown that N-terminal PKG-Iα is a homodimer in solution (7-9). Recently we have demonstrated that the LZ region of PKG-Iα, PKG-Iα1-59, is indeed a coiled coil (CC) LZ dimer of ∼38 residues commencing at residue Ala9 and extending through to Gln44 on the basis of our 15N R1 and R2 relaxation data (10). Our NMR structure of this region of PKG-Iα was determined using residual dipolar couplings as structural restraints, whereas the parallel orientation of this homodimer was determined using Prediction of Alignment from Structure (PALES), a method that evaluates the structural charge distribution of the monomer and dimer structures (10).
The interaction between PKG-Iα1-59 and the CC and/or LZ domain of MBSct180 has recently been documented by several workers (8, 11, 12). MBSct180 is comprised of a predicted binding domain, residues 888-928; a predicted CC domain, residues 929-969; and a C-terminal LZ domain, residues 1013-1039 (12). Huang et al. (11) demonstrated that the interaction between PKG and MBS is independent of the MBS LZ domain and is mediated through the binding domain that was identified previously as a CC domain (13). Given et al. (12) later demonstrated that MYPT1 fragments that are either LZ-positive (MYPT1 LZ+; residues 500-1039) or LZ-negative (MYPT1 LZ-; residues 500-1010) are capable of forming a complex with PKG-Iα in avian smooth muscle tissue lysates. From these studies it was concluded that the LZ domain of MBS although involved in the interaction is not essential. Recently Lee et al. (8) have published data that support that the N-terminal LZ of PKG-Iα binds to both the LZ domain of MYPT1 and/or the CC domain in the context of additional upstream regions of MYPT1 to form a heterotetramer. Taken together these studies provide data in support of the importance of both the CC and/or LZ domain of MBS in the formation of the PKG-Iα·MBS complex; however, there are differences that may be attributed to the different approaches used.
Importantly some of the studies evaluating the interaction between PKG-Iα and MBS have been performed in the presence of both the LZ and CC domains of MBS; this has prevented a detailed understanding of the individual contributions made by each domain, including the CC domain, from being definitively ascribed (8, 11, 12). To support a role for this MBS CC domain, we present data identifying a functional interaction between this domain (residues 929-970), which we refer to as MBSct42, and the N-terminal LZ of PKG-Iα, PKG-Iα1-59. Using GST pulldowns, chemical cross-linking, size exclusion chromatography (SEC), circular dichroism (CD), and isothermal titration calorimetry (ITC) we identified and characterized this interaction between PKG-Iα1-59 and MBSct42 that supports the formation of a dimer·dimer interaction complex. Further 1H-15N HSQC NMR chemical shift perturbation data identify that this interaction is mediated by several PKG-Iα residues that are on the hydrophobic and electrostatic interface of the C-terminal heptad layers of PKG-Iα.
EXPERIMENTAL PROCEDURES
Expression and Purification of PKG-Iα LZ Domain (PKG-Iα1-59) and MBS LZ Domains—Plasmids comprised of the PKG-Iα LZ domain (PKG-Iα1-59) and the C-terminal 100 amino acids (residues 930-1030) of MBS (MBSct100) were used for this investigation. Briefly the 177-bp cDNA insert for PKG-Iα1-59 was cloned into the pGEX-2T vector (GE Healthcare) and expressed with glutathione S-transferase (GST) fusion tag at its N terminus as described previously (10). The 300-bp cDNA insert representative of MBSct100 was cloned into the pQE-30 vector (Qiagen) and expressed with His6 tag at its N terminus. 15N and/or 13C/15N isotopically enriched PKG-Iα1-59 proteins were expressed and purified as described previously (1, 2, 10).
GST-tagged PKG-Iα1-59 was batch-purified on glutathione-Sepharose 4B beads (GE Healthcare) as described previously (1, 2, 10). PKG-Iα fusion protein was incubated with thrombin at 22 °C for 2.5 h. The reaction was quenched by adding 1 mm phenylmethylsulfonyl fluoride, and the cleaved PKG-Iα1-59 was separated from the GST-tagged resin using a low speed spin at 500 × g for 5 min. Supernatant containing PKG-Iα1-59 was extensively dialyzed at 4 °C and purified further using SEC with a HiLoad 16/60 Super-dex 75 Prep Grade column (Amersham Biosciences). The eluted fractions containing PKG-Iα1-59 were pooled, concentrated, and verified using SDS-PAGE. His-tagged MBSct100 was expressed as described above and purified under native conditions using Ni2+-nitrilotriacetic acid (Ni2+-NTA)-agarose (Qiagen) affinity chromatography as described elsewhere (1, 2).
GST-PKG-Iα1-59 His-MBSct100 Pulldown Experiments—Escherichia coli XL-10 Gold cells were transformed with His6-MBSct100 cDNA cloned into the pQE30 vector as described elsewhere (1, 2). Following cell growth and lysis the supernatant was incubated with Ni2+-NTA resin (Invitrogen) and rocked at 4 °C for 16 h. Ni2+-NTA-agarose alone (control) or Ni2+-NTA-agarose-His6-MBSct100 was washed with Triton lysis buffer prior to loading equal amounts of purified GST or GST-PKG-Iα1-59. The resin was incubated overnight at 4 °C prior to elution, and the proteins were eluted with 500 mm imidazole. Binding of GST or GST-PKG-Iα1-59 to the beads was assessed using anti-GST antibodies.
Defining the CC in MBSct180—Sequence-based prediction of the CC region localized within MBSct180 was made using the programs COILS (14) and PAIRCOIL (15).
Chemical Synthesis of MBSct42—The MBSct42 peptide (residues Lys929-Phe970) (KKLYEQILAENEKLKAQLHDTNMELTDLKLQLEKATQRQERG) was synthesized using standard Fmoc (N-(9-fluorenyl)methoxycarbonyl) chemistry as we have described previously (16, 17). The cleaved peptide was purified using C8 preparative reversed-phase high pressure liquid chromatography, and the eluted peptide was purified to homogeneity (>95% pure).
Chemical Cross-linking—Cross-linking experiments were performed with a homobifunctional cross-linker, DSP (Pierce). Time course cross-linking reactions were carried out for 3, 8, and 15 min at 25 °C. DSP was incubated with PKG-Iα1-59 and/or MBSct42 (concentrations, 0.5-1.0 mg/ml) in separate reaction vessels at a molar ratio of 20:1 according to the instructions. To each cross-linked sample, Laemmli buffer (without reducing agent) was added, and the sample was split into two equal fractions. To one set of fractions, DTT was added to a final concentration of 50 mm. The other set of fractions was treated identically without DTT (sample volume maintained equal). Following a predefined incubation period, each reaction was quenched by the addition of 1 m Tris, pH 7.5. Samples were analyzed by SDS-PAGE (16.5% gel).
Size Exclusion Chromatography—SEC was carried out on a Superdex 75 column with a flow rate of 1 ml/min. The column was precalibrated with molecular weight standards (Bio-Rad). Samples were dialyzed in buffer A (20 mm potassium phosphate, 10 mm NaCl, 1 mm EDTA, and 5 mm DTT, pH 7.0) and centrifuged prior to loading onto the column.
CD Spectroscopy—Far-UV CD (260-195 nm) was carried out at 25 °C on a Jasco-810 spectropolarimeter (Jasco, Easton, MD) purged with nitrogen gas. Data were acquired in 1-mm quartz cuvette with 1-nm bandwidth, 2-s response time, 10 nm/min scan speed, and four scans. Samples were dialyzed at 4 °C and centrifuged prior to data acquisition to remove compounds that scatter light. Spectra were recorded using 24 μm PKG-Iα1-59 and 34 μm MBSct42. Data were collected at molar ratios of 1:1 and 1:2 (PKG-Iα1-59 and MBSct42), respectively. These raw data were converted into molar ellipticity (θ × 10-3) (degree cm2 dmol-1), and percent secondary structure was calculated using the program K2D (18, 19).
Isothermal Titration Calorimetry—ITC was carried out at 25 °C with a VP-ITC titration calorimeter (Microcal Inc., Northampton, MA). Samples were centrifuged and degassed prior to use. Binding isotherms were recorded for PKG-Iα1-59 following the injection of 8-μl aliquots of MBSct42 (stock concentration, 630 μm) into a 1.4-ml sample cell. Each experiment consisted of 29 injections performed with a 4-min delay between each titration point to allow the sample to reach equilibrium. We also performed experiments in which MBSct100 (160 μm) was titrated with PKG-Iα1-59 using identical acquisition parameters. These data were analyzed using a non-linear least square program (Microcal, Origin) and fit to a single site or sequential two-site binding model permitting us to determine a dissociation binding constant (Kd), change in enthalpy (ΔH), and/or change in entropy (ΔS). Each experiment was performed in triplicate.
NMR Spectroscopy—Isotopically enriched samples of PKG-Iα1-59 were concentrated to ∼0.3 mm 15N-labeled or 0.8-1 mm 2H/13C/15N-labeled PKG-Iα1-59 (∼90% perdeuteration). 15N-PKG-Iα1-59 samples were prepared in 90% H2O, 10% D2O in the presence of 1 mm NaN3 and 1 mm DSS as an internal standard. NMR data were acquired on a Bruker Avance 500-MHz spectrometer equipped with a 5-mm triple resonance pulsed field gradient (z axis) probe or on a Varian Unity INOVA 500-MHz spectrometer equipped with 5-mm triple resonance pulsed field gradient (z axis) cold probe at 25 and 30 °C. Standard two-dimensional 1H-15N HSQC and three-dimensional deuterium-decoupled triple resonance spectra including 15N-edited three-dimensional nuclear Overhauser effect spectroscopy-HSQC (mixing time, 120 ms) were recorded (10). Chemical shift perturbation studies were carried out by collecting a series of two-dimensional 1H-15N HSQC spectra at 25 °C on uniformly 15N-labeled PKG-Iα1-59 as a function of increasing MBSct42 concentrations until a stoichiometric ratio of 1:2 was reached. Beyond this ratio, no further chemical shift change or peak intensity perturbation was noticed. All NMR data were processed using NMRPipe (20). Chemical shifts were referenced to DSS, and all heteronuclear data were analyzed using Assignment of NMR Spectra by Interactive Graphics (ANSIG) (21, 22). Figures were generated using MOLMOL (23).
RESULTS
Expression and Purification of LZ Domain of PKG-Iα (PKG-Iα1-59) and CC LZ Domain of MBS (MBSct100)
Fig. 1A illustrates the domain architecture of PKG-Iα. The amino acid sequence of PKG-Iα1-59 (Fig. 1A) highlights the leucine/isoleucine residues (rendered in green) positioned in register at the a and d interface of the heptad repeat (abcdefg)n. Two positively charged lysine residues located at position d are shown in blue. Fig. 1B shows the SDS-PAGE of recombinant PKG-Iα1-59 in the presence of 50 mm DTT. Expression of recombinant GST-PKG-Iα1-59 (33.4 kDa) was identified by a band corresponding to this molecular mass (lane 2). Following thrombin cleavage bands corresponding to GST tag (26 kDa) and PKG-Iα1-59 (7.4 kDa) were observed (lane 3). Purified recombinant PKG-Iα1-59 is identified by the monomeric band (labeled “p”) and a low intensity dimeric band (labeled “2p”), which is present in lane 4. Amino acid sequence analysis confirmed that the recombinantly expressed monomeric band at 7.4 kDa corresponds to PKG-Iα1-59 and an additional 8 residues from the GST tag. Electron spray mass spectrometry confirmed the molecular size of PKG-Iα1-59. During our SEC, PKG-Iα1-59 was eluted as a single band with a retention time corresponding to an estimated mass of ∼14.8 kDa, which suggested the presence of a homodimer.
FIGURE 1.

Schematic illustration of PKG-Iα1-59 and its expression and purification. A, the domain structure of PKG-Iα including the N-terminal LZ (PKG-Iα1-59), two cGMP binding sites (G domains), and a C-terminal catalytic domain (CD) is shown. Polar and non-polar residues that are in the a and d positions of each heptad layer are highlighted in blue and green, respectively. B, expression and purification of PKG-Iα1-59 analyzed using 15% (w/v) SDS-PAGE in the presence of 50 mm DTT. Lane 1, protein standards; lane 2, cell lysate demonstrating expression of GST-PKG-Iα1-59 fusion protein (33.4 kDa); lane 3, separation of GST tag (26 kDa) from PKG-Iα1-59 following thrombin cleavage; lane 4, purified PKG-Iα1-59 depicting monomer and dimer fractions, which are labeled as p and 2p, respectively.
NMR Data of PKG-Iα1-59
Using triple resonance NMR experiments as described previously (10), we assigned many of the amino acid residues; however, residues from the GST tag and the residues at the N terminus as well as three C-terminally positioned residues could not be assigned. 1H-15N correlations assigned in two-dimensional 1H-15N HSQC of PKG-Iα1-59 are shown (supplemental Fig. SI1A). Residues from Lys39 to Leu47 (excluding Cys43) were not observed in our non-deuterated 1H-15N HSQC; however, these residues were seen and easily assigned in the data acquired with 2H/15N/13C-labeled PKG-Iα1-59. We believe this is because of the fact that perdeuteration of PKG-Iα1-59 likely slows the relaxation pathways by attenuating 13C-1H dipolar interactions (by a factor of ∼15), which facilitates increased efficiency of magnetization transfer and an improvement in sensitivity and resolution as others have documented (24, 25). 1H and 15N chemical shifts for the portions of heptad layers 3, 4, and 5 and their sequential 15N slices are illustrated as nuclear Overhauser effect strips from three-dimensional 1H-15N nuclear Overhauser effect spectroscopy-HSQC (supplemental Fig. SI1B). These data clearly identify dnn(i, i + 1) and dnn(i, i + 2) contacts. Taken together, these data and previously assigned Cα and Cβ chemical shifts support the presence of an α-helical structure that commences at Ala9 and continues through Gln44 (10).
Interaction between the LZ Domain of PKG-Iα and MBS
GST Pulldown of LZ PKG-Iα1-59 and CC LZ MBSct100—Fig. 2A identifies that neither GST nor GST-PKG-Iα1-59 is bound to the Ni2+ beads. GST-PKG-Iα1-59 but not GST bound to Ni2+-MBSct100 beads identifying a direct interaction between PKG-Iα1-59 and MBSct100 (residues 930-1030).
FIGURE 2.

Characterization of MBS C-terminal domain. A,
GST-PKG-Iα1-59 incubated with Ni2+-NTA-linked
His6-MBSct100 and as a control with
Ni2+-NTA-agarose alone (Invitrogen). Associated proteins were
transferred to nitrocellulose and immunoblotted with mouse anti-GST antibody
(Santa Cruz Biotechnology, Inc.). Membranes were developed with ECL (Amersham
Biosciences). A band representative of a complex between
His6-MBSct100 and
GST-PKG-Iα1-59 is identified, supporting an interaction
between these two proteins. B, CC score prediction of the C-terminal
180 residues of MBS scaled between 0 and 1.0 using the COILS server. These
data were used to define the region of MBS that had the highest probability of
being a truly structured CC domain within MBSct100. The
predicted CC and LZ CC domains of MBS are labeled with
 and #, respectively, within
the C-terminal 100 residues, MBSct100.
and #, respectively, within
the C-terminal 100 residues, MBSct100.
Identification of CC Segment in MBSct180—Fig. 2B illustrates the region of MBS that commences at residue Ser928 (residue 81 using Fig. 2B nomenclature) through residue Lys1030 (of MBSct100 as labeled) that is within MBSct180. The CC domain with the highest probability (∼1.0) is illustrated for MBSct42. To evaluate individual roles for the CC domain versus the LZ domain of MBS within MBSct100 we performed interaction studies with this predicted CC domain, MBSct42 (residues 82-123 in Fig. 2B).
Chemical Cross-linking
Fig. 3A illustrates the cross-linking profile of PKG-Iα1-59. In the presence of 50 mm DTT a monomer of PKG-Iα can be seen (lanes 2, 3, and 4), whereas in the absence of DTT PKG-Iα1-59 forms a dimer (lanes 5, 6, and 7), corresponding to a band migrating with approximately the same mobility as the 15-kDa molecular mass marker. Over the time course studies no change in the oligomeric state was seen except for a slight increase in band intensities with increasing time. In the absence of both DSP and DTT, PKG-Iα1-59 shows a single dimeric band (lane 8). These data suggest that native PKG-Iα1-59 is a homodimer in solution, a finding that was recently supported through analytical ultracentrifugation studies (8).
FIGURE 3.

Chemical cross-linking of PKG-Iα1-59, MBSct42, and the PKG-Iα1-59·MBSct42 complex. A, time course analysis of DSP-cross-linked PKG-Iα1-59. Lane 1, protein standards. Lanes 2, 3, and 4, represent cross-linked PKG-Iα1-59 in the presence of 50 mm DTT at 3, 8, and 15 min, respectively, with monomeric PKG-Iα1-59 (p). Lanes 5, 6, and 7 represent cross-linked PKG-Iα1-59 in the absence of DTT at 3, 8, and 15 min, respectively; the dimer (2p) band is seen. Lane 8, purified PKG-Iα1-59 sample without cross-linker in the absence of DTT shows dimer (2p) conformation. B, time course analysis of DSP-cross-linked MBSct42. Lanes 1, 2, and 3 represent cross-linked MBSct42 in the presence of 50 mm DTT at 3, 8, and 15 min, respectively; monomer (m) and dimer bands (2m) are seen. Lanes 4, 5, and 6 represent cross-linked MBSct42 in the absence of DTT at 3, 8, and 15 min, respectively; bands corresponding to dimer (2m) and a tetramer (4m) are seen. Lane 7, purified MBSct42 sample without cross-linker in the absence of DTT shows monomer (m) conformation. Lane 8, protein standards. C, time course analysis of PKG-Iα1-59·MBSct42 complex. Lane 1, protein standards. Lanes 2, 3, and 4 illustrate cross-linked complex in the presence of 50 mm DTT at 3, 8, and 15 min, respectively; bands corresponding to PKG-Iα1-59 (p) and MBSct42 (m) monomers are seen. Lanes 5, 6, and 7 represent cross-linked complex in the absence of DTT at 3, 8, and 15 min, respectively; bands corresponding to monomer (p) PKG-Iα1-59 and dimer (2m) and tetramer (4m) MBSct42 are seen, and the complex band at ∼25 kDa is labeled with *. Lane 8, purified complex sample without cross-linker in the absence of DTT shows the dimer (2p) of PKG-Iα1-59 and monomer (m) of MBSct42. D, SEC of PKGIα1-59 and MBSct42 complex. SEC of the complex sample (molar ratio of 1:2 for PKG-Iα1-59:MBSct42) performed at 25 °C on a Superdex 75 (120-ml bed volume) column. The chromatogram illustrates the elution profile of the complex, which consisted of three peaks. Of these, two represent the non-interacting proteins, and the third non-Gaussian shaped peak represents the complex. These eluted peaks were identified and correspond to MBSct42 homodimer (10 kDa) (MBSct42d), PKG-Iα homodimer (14.8 kDa) (PKG-Iα1-59D), and non-covalent complex (dimer-dimer interaction) of ∼25 kDa that is labeled with * in the chromatogram. D, dimer. The inset is the SDS-PAGE of this eluted complex sample (lane 2) supporting the presence of a band corresponding to its ∼25-kDa size (marked with *). Lane 1 shows protein standards as indicated in A. As a reference, a chromatogram of bovine aprotinin (6.5 kDa) is also shown.
Fig. 3B shows the cross-linking time course of MBSct42 alone. In the presence of 50 mm DTT, MBSct42 forms both a monomer (weak band) and dimer (increased intensity) (lanes 1, 2, and 3). In the absence of DTT, the dimer conformation was the predominant species with traces of a tetramer conformation present (lanes 4, 5, and 6). In addition, Fig. 3B illustrates a modest increase in band intensity for the dimer conformation with time in the presence of DTT. In control experiments in the absence of DSP and DTT, we identified a single MBSct42 band representative of the monomeric state (lane 7). These data suggest that MBSct42 can also form a homodimer in solution, results that are supported by our SEC studies (Fig. 3D).
These and other biophysical studies identified that a molar ratio of 1:2 (PKG-Iα1-59 to MBSct42) is optimal for the generation of this non-covalent complex (Fig. 3, C and D). In cross-linking studies evaluating the stoichiometry of the complex we determined that in the presence 50 mm DTT monomeric states for both PKG-Iα1-59 and MBSct42 are observed (Fig. 3C, lanes 2, 3, and 4) (supporting Fig. 3, A and B). In the absence of DTT bands for the PKG-Iα1-59 dimer as well as MBSct42 dimer and traces of a tetramer complex (Fig. 3C, lanes 5, 6, and 7) were observed. This tetramer band migrates with a molecular mass of approximately ∼25 kDa (marked with * in lanes 5, 6, and 7). The presence of this ∼25-kDa band is suggestive of a dimer-dimer interaction between homodimers of PKG-Iα1-59 and MBSct42. In the absence of both DTT and DSP, bands corresponding to PKG-Iα1-59 dimer and MBSct42 dimer (lane 8) were observed.
Size Exclusion Chromatography of PKG-Iα1-59·MBSct42 Complex
Our cross-linking studies suggested the presence of a PKG-Iα1-59·MBSct42 non-covalent complex of moderate to weak affinity. These SEC data further verify the presence of a dimer·dimer complex. SEC was performed on a sample prepared by adding a 2:1 excess of MBSct42 to PKG-Iα1-59. Prior to loading onto the column, the sample was gently rocked for 1 h to allow complete mixing (26). Fig. 3D illustrates our SEC data for the PKG-Iα1-59·MBSct42 complex identifying the elution of PKG-Iα1-59 and MBSct42 dimers as single peaks at their respective retention times along with an additional peak with a shorter retention time corresponding to a complex of ∼25 kDa (labeled with * in Fig. 3, C and D) as determined using several internal molecular mass standards including aprotinin. To characterize the composition of the proteins in this peak that elutes with a molecular mass of ∼25 kDa we performed SDS-PAGE and then performed tandem liquid chromatography-mass spectroscopy on this band following tryptic digestion. SDS-PAGE of this band is presented in Fig. 3D, inset. These liquid chromatography-mass spectroscopy data clearly demonstrate that this peak is comprised of amino acid sequence fragments representative of both PKG-Iα1-59 and MBSct42 (supplemental Fig. SI2). Taken together, these data support that the band migrating at ∼25 kDa represents a non-covalent interaction between a dimer of the LZ domain of PKG-Iα1-59 and a dimer of the CC domain of MBS (MBSct42) that results in a non-covalent dimer of dimers. In additional studies we observed that increasing salt concentration from 10 to 100 mm had no effect on complex formation, supporting the specificity of this complex and further ruling out the presence of a nonspecific electrostatic interaction (data not shown).
Circular Dichroism
CD spectra recorded for PKG-Iα1-59, MBSct42, and PKG-Iα1-59·MBSct42 complex are shown in Fig. 4A. Each CD spectrum shows minimum ellipticity at 222- and 208-nm wave-length, supporting the presence of significant α-helical secondary structure in each protein. The estimated percentage of helical content was found to be 74, 99, and 100% in PKG-Iα1-59, MBSct42, and the formed 1:2 (PKG:MBS) complex, respectively. These data identify that upon forming the non-covalent complex there is a modest increase in helicity, which we believe may be the result of this non-covalent interaction between the respective domains. The ratio of molar ellipticity at 222 and 208 nm (θ222:θ208) is crucial in describing CC structures, and it approximates 1.0 for stable CC structures (27-29). The ratio of θ222:θ208 for PKG-Iα1-59 and MBSct42 as well as the 1:2 complex were determined to be 0.95, 1.00, and 0.98, respectively. These data support our 15N T1/T2 data (10) that identified that the CC region of PKG-Iα commences at Ala9 and continues through Gln44. Residues beyond this highly structured region are likely in a random coil conformation.
FIGURE 4.

Characterization of PKG-Iα1-59 and MBSct42 interaction. A, CD spectra of PKG-Iα1-59, MBSct42, and its complex. An overlay of CD spectra from PKG-Iα1-59, MBSct42, and complex collected at 25 °C is shown. Data points for each spectrum are highlighted in symbols as indicated at the top right of the spectra panel. deg, degree. B, ITC of the interaction between MBSct42 and PKG-Iα1-59. Top panel, heat absorbed (μcal/s) (endothermic binding) for each isotherm was plotted against titration time (min). Bottom panel, integrated heats (kcal/mol) were plotted against peptide/protein molar ratio.
Isothermal Titration Calorimetry Studies of PKG-Iα1-59 Interacting with MBSct42
The top panel in Fig. 4B shows the binding isotherms of PKG-Iα1-59 titrated with MBSct42. These data support that the binding between these two molecules is endothermic in nature. These integrated heats were fit to a single site binding model with a stoichiometry of 1:1 (n = 0.91) and an equilibrium dissociation constant (Kd) of 178 ± 1.45 μm. The bottom panel of Fig. 4B supports the presence of a weak endothermic interaction. The endothermic binding enthalpy for this interaction is high (ΔH = 32.90 kcal/mol) with a favorable ΔG of -5.24 kcal/mol. These data support our cross-linking and SEC results that also identified a weak protein-protein interaction between these domains as has been described for many other protein·protein complexes (8, 30-34).
To further understand the complexity of the interaction between PKG-Iα and MBS we performed additional ITC studies to probe the interaction between PKG-Iα1-59 and MBSct100, which is a larger MBS fragment comprised of the CC domain (MBSct42) as well as the LZ domain of MBS (supplemental Fig. SI3). The exothermic/endothermic binding isotherms for this complex fit to a sequential two-site model of interaction, which provided two Kd values, Kd1 of 10.8 ± 1.75 μm and Kd2 of 34 ± 0.61 μm, which are labeled as sites I and II, respectively (supplemental Fig. SI3). These data identify that the second site of interaction is ∼3-fold lower in affinity than the first binding site, which is initiated following saturation of the first site (supplemental Fig. SI3). This type of mixed exothermic/endothermic binding has also been reported previously for other protein-protein interactions including the c-C1C2 interaction with myosin-S2 domain (35), an interaction that is also regulated by phosphorylation. A comparison of the binding affinities obtained for PKG-Iα1-59·MBSct100 and PKG-Iα1-59/MBSct42 clearly supports that the inclusion of the MBS LZ domain with the CC domain improves the affinity of the interaction severalfold. This model of sequential two-site interactions has been well documented for several other novel protein-protein interactions (36-39).
PKG-Iα1-59·MBSct42 Interaction Using Heteronuclear NMR
Cross-linking, SEC, CD, and ITC data support the presence of a weak dimer-dimer interaction between PKG-Iα1-59 and the CC MBSct42. To probe this interaction at the amino acid level, we performed chemical shift perturbation analysis using two-dimensional 1H-15N HSQC titration experiments. These data identified only modest spectral changes at a molar ratio of 1:1 (PKG-Iα1-59:MBSct42). However at a 2:1 ratio of MBSct42 to PKG-Iα1-59 significant amino acid perturbations within PKG-Iα1-59 were observed. Our 1H-15N HSQC spectra of this titration series supported saturation of binding at this stoichiometric ratio, a conclusion observed in our experiments. Fig. 5A illustrates the superposition of 1H-15N HSQC spectra in the absence (pink) and presence of MBSct42 (cyan) at this molar ratio. These data highlight those residues that are perturbed. In the absence of deuteration, the 1H-15N cross-peaks for Lys39, Leu40, Lys42, Gln44, Ser45, Val46, and Leu47 were not observed in the absence of MBSct42. Following the addition of MBSct42 these 1H-15N cross-peaks appeared as a result of altered conformational exchange and/or dipolar relaxation upon complex formation. Interestingly these data suggest that these residues may be within or in close proximity to the interacting interface of PKG-Iα1-59·MBSct42.
FIGURE 5.

Residue perturbation analysis of PKG-Iα1-59 following the interaction with MBSct42. A, two-dimensional 1H-15N HSQC superimposition of uniformly 15N-labeled PKG-Iα1-59 (pink contours) and PKGIα1-59 titrated with MBSct42 (cyan contours) in a molar ration of 1:2. PKG-Iα1-59 residues that show significant perturbation upon titration with MBSct42 (either have shown a chemical shift change or a decrease in peak intensity) are highlighted in red with a single letter amino acid code followed by its position in primary sequence. B, chemical shift perturbation of PKG-Iα1-59. 1H and 15N weighted chemical shifts, Δδweighted = [(Δ1H)2 + (0.1 × Δ15N)2]½ were plotted versus residue number. The perturbation cutoff value is shown by a horizontal line (dashed). Perturbed residues are highlighted as red bars. C, chemical shift perturbation of PKG-Iα1-59. The peak intensity ratios (IComplex/IPKG-Iα1-59) for each residue were plotted against residue number. The perturbation cutoff value is shown by a horizontal line (dashed). Perturbed residues are highlighted as red bars. The amide proton of His41 was not assignable.
The residual perturbation in PKG-Iα1-59 was measured by examining chemical shift change as well as decrease in peak intensity. Chemical shift changes were observed for 5 residues, viz. Leu36, Lys37, Leu40, Ile54, and Gly55, whereas a significant decrease in peak intensity was observed for 8 residues, viz. Leu22, Ile33, Leu36, Lys37, Lys39, Leu40, Cys43, and Gln44 (Fig. 5A). Decreased peak intensity for these residues is likely because of the slowed rotational tumbling of the PKG-Iα1-59·MBSct42 complex, which although estimated at ∼25 kDa will tumble as a much larger complex because of the elongated nature of the CC structure, which tends to have a much larger Stokes radius (40, 41). We also observed chemical exchange broadening following complex formation as measured by the increased line widths within the complex (Fig. 6A). These increased line widths parallel the increase in molecular mass following complex formation and decreased peak intensity, which all support of a role for these residues in the interaction with MBSct42 (40-43).
FIGURE 6.

CC LZ tertiary structure of parallel conformation of PKG-Iα1-59 upon binding to MBSct42. A, helical wheel representation of residues 12-46 of LZ PKG-Iα1-59. Residues in the first heptad layer (from the N terminus) are circled. The repeating heptad positions are labeled a through g with residues placed at their position. Perturbed PKG-Iα1-59 residues following the addition of MBSct42 are highlighted in red. B, lateral view of PKG-Iα1-59 along the principal axis. Side chains of the perturbed residues are highlighted in red.
These findings are clearly present in Fig. 5, B and C, which illustrate the weighted chemical shift change (Δδ) and changed cross-peak intensity ratio, respectively, for the CC LZ of PKG-Iα1-59 (residues 9-44) following the addition of MBSct42. The residual bars above the cutoff value (0.01) in Fig. 5B show the effective chemical shift change (highlighted in red), and the residual bars below the cutoff value (0.57) in Fig. 5C show the effective decrease in cross-peak intensity (highlighted in red). Based on these analyses, residues Leu36, Lys37, Leu40, Ile54, and Gly55 support both chemical shift perturbation and peak intensity decreases, whereas residues Leu22, Cys43, and Gln44 only support significantly decreased peak intensities. Chemical shift changes (Hz) and/or peak intensity (ΔI) decreases for residues Leu22, Leu36, Lys37, and Cys43 have been plotted as two-dimensional contour plots overlaid with their one-dimensional traces to illustrate these perturbations (supplemental Fig. SI4). Taken together, these data identify that residues Leu22, Leu36, Lys37, Leu40, Cys43, Gln44, Ile54, and Gly55 are most affected by the interaction with MBSct42; this suggests that these residues are either within close proximity to and/or involved in the formation of the interaction interface. These MBSct42 interactions are within the intermediate exchange regime on the NMR time scale (Fig. 5A) (41).
When these residues are mapped onto the PKG-Iα1-59 structure, they are localized at a, d, e, and g positions of the heptad repeats as illustrated by a helical wheel representation (shown in red in Fig. 6A). Interestingly many of interacting residues are positioned within the C-terminal fourth and fifth heptad repeats (Fig. 6A). Fig. 6B illustrates the lateral orientation of the homodimer backbone of PKG-Iα1-59 along the C2 axis of symmetry (rendered in cyan) highlighting the side chains of the perturbed residues (rendered in red).
DISCUSSION
The N-terminal residues (∼60) of PKG-Iα represent a canonical CC,
LZ homodimer which utilizes five heptad repeats as illustrated using a
two-dimensional helical wheel representation
(Fig. 6A). This
structure is formed and stabilized by the packing of hydrophobic residues at
the a and d positions of two
PKG-Iα1-59 monomers that associate to form a homodimer
creating an extended interface with a slight left-handed helical twist or
supercoil (see Protein Data Bank code 1ZXA). The register of the
PKG-Iα1-59 CC that we have proposed using residual dipolar
coupling data positions the Leu/Ile residues in the a position
of the repeating heptad unit while aligning additional Leu residues within the
d position. Although most of the a and
d residues are hydrophobic in nature, the two lysine residues
(Lys15 and Lys29) in the d position are
tolerated because of stabilization of these residues through an extensive
hydrogen bonding network between adjacent helical monomers as well as a
hydrophobic contribution of the side-chain atoms with other hydrophobes at the
interface (44). The polar
residues that flank this predominantly hydrophobic face (e and
g positions) provide additional electrostatic interactions
that contribute to the stability of the PKG-Iα1-59 dimer.
These interactions likely contribute through the formation of interhelical ion
pairs between residues in the gi and
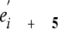 positions including Arg18/Glu23,
Arg25/Glu30, and Glu32/Lys37
(Fig. 6A) as have been
observed by others
(45-49).
positions including Arg18/Glu23,
Arg25/Glu30, and Glu32/Lys37
(Fig. 6A) as have been
observed by others
(45-49).
A better understanding of the PKG-Iα1-59 dimer structure has permitted us to query its interaction with MBS at the residue level. We and others have reported an interaction between PKG-Iα and MBS that is mediated via an interaction involving the N-terminal CC LZ of PKG-Iα and the region of MBS that is within the MBSct180 or AL9 (1, 12). Although having established the importance of the N-terminal LZ domain of PKG-Iα, PKG-Iα1-59, in this interaction, the role of the LZ, CC, and/or binding domain of MBS in this interaction remains under investigation (1, 8, 11, 12).
Our data support a functional interaction between the N-terminal LZ of PKG-Iα and the CC motif of MBS (residues 929-970), MBSct42. Using several biophysical approaches complemented with heteronuclear NMR methods we identified residues within the CC as well as outside of this region of the PKG LZ PKG-Iα1-59 requisite for mediating the interaction between PKG-Iα1-59 and the MBS CC domain.
The possibility of this domain-domain interaction was recently proposed by Lee et al. (8); however, their approach, which involved the use of analytical ultracentrifugation and surface plasmon resonance, precluded a definitive answer because of the experimental limitations of these techniques as they noted. It should be noted that their comprehensive study identified several MBS domains that were critical for this interaction, including the C-terminal LZ domain and the CC domain that we have studied, MBSct42 (8). MBSct100, which interacts with PKG-Iα1-59 as observed in our GST pulldown experiments (Fig. 2A), is comprised of both the CC domain studied here and the canonical LZ domain of MBS at the C terminus. Analogous to their surface plasmon resonance data, our ITC experiments in the presence of this extended MBS fragment (MBSct100) resulted in a similar binding affinity. Our ITC data suggest that inclusion of the LZ domain with the CC domain of MBS decreases the Kd, which argues in favor of a cooperative binding model for these two domains when binding the LZ of PKG-Iα1-59. Taken together these data support a role for both of these domains, a conclusion that is supported by other recent findings (8, 11, 12).
To further probe this interaction we used 1H-15N HSQC perturbation analysis, which provides a weighted chemical shift (Δδ) (Fig. 5) and peak intensity ratio (ΔI) of bound versus free (supplemental Fig. SI4) to identify those residues of PKG-Iα1-59 that are within the interaction interface of this domain-domain interaction. These studies identified that PKG-Iα1-59 residues Leu22, Ile33, Leu36, Lys37, Lys39, Leu40, Cys43, Gln44, Ile54, and Gly55 are critical for mediating this dimer-dimer interaction (Fig. 6B). Interestingly many of the interaction interface residues, Lys37, Lys39, Cys43, Gln44, Ile54, and Gly55, are unique to the PKG-Iα isoform suggesting the specificity of these residues in the PKG-Iα and MBS interaction because the PKG-Iβ isoform does not interact with MBS (50, 51). The hydrophobic residues that are conformationally affected upon complex formation, Leu22, Ile33, Leu36, and Leu40, are localized within the a/d hydrophobic core of the heptad repeat and are shared by both PKG-Iα and PKG-Iβ supporting a structural role for these residues, a notion that has recently been supported by the extensive structural studies of GCN4 and LZ variants (52, 53). In addition, the interaction interface of PKG-Iα1-59·MBSct42 contains two charged lysines (Lys37 and Lys39) that are in the e and g positions of the fourth heptad repeat of PKG-Iα1-59, respectively. Based on our structure (10) these lysines are partially exposed to the solvent extending out from the CC structure, which may permit them to form salt bridges with the Glu residues at g position in MBS. It is interesting that Harbury et al. (54) have reported that upon tetramer formation (a dimer of dimers) as we identified here the interacting dimers do improve their packing density at the intermolecular interface through perturbations within the a, d, e, and g positions. However, although interesting, these details remain speculative in the absence of a structure of this complex.
In a study that was recently published, we further evaluated the biological role of MBSct100 in mediating the interaction with PKG-Iα1-59 (55). Using “knock-in” mice that possessed critical leucine/isoleucine residues within the LZ heptad repeat mutated to alanine in full-length PKG-Iα, it was demonstrated that the direct interaction between PKG-Iα and myosin phosphatase is fully disrupted; however, the mutant PKG-Iα protein in these vascular smooth muscle cells retained its ability to respond to cGMP (55). Although we did not directly evaluate whether this PKG-Iα mutant exists as a monomer or a dimer in this study, we have extensively characterized similar mutations within PKG-Iα previously and determined that these alanine mutations did not disrupt dimer formation of the PKG-Iα leucine zipper domain in vitro but did result in a loss of the protein-protein interaction between PKG-Iα and AL9 (MBSct180) (2). Differential binding of PKG-Iα to MBS in mouse vascular smooth muscle cells from wild type mice versus mice in which the PKG-Iα LZ domain was mutated was validated using GST fusion protein pulldown experiments. In support of the interaction interface proposed here wild type PKG-Iα bound MBS as expected, but PKG-Iα from vascular smooth muscle cells in which the LZ CC domain harbored L/IΔA mutations failed to interact with GST-MBS as reported previously (55).
Burgoyne et al. (9) recently demonstrated that endogenous hydrogen peroxide caused an interprotein disulfide bond between cysteine residues (Cys43) of each monomer of PKG-Iα. This rendered the kinase catalytically active independent of cGMP. It was demonstrated further that PKG-Iα wild type, which is a dimer, but not PKG-Iα in which the Cys43 has been mutated to Ser43, was co-purified with MBS (9). Importantly all of our NMR data were collected in the presence of 5 mm DTT (an ∼10-fold excess with respect to protein concentration), and under these conditions we determined that Cys43 of PKG-Iα1-59 is predominantly (but not entirely) in the oxidized conformation (13Cβ chemical shift of 43.4 ppm) in agreement with the results obtained from other experimental approaches used in this study. In their supplemental data they identify that PKG-Iα activity is reduced ∼4-5-fold when the protein is subjected to oxidation and then reduction, an event that may be occurring in our binding experiments and may help explain the binding constant we determined. This process of reversible oxidation is enhanced when the cysteine of interest is in the reactive thiolate state, a physiochemical event that is facilitated by the presence of a basic environment such as that provided by neighboring lysine and arginine residues around Cys43. Our structural studies both in this current investigation and in a previous publication (10) strongly support that Cys43 is juxta-posed to several lysine and arginine residues, which contribute to lowering the pKa of PKG-Iα and generating the reactive thiolate species of this cysteine residue, which may contribute to the presence of the oxidized form in the presence of DTT.
1H-15N HSQC chemical shift perturbation analysis identified a modest chemical shift perturbation of the backbone amide of Cys43 upon complex formation with the CC of MBS, suggesting its involvement in this interaction (supplemental Fig. SI4). However, although interesting for probing protein-protein interactions by evaluating backbone (or NH-bearing side-chain) conformational changes, these 1H-15N chemical shift data do not permit us to speculate on side-chain conformations of all PKG-Iα1-59 residues in this dimer·dimer complex. Complete 13C and 15N spectral assignment are needed to comment on this further.
In conclusion, we have significantly extended our structural studies of the CC LZ domain of PKG-Iα, PKG-Iα1-59. Through the use of chemical cross-linking, GST pulldown, size exclusion chromatography, tandem mass spectrometry, circular dichroism, and isothermal titration calorimetry experiments, we demonstrated that 1) the N-terminal LZ domain of PKG-Iα, PKG-Iα1-59, and the CC MBSct42 domain both form homodimers in solution, 2) these homodimers interact to form a non-covalent dimer of dimers or a heterotetramer, and 3) many of the residues that form the interaction interface are unique to the PKG-Iα LZ. Taken together, these data provide important details necessary to support a role for the CC domain of MBS in the formation of the PKG-Iα-MBS interaction.
The loss of this spatial and temporal regulatory interaction between PKG-Iα·MBS may contribute to cardiovascular diseases including hypertension and atherosclerosis (56). Through the identification of these and other molecular details of the complex interaction between PKG-Iα and MBS we may be in a position to provide additional information requisite to understanding the importance of this interaction in cardiovascular disease.
Supplementary Material
Acknowledgments
We thank Drs. Marianne Grant and Mingdong Huang for assisting with the expression of PKG-Iα and helpful discussion. We acknowledge support from Dr. James D. Baleja (Tufts University School of Medicine, Boston, MA) for providing access to CD and ITC instruments.
This work was supported, in whole or in part, by National Institutes of Health Grants 1PO1HL077378 (to M. E. M.) and 1PO1HL077378 and 1RO1HL74069 (to H. K. S.). This work was also supported by an Atorvastatin Research Award (to A. C. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. SI1-SI4.
Footnotes
The abbreviations used are: MLCP, myosin light chain phosphatase; LZ, leucine zipper; CC, coiled coil; PKG-Iα1-59, N-terminal leucine zipper domain of 59 residues in PKG-Iα; MBSct42, C-terminal 42 amino acids of MBS; HSQC, heteronuclear single quantum correlation; DSS, sodium 2,2-dimethyl-2-silapentane-5-sulfonate; DSP, dithiobis(succinimidyl propionate); MBS, myosin binding subunit; GST, glutathione S-transferase; MBSct180, C-terminal 180 residues of MBS; PKG, cGMP-dependent protein kinase; SEC, size exclusion chromatography; ITC, isothermal titration calorimetry; MBSct100, C-terminal 100 amino acids of MBS; Ni2+-NTA, Ni2+-nitrilotriacetic acid; DTT, dithiothreitol.
References
- 1.Surks, H. K., Mochizuki, N., Kasai, Y., Georgescue, S. P., Tang, K. M., Ito, M., Lincoln, T. M., and Mendelsohn, M. E. (1999) Science 286 1583-1587 [DOI] [PubMed] [Google Scholar]
- 2.Surks, H. K., and Mendelsohn, M. E. (2003) Cell. Signal. 15 937-944 [DOI] [PubMed] [Google Scholar]
- 3.Mendelsohn, M. E. (2005) J. Clin. Investig. 115 840-844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Somlyo, A. P., and Somlyo, A. V. (1994) Nature 372 231-236 [DOI] [PubMed] [Google Scholar]
- 5.Hartshorne, D. J., and Hirano, K. (1999) Mol. Cell. Biochem. 190 79-84 [PubMed] [Google Scholar]
- 6.Mendelsohn, M. E. (2005) Nat. Med. 11 115-116 [DOI] [PubMed] [Google Scholar]
- 7.Atkinson, R., Saudek, V., Huggins, J., and Pelton, J. (1991) Biochemistry 30 9387-9395 [DOI] [PubMed] [Google Scholar]
- 8.Lee, E., Hayes, D. B., Langsetmo, K., Sundberg, E. J., and Tao, T. C. (2007) J. Mol. Biol. 373 1198-1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgoyne, J. R., Madhani, M., Cuello, F., Charles, R. L., Brennan, J. P., Schroder, E., Browning, D. D., and Eaton, P. (2007) Science 317 1393-1397 [DOI] [PubMed] [Google Scholar]
- 10.Schnell, J. R., Zhou, G.-P., Zweckstetter, M., Rigby, A. C., and Chou, J. J. (2005) Protein Sci. 14 2421-2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang, Q. Q., Fisher, S. A., and Brozovich, F. V. (2004) J. Biol. Chem. 279 597-603 [DOI] [PubMed] [Google Scholar]
- 12.Given, A. M., Ogut, O., and Brozovich, F. V. (2007) Am. J. Physiol. 292 C432-C439 [DOI] [PubMed] [Google Scholar]
- 13.Langsetmo, K., Stafford, W. F., III, Mabuchi, K., and Tao, T. (2001) J. Biol. Chem. 276 34318-34322 [DOI] [PubMed] [Google Scholar]
- 14.Lupas, A., Van, D. M., and Stock, J. (1991) Science 252 1162-1164 [DOI] [PubMed] [Google Scholar]
- 15.Berger, B., Wilson, D. B., Wolf, E., Tonchev, T., Milla, M., and Kim, P. S. (1995) Proc. Natl. Acad. Sci. U. S. A. 92 8259-8263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falls, L. A., Furie, B. C., Jacobs, M., Furie, B., and Rigby, A. C. (2001) J. Biol. Chem. 276 23895-23902 [DOI] [PubMed] [Google Scholar]
- 17.Grant, M. A., Hansson, K., Furie, B. C., Furie, B., Stenflo, J., and Rigby, A. C. (2004) J. Biol. Chem. 279 32464-32473 [DOI] [PubMed] [Google Scholar]
- 18.Andrade, M. A., Chacón, P., Merelo, J. J., and Morán, F. (1993) Protein Eng. 6 383-390 [DOI] [PubMed] [Google Scholar]
- 19.Merelo, J. J., Andrade, M. A., Prieto, A., and Morán, F. (1994) Neurocomputing 6 443-454 [Google Scholar]
- 20.Delaglio, F., Grzesiek, S., Vuister, G. W., Zhu, G., Pfeifer, J., and Bax, A. (1995) J. Biomol. NMR 6 277-293 [DOI] [PubMed] [Google Scholar]
- 21.Kraulis, P. J. (1989) J. Magn. Reson. 24 627-633 [Google Scholar]
- 22.Kraulis, P. J., Domaille, P. J., Campbell-Burk, S. L., van Aken, T., and Laue, E. D. (1994) Biochemistry 33 3515-3531 [DOI] [PubMed] [Google Scholar]
- 23.Koradi, R., Billeter, M., and Wüthrich, K. (1996) J. Mol. Graph. 14 51-55 [DOI] [PubMed] [Google Scholar]
- 24.Gardner, K. H., and Kay, L. E. (1998) Annu. Rev. Biophys. Biomol. Struct. 27 357-406 [DOI] [PubMed] [Google Scholar]
- 25.Goto, N. K., and Kay, L. E. (2000) Curr. Opin. Struct. Biol. 10 585-592 [DOI] [PubMed] [Google Scholar]
- 26.Malia, T. J., and Wagner, G. (2007) Biochemistry 46 514-525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishigaki, T., Ohki, I., Utsunomiya-Tate, N., and Tate, S. (2007) J. Biochem. 141 855-866 [DOI] [PubMed] [Google Scholar]
- 28.Cooper, T. M., and Woody, R. W. (1990) Biopolymers 30 657-676 [DOI] [PubMed] [Google Scholar]
- 29.Zhou, N. E., Kay, C. M., and Hodges, R. S. (1992) J. Biol. Chem. 267 2664-2670 [PubMed] [Google Scholar]
- 30.Noble, C. G., Hollingworth, D., Martin, S. R., Ennis-Adeniran, V., Smerdon, S. J., Kelly, G., Taylor, I. A., and Ramos, A. (2005) Nat. Struct. Mol. Biol. 12 144-151 [DOI] [PubMed] [Google Scholar]
- 31.Vaynberg, J., and Qin, J. (2006) Trends Biotechnol. 24 22-27 [DOI] [PubMed] [Google Scholar]
- 32.Collins, B. M., Praefcke, G. J. K., Robinson, M. S., and Owen, D. J. (2003) Nat. Struct. Biol. 10 607-613 [DOI] [PubMed] [Google Scholar]
- 33.Kang, R. S., Daniels, C. M., Francis, S. A., Shih, S. C., Salerno, W. J., Hicke, L., and Radhakrishnan, I. (2003) Cell 113 621-630 [DOI] [PubMed] [Google Scholar]
- 34.Zhang, T., and Johansson, J. S. (2003) Biophys. J. 85 3279-3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gruen, M., Prinz, H., and Gautel, M. (1999) FEBS Lett. 453 254-259 [DOI] [PubMed] [Google Scholar]
- 36.Lamb, H. K., Thompson, P., Elliot, C., Charles, I. G., Richards, J., Lockyer, M., Watkins, N., Nichols, C., Stammers, D. K., Bagshaw, C. R., Cooper, A., and Hawkins, A. R. (2007) Protein Sci. 16 2391-2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soupene, E., Serikov, V., and Kuypers, F. A. (2008) J. Lipid Res. 49 1103-1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Green, S. N., Foss, T. R., and Kelly, J. W. (2005) Proc. Natl. Acad. Sci. U. S. A. 102 14545-14550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foss, T. R., Kelker, M. S., Wiseman, R. L., Wilson, I. A., and Kelly, J. W. (2005) J. Mol. Biol. 347 841-854 [DOI] [PubMed] [Google Scholar]
- 40.Matsuo, H., Walters, K. J., Teruya. K., Tanak, T., Gassner, G. T., Lippard, S. J., Kyogoku, Y., and Wagner, G. (1999) J. Am. Chem. Soc. 121 9903-9904 [Google Scholar]
- 41.Shekhtman, A., Ghose, R., Wang, D., Cole, P. A., and Cowburn, D. (2001) J. Mol. Biol. 314 129-138 [DOI] [PubMed] [Google Scholar]
- 42.Amezcua, C. A., Harper, S. M., Rutter, J., and Gardner, K. H. (2002) Structure (Lond.) 10 1349-1361 [DOI] [PubMed] [Google Scholar]
- 43.Hamilton, K. S., Ellison, M. J., and Shaw, G. S. (2000) J. Biomol. NMR 18 319-327 [DOI] [PubMed] [Google Scholar]
- 44.O'Shea, E. K., Lumb, K. J., and Kim, P. S. (1993) Curr. Biol. 3 658-667 [DOI] [PubMed] [Google Scholar]
- 45.Monera, O. D., Kay, C. M., and Hodges, R. S. (1994) Biochemistry 33 3862-3871 [DOI] [PubMed] [Google Scholar]
- 46.Mason, J. M., and Arndt, K. M. (2004) Chembiochem 5 170-176 [DOI] [PubMed] [Google Scholar]
- 47.O'Shea, E. K., Rutkowski, R., and Kim, P. S. (1992) Cell 68 699-708 [DOI] [PubMed] [Google Scholar]
- 48.Junius, F. K., Mackay, J. P., Bubb, W. A., Jensen, S. A., Weiss, A. S., and King, G. F. (1995) Biochemistry 34 6164-6174 [DOI] [PubMed] [Google Scholar]
- 49.Freedman, S. J., Song, H. K., Xu, Y., Sun, Z. Y., and Eck, M. J. (2003) J. Biol. Chem. 278 13462-13467 [DOI] [PubMed] [Google Scholar]
- 50.Schlossmann, J., Ammendola, A., Ashman, K., Zong, X., Huber, A., Neubauer, G., Wang, G. X., Allescher, H. D., Kroth, M., Wilm, M., Hofmann, F., and Ruth, P. (2000) Nature 404 197-201 [DOI] [PubMed] [Google Scholar]
- 51.Surks, H. (2007) Circ. Res. 101 1078-1080 [DOI] [PubMed] [Google Scholar]
- 52.Deng, Y., Zheng, Q., Liu, J., Cheng, C. S., Kallenbach, N. R., and Lu, M. (2007) Protein Sci. 16 323-328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steinmetz, M. O., Jelesarov, I., Matousek, W. M., Honnappa, S., Jahnke, W., Missimer, J. H., Alexandrescu, A. T., and Kammerer, R. A. (2007) Proc. Natl. Acad. Sci. U. S. A. 104 7062-7067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Harbury, P. B., Zhang, T., Kim, P. S., and Alber, T. (1993) Science 262 1401-1407 [DOI] [PubMed] [Google Scholar]
- 55.Michael, S. K., Surks, H. K., Wang, Y., Zhu, Y., Blanton, R., Jamnongjit, M., Aronovitz., M., Baur, W., Ohtani, K., Wilkerson, M. K., Bonev, A. D., Nelson, M. T., Karas, R. H., and Mendelsohn, M. E. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 6702-6707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tang, K. M., Wang, G. R., Lu, P., Zhu, Y., and Mendelsohn, M. E. (2003) Nat. Med. 9 1506-1512 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.