

Abstract
The yeast nucleosome assembly protein 1 (yNap1) plays a role in chromatin maintenance by facilitating histone exchange as well as nucleosome assembly and disassembly. It has been suggested that yNap1 carries out these functions by regulating the concentration of free histones. Therefore, a quantitative understanding of yNap1-histone interactions also provides information on the thermodynamics of chromatin. We have developed quantitative methods to study the affinity of yNap1 for histones. We show that yNap1 binds H2A/H2B and H3/H4 histone complexes with low nm affinity, and that each yNap1 dimer binds two histone fold dimers. The yNap1 tails contribute synergistically to histone binding while the histone tails have a slightly repressive effect on binding. The (H3/H4)2 tetramer binds DNA with higher affinity than it binds yNap1.
Histone chaperones are a diverse group of acidic proteins that bind histones and participate in chromatin assembly and disassembly during replication and transcription. Many members of the various chaperone families play additional and often ill-described roles in cell cycle regulation, apoptosis, and DNA damage repair. Several histone chaperones exhibit tissue-specific functions in transcription regulation (recently reviewed in Refs. 1, 2). As the dynamic nature of nucleosomes and chromatin has become evident, the role of histone chaperones in the modulation of chromatin structure is increasingly recognized. However, little mechanistic insight into the processes of chaperone-mediated nucleosome assembly and disassembly is available. In particular, no quantitative information exists on histone binding, the most fundamental function of histone chaperones.
Nucleosome assembly protein 1 (Nap1)4 was one of the first histone chaperones to be identified (reviewed in Refs. 3, 4). The structure of yeast Nap1 (yNap1) reveals a novel fold that is likely conserved among other members of the Nap1 family (5). At low micromolar concentrations yNap1 exists as a homodimer (6, 7). The large dimer interface is predominantly hydrophobic in character and buries ∼22% of the overall surface of yNap1(5); it is therefore unlikely that yNap1 exists in a monomeric state except for perhaps under the most dilute conditions (6). All Nap1 family members have a C-terminal acidic domain (CTAD) of varying length that is not required for histone binding and chromatin assembly, and a variable N-terminal tail of unknown function (3).
Nap1 binds all four core histones as well as the linker histone H1 (8–10). Nap1-mediated nucleosome formation in vitro is characterized by the transfer of a (H3/H4)2 tetramer onto DNA, followed by the incorporation of H2A/H2B dimer (8). Nakagawa et al. (11) have qualitatively shown that the affinity of the (H3/H4)2 tetramer for DNA exceeds its affinity for Nap1, and that the affinity of H2A/H2B for a DNA-bound (H3/H4)2 tetramer (a tetrasome) is greater than its affinity for Nap1.
Our understanding of the thermodynamics of chaperonehistone interactions in general, and of Nap1-histone interactions in particular is very limited. Qualitative data have been obtained from gel-shift and pull-down experiments. Depending on input stoichiometries, yNap1-histone complexes appear on gels shifts in at least two distinct complexes, which complicates the interpretation of these data (9). Pull-down experiments are notoriously difficult to quantify. This has resulted in difficulty interpreting results especially regarding the contributions of histone tails and yNap1 tails on yNap1-histone interaction (9).
We and others (10, 12) have previously found that yNap1 promotes nucleosome disassembly under certain conditions, and that it is capable of exchanging histones and their variants into nucleosomes. The latter function is of particular interest, because it is currently not well understood how histone variants and replacement histones (non-allelic isoforms of major type histones with distinct amino acid sequence, expression patterns, and chromosomal locations) are incorporated into chromatin in a replication-independent manner. Furthermore, these two noncanonical chaperone functions require the CTAD (9, 10). The simplest model that explains the diverse roles of yNap1 is that the chaperone prevents the incorrect binding of histones to DNA or other histones, thereby promoting formation of canonical chromatin. This model of yNap1 function would also allow for histone exchange through establishing an equilibrium between yNap1-bound and nucleosomal histones. This model predicts a thermodynamic link between the nucleosome and yNap1. Thus, a quantitative and systematic exploration of the interactions of yNap1 with histones not only forms the basis for our understanding of the many functions of yNap1, but will also provide information on the extreme upper limits of nucleosome thermodynamic constants.
EXPERIMENTAL PROCEDURES
Reagents—Wild-type, mutant, and tail-less Xenopus laevis histones (amino acids 14–118 for H2A, 24–122 for H2B, 27–135 for H3, and 20–102 for H4) histones were prepared as described (12, 13). The H2B(T112C) and H4(E63C) were prepared according to Ref. 14. Cysteine-to-alanine mutagenesis of yNap1 was done using QuikChange mutagenesis. The resulting triple mutant, C200A/C249A/C272A, further referred to as (yNap1m), leaves only the Cys-414 position available for labeling. Complex formation between H2A/H2B dimer and yNap1 was tested using native gel electrophoresis. All mutant yNap1 constructs interact with H2A/H2B dimer in the same manner as wild-type yNap1, as checked by gel-shift. Both Alexa 488 and 546 were purchased from Invitrogen, and labeling was done as described (14). Biotin polyethyleneoxide iodoacetamide was purchased from Sigma-Aldrich and attached to the proteins via the same protocol as the Alexa dyes.
Fluorescence Titrations—Fluorescence was measured using either an AVIV model ATF105 or a Horiba Jobin Yvon Fluorolog-3 spectrofluorometer. Labeled protein was added to both the sample and the reference cuvette, with non-labeled protein added to the sample cuvette and buffer added to the reference. Varying incubation times (2–15 min) confirmed that the fluorescence signal had reached equilibrium. The normalized fluorescence change was determined by Equation 1,
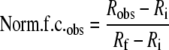 |
(Eq. 1) |
where Robs is the observed ratio of the fluorescence signal (sample cuvette signal/reference cuvette signal), Ri is ratio of the fluorescence initial, and Rf is the ratio of the fluorescence final or where saturation is reached. While the magnitude of the signal change was constant for each experiment, it varied from 10 to 30% between different experiments (i.e. labeled yNap1 binding H2A/H2B versus labeled H2A/H2B binding to yNap1) and with the label used (546 or 488 Alexa). We also monitored the normalized fluorescence ratio of protein titrated into its corresponding binding partner in either buffer or 5 m guanidium-HCl. The presence of guanidium-HCl did not alter the initial signal (pre-addition of the binding partner) nor did the signal change with the addition of μm concentrations of the binding partner in guanidium HCl.
Biotin Pull-down Experiments—Biotin-tagged yNap1 was used to pull Alexa-labeled histones out of solution to confirm that we were monitoring binding under low protein concentrations. Trace amounts (5–30 × 10–9 m) of Alexa-labeled protein were mixed with either biotin-tagged protein at concentration 5–10-fold above the observed Kd or buffer and incubated for 5–20 min. The reaction mix was added to BioMag streptavidin (Qiagen) in a 100-fold excess and incubated for 30 min, while shaking in a 96-well plate. The plate was then placed on a 96-well magnet (Qiagen) and incubated for 15 min. Once the beads were pulled-down, the buffer was removed and checked for total fluorescence. Beads were then washed extensively, and the wash was also checked for fluorescence.
Data Analysis—Affinity measurements were done using concentrations of labeled protein (P) that were at least 5–10-fold less than the Kappd. Kappd was determined by fitting Equation 2 derived from Reaction Scheme 1 to the normalized f.c.,
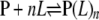 |
REACTION SCHEME 1 |
 |
(Eq. 2) |
which was observed as a function of L, where Lt is the total concentration of protein titrated, nH is the Hill coefficient, and Kd is the apparent dissociation constant. The nH was assumed to be one unless the data dictated otherwise. When the nH was determined not to be equal to one, the linearized form of Equation 2 was used as Equation 3,
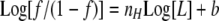 |
(Eq. 3) |
where f is equal to the normalized f.c. divided by the normalized f.c.max. A dimerization model (Reaction Scheme 2) was also used to determine if this could explain the observed cooperativity.
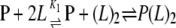 |
REACTION SCHEME 2 |
In this model, the normalized f.c. is still described by Equation 2 except nH = 1 and Lt is replaced by (L)2, where (L)2 is defined in Equation 4.
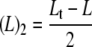 |
(Eq. 4) |
In Equation 4, Lt is total concentration of protein titrated, and L is calculated in Equation 5,
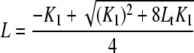 |
(Eq. 5) |
where K1 is the dimerization constant, and Lt is total concentration of protein titrated. Stoichiometries were determined by fluorescence titrations with labeled protein concentration increased to >10-fold higher the Kd. The fluorescence ratio was plotted as a function of the ratio of protein titrated to labeled protein. Under these conditions, the protein ratio at which the normalized f.c. leveled off is equal to the stoichiometry. All data analysis was performed using either Kaleidagraph or Graphpad Prism.
RESULTS
A Quantitative Assay to Study Nap1-Histone Interactions—To determine the binding affinity of yNap1 to histones we developed a series of fluorescence binding assays. yNap1 contains four cysteines in the structured region of yNap1. These were changed to alanine (yNap1m), while the cysteine that is located in the unstructured CTAD was labeled with either Alexa-546 or -488. With the exception of Cys-272 (at the tip of β4), these residues are not conserved among the various members of the family. The addition of unlabeled H2A/H2B dimer to a preparation of yNap1m labeled with Alexa-488 (yNap1m*) resulted in a decrease in fluorescence (Fig. 1A). To exclude the possibility that either mutation or derivatization of Cys-414 have an impact on the affinity of yNap1 for histones, we measured the interaction of an H2A/H2B(T112C) dimer in which H2B had been labeled with Alexa-546. H2BT112 is located in the well-defined αC-helix and is solvent-exposed; the attachment of a fluorescent label to this position is not expected to disrupt the structure of the histone dimer (15). Addition of the unlabeled protein (in this case wild-type yNap1) resulted in a change in fluorescence (Fig. 1).
FIGURE 1.

Fluorescence change as a function of protein binding. A, fluorescence (A.U.) as a function of wavelength (nm), with closed circles/squares representing the signal from the excitation of Alexa-488 (yNap1m*) in the presence (closed squares) or absence (closed circles) of H2A/H2B and the open circles/squares representing Alexa-546 (H2A/H2B(T112C)) in the presence (open squares) or absence (open circles) of yNap1. B, normalized fluorescence change as a function of either H2A/H2B titrated into Alexa-546-labeled yNap1 (closed squares) or yNap1 titrated into Alexa-546 labeled H2A/H2B (open circles). C, change in fluorescence plotted as a function of the ratio of H2A/H2B to yNap1. The intersection of the linear phase with the plateau is equal to the molar ratio at which yNap1 is saturated. These data suggest that two H2A/H2B dimers bind one yNap1 dimer or that one H2A/H2B binds one yNap1 monomer. The conditions were 20–50 mm Tris, pH 7.5, 1 mm dithiothreitol, 0.1 mg/ml bovine serum albumin, 90–120 mm NaCl, (1–5) × 10–10 m Alexa-labeled protein, and (0–5) × 10–7 m non-labeled protein. For results of the fit see Table 1.
The quantitative analysis of yNap1-histone interactions is complicated by the oligomeric nature of all interaction partners. The minimal unit of yNap1 is very likely a homodimer, but we cannot completely exclude a monomer-dimer equilibrium at the low concentrations used in some of the assays (7). H2A/H2B very likely exists exclusively as a heterodimer under physiological conditions (16), whereas the (H3/H4)2 tetramer may dissociate into two half-tetramers (consisting of a histone fold dimer of one copy each of H3 and H4) under certain conditions, although this has not been investigated systematically (17, 18). When either histone complex is refolded, it elutes as an H2A/H2B dimer and (H3/H4)2 tetramer, respectively (13). For ease of comparison, all numbers given here assume a single chain of yNap1, and one histone fold dimer for either H2A/H2B or H3/H4, unless otherwise stated.
The labeled protein was kept at ∼0.5–0.05 nm (or
10-fold below the 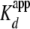 ) while the
binding partner was titrated to ∼10–9 to
10–6 m. The ratio of these titrations to a
titration of equivalent buffer volumes is plotted in
Fig. 1B. The
fluorescence change is reversed with the addition of high concentrations of
salt (>850 mm final concentration, data not shown).
Fig. 1B shows the
normalized fluorescence change as a function of either H2A/H2B dimer titrated
into Alexa-546-labeled yNap1 (yNap1m*) or wild-type yNap1 titrated
into Alexa-546 labeled H2A/H2B (T112C) dimer. The two curves (assuming yNap1
as a monomer) superimpose almost exactly, clearly demonstrating the validity
of our assay. At concentrations above 10–5 m, a
second phase was observed that did not plateau at the maximal concentrations
testable (>10–4 m, not shown). This second
phase is consistent with gel-shift experiments, where multiple higher shifts
were observed at simular concentrations
(9). We assume that the higher
order gel-shifts and the second phase of fluorescence seen at high
concentrations are due to nonspecific interactions between histones and the
chaperone.
) while the
binding partner was titrated to ∼10–9 to
10–6 m. The ratio of these titrations to a
titration of equivalent buffer volumes is plotted in
Fig. 1B. The
fluorescence change is reversed with the addition of high concentrations of
salt (>850 mm final concentration, data not shown).
Fig. 1B shows the
normalized fluorescence change as a function of either H2A/H2B dimer titrated
into Alexa-546-labeled yNap1 (yNap1m*) or wild-type yNap1 titrated
into Alexa-546 labeled H2A/H2B (T112C) dimer. The two curves (assuming yNap1
as a monomer) superimpose almost exactly, clearly demonstrating the validity
of our assay. At concentrations above 10–5 m, a
second phase was observed that did not plateau at the maximal concentrations
testable (>10–4 m, not shown). This second
phase is consistent with gel-shift experiments, where multiple higher shifts
were observed at simular concentrations
(9). We assume that the higher
order gel-shifts and the second phase of fluorescence seen at high
concentrations are due to nonspecific interactions between histones and the
chaperone.
To confirm that the first change in fluorescence was indeed due to the
interaction between histones and yNap1, we used biotin-tagged histones at
concentrations above the first plateau (10-fold above the
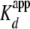 ; ∼100 nm) and
200-fold lower than the second transition to pull-down
(yNap1m*)-Alexa-546 (10 nm). Under these conditions, we
were able to reduce the amount of unbound (yNap1m*)-Alexa-546 to
undetectable levels (data not shown), confirming that the first phase of
fluorescence change indeed represents histone binding by yNap1.
; ∼100 nm) and
200-fold lower than the second transition to pull-down
(yNap1m*)-Alexa-546 (10 nm). Under these conditions, we
were able to reduce the amount of unbound (yNap1m*)-Alexa-546 to
undetectable levels (data not shown), confirming that the first phase of
fluorescence change indeed represents histone binding by yNap1.
One yNap1 Dimer Binds Two H2A/H2B Dimers with High
Affinity—The change in fluorescence of H2A/H2B (T112C)-Alexa-488
(<5-fold the 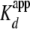 ) was measured as a
function of yNap1 (Fig.
1C and Table
1). The observed change in fluorescence followed a cooperative
dependence on the concentration of yNap1 added. The data were plotted as a
function of yNap1 concentration assuming either a yNap1 monomer or yNap dimer
(Table 1). The
) was measured as a
function of yNap1 (Fig.
1C and Table
1). The observed change in fluorescence followed a cooperative
dependence on the concentration of yNap1 added. The data were plotted as a
function of yNap1 concentration assuming either a yNap1 monomer or yNap dimer
(Table 1). The
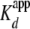 and Hill coefficient
(nH) was calculated from the fit of a binding isotherm
(Equation 2) to fluorescence
change (f.c.) (Fig.
1B). The
and Hill coefficient
(nH) was calculated from the fit of a binding isotherm
(Equation 2) to fluorescence
change (f.c.) (Fig.
1B). The 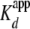 of
H2A/H2B dimer for a yNap1 monomer was determined to be 1.3 ± 0.1
nm with a nH of 2.3 ± 0.3, and as
expected, the
of
H2A/H2B dimer for a yNap1 monomer was determined to be 1.3 ± 0.1
nm with a nH of 2.3 ± 0.3, and as
expected, the 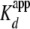 was ∼2-fold
lower (
was ∼2-fold
lower (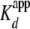 = 0.7 ± 0.1
nm, nH = 2.6 ± 0.3) when yNap1 was fit
as a dimer (Table 1).
= 0.7 ± 0.1
nm, nH = 2.6 ± 0.3) when yNap1 was fit
as a dimer (Table 1).
TABLE 1.
Values of the observed disassociation constants calculated with yNap1 as either a monomer or dimer at low ionic strength (0.15 m)
| Protein titrated | Kdapp | Hill coefficient | Labeled protein | pH |
|---|---|---|---|---|
| ×10-9m | ||||
| yNap1 - Monomer | 1.3 ± 0.1 | 2.3 ± 0.3 | H2A/H2B(T112C) | 7.5 |
| Dimer | 0.7 ± 0.1 | 2.6 ± 0.3 | H2A/H2B(T112C) | 7.5 |
| yNap1(74-365) - Monomer | 10 ± 0.3 | 2.0 ± 0.1 | H2A/H2B(T112C) | 7.5 |
| Dimer | 5.4 ± 1.3 | 2.0 ± 0.1 | H2A/H2B(T112C) | 7.5 |
| yNap1 (74-417) - Monomer | 1.7 ± 0.1 | 2.4 ± 0.2 | H2A/H2B(T112C) | 7.5 |
| Dimer | 0.9 ± 0.1 | 2.5 ± 0.2 | H2A/H2B(T112C) | 7.5 |
| yNap1(1-365) - Monomer | 2.1 ± 0.1 | 2.1 ± 0.2 | H2A/H2B(T112C) | 7.5 |
| Dimer | 1.0 ± 0.1 | 2.2 ± 0.1 | H2A/H2B(T112C) | 7.5 |
| H2A/H2B | 1.2 ± 0.1 | 1.9 ± 0.4 | yNap1m* | 7.5 |
The fit to quenching data obtained from the converse experiment
(i.e. labeled yNap1 and unlabeled H2A/H2B dimer,
Fig. 1 and
Table 1) resulted in a
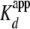 of 1.2 ± 0.1 ×
10–9 m and a nH of 1.9
± 0.4 (Fig.
1B). Taken together these data indicate yNap1 has a high
affinity for the H2A/H2B dimer and that neither the point mutations in H2B or
yNap1, nor the addition of fluorescent labels to defined regions in either
binding partner has a measurable effect on the interaction. Our data further
suggest that each yNap1 monomer (irrespective of its oligomeric state) has one
binding site for an H2A/H2B dimer.
of 1.2 ± 0.1 ×
10–9 m and a nH of 1.9
± 0.4 (Fig.
1B). Taken together these data indicate yNap1 has a high
affinity for the H2A/H2B dimer and that neither the point mutations in H2B or
yNap1, nor the addition of fluorescent labels to defined regions in either
binding partner has a measurable effect on the interaction. Our data further
suggest that each yNap1 monomer (irrespective of its oligomeric state) has one
binding site for an H2A/H2B dimer.
To determine the stoichiometry of the H2A/H2B-yNap1 complex, we used
(yNap1m*)-Alexa-546 at concentrations 10-fold above the
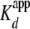 and monitored the change in signal
as a function of H2A/H2B dimer added. The signal increased linearly until
saturation was reached. The molar ratio at which saturation was reached was
equal to one mole of H2A/H2B dimer bound to one mole of yNap1 monomer, or of
two H2A/H2B dimers bound to the symmetric yNap1 dimer
(Fig. 1C).
and monitored the change in signal
as a function of H2A/H2B dimer added. The signal increased linearly until
saturation was reached. The molar ratio at which saturation was reached was
equal to one mole of H2A/H2B dimer bound to one mole of yNap1 monomer, or of
two H2A/H2B dimers bound to the symmetric yNap1 dimer
(Fig. 1C).
yNap1 C- and N-terminal Tails Contribute Synergistically to Histone Binding—It has been reported that the N- and C-terminal tails of yNap1 contribute to histone binding based on gel-shift and pull-down experiments (9, 10). To quantify the contribution of the disordered N- and C-terminal tails of yNap1, we prepared constructs of yNap1 in which either the N-terminal tail, the CTAD, or both are deleted (yNap1(74–417), yNap1(1–365), and yNap1(74–365), respectively). We measured binding to fluorescently labeled H2A/H2B dimer (Fig. 2 and Table 1). The individual deletion of the N- or C-terminal tail reduced the affinity by less than ∼1.5-fold, whereas yNap1(74–365) exhibited ∼8-fold reduced affinity for the H2A/H2B dimer (10 ± 0.3 × 10–9 m) compared with wild-type yNap1.
FIGURE 2.

Affinity and stoichiometry of the yNap1-H2A/H2B dimer complex. Normalized change in fluorescence as a function of yNap1 (open circles), yNap1(74–417) (open squares), yNap1(1–365) (closed triangles), and yNap1(74–365) (closed circles), all as monomers. The conditions were 20–50 mm Tris, pH 7.5, 1 mm dithiothreitol, 0.1 mg/ml bovine serum albumin, 90–120 mm NaCl, (0.5–1) × 10–10 m Alexa-488-H2A/H2B(T112C), and (0–5 × 10–7 m yNap1 or its truncations. For results of the fit see Table 1.
The Cooperative Nature of the H2A/H2B-yNap1 Interaction Is Dependent on the Ionic Strength and on the Histone Tails—Given the highly charged character of yNap1 and histone H2A/H2B dimer, we hypothesized that the affinity of histones for yNap1 would decrease with increasing ionic strength. We also wanted to investigate the effect of ionic strength on the cooperative nature of histone binding. We measured the affinity of yNap1 to H2A/H2B at 0.15, 0.35, and 0.55 m total ionic strength (Tables 1 and 2). While the Kd increased from 1.3 ± 0.1 to 7.8 ± 1.5 × 10–9 m upon increasing the ionic strength from 0.15 to 0.35 and 0.55 m, surprisingly the cooperativity also changed from a Hill coefficient (nH) of ∼2 to 1 (Fig. 3A and Tables 1 and 2).
TABLE 2.
Values of the observed dissociation constants for major type and variant histones at 0.35 m ionic strength
| Protein titrated | Kdapp | Hill coefficient | Labeled protein | pH |
|---|---|---|---|---|
| ×10-9m | ||||
| H2A/H2B | 7.8 ± 0.4 | n.a.a | yNap1m* | 7.5 |
| Tailless H2A/H2B | 2.1 ± 0.2 | 1.7 ± 0.2 | yNap1m* | 7.5 |
| H2A.BBd/H2B | 4.2 ± 0.5 | n.a. | yNap1m* | 7.5 |
| H2A.Z/H2B | 7.4 ± 0.4 | n.a. | yNap1m* | 7.5 |
| H3/H4-tetramer | 10.0 ± 0.6 | 1.4 ± 0.1 | yNap1m* | 7.5 |
| Dimer | 20.2 ± 0.7 | 1.5 ± 0.1 | ||
| Tailless H3/H4-tetramer | 4.0 ± 0.2 | 1.5 ± 0.1 | yNap1m* | 7.5 |
| Dimer | 8.8 ± 0.7 | 1.3 ± 0.1 | ||
| H3(H113A)/H4-tetramer | 7.9 ± 0.4 | 1.3 ± 0.1 | yNap1m* | 7.5 |
| Dimer | 16 ± 0.7 | 1.3 ± 0.1 | ||
| H1 | 3.0 ± 0.1 | 1.4 ± 0.1 | yNap1m* | 7.5 |
| H2A | 3.5 ± 0.5 | n.a. | yNap1m* | 7.5 |
| H3 | >104 | n.a. | yNap1m* | 7.5 |
| Archaeal Histones | d.n.b. | n.a. | yNap1m* | 7.5 |
| DNA | 1.6 ± 0.2 | n.a. | H3/H4(E63C) | 7.5 |
| H2A/H2B | 7.8 ± 1.5 | n.a. | yNap1m* | 7.5 |
| H2A/H2B | 21.6 ± 1.8 | n.a. | yNap1m* | 8.8 |
| yNap1 - Monomer | 20.1 ± 1.4 | n.a. | H3/H4(E63C) | 7.5 |
| Dimer | 10.1 ± 0.7 | n.a. |
n.a., not applicable.
FIGURE 3.

Cooperativity is dependent on both the ionic strength and histone tails. A, Hill plot of H2A/H2B binding yNap1 at ∼0.15 m (closed circles) and ∼0.35 m (open squares-same data in all plots) ionic strength. The slopes of these data result in a nH of 1 ± 0.1 and 2.4 ± 0.2, respectively. B, normalized fluorescence change as a function of histone dimer. Closed circles are tail-less dimers, and open squares are major type histone dimer. Inset of B is the Hill plot of the tail-less (closed circles) and major type (open squares) dimer. The slopes of these data result in an nH of 1.5 ± 0.1 and 1.0 ± 0.1, respectively.
It has been proposed that the histone tails contribute to the interaction
with yNap1 (9). To quantitate
this contribution we measured the affinity of tail-less H2A/H2B to
(yNap1m*)-Alexa-546 (<5-fold
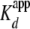 ). Because tail-less histones are
notoriously sticky at low ionic strength, especially when fluorescently
labeled, all subsequent measurements were performed at 0.35 m NaCl
(Table 2). The data for the
interaction of tail-less H2A/H2B with yNap1 were best described by a
cooperative model with a
). Because tail-less histones are
notoriously sticky at low ionic strength, especially when fluorescently
labeled, all subsequent measurements were performed at 0.35 m NaCl
(Table 2). The data for the
interaction of tail-less H2A/H2B with yNap1 were best described by a
cooperative model with a 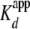 of 2.1
± 0.2 × 10–9 m and a
nH of 1.7 ± 0.2. Thus, these histone constructs
exhibit cooperativity under conditions where wild type histones bind in a
non-cooperative manner (Fig.
3B, inset). We conclude that the tails of H2A
and H2B contribute negatively to the interaction with the chaperone, and also
are responsible for preventing cooperative binding under these conditions.
of 2.1
± 0.2 × 10–9 m and a
nH of 1.7 ± 0.2. Thus, these histone constructs
exhibit cooperativity under conditions where wild type histones bind in a
non-cooperative manner (Fig.
3B, inset). We conclude that the tails of H2A
and H2B contribute negatively to the interaction with the chaperone, and also
are responsible for preventing cooperative binding under these conditions.
Two Copies of H3/H4 Bind One yNap1 Dimer with High
Affinity—To determine the affinity of yNap1 to (H3/H4)2
we titrated (yNap1m*)-Alexa-546 (<5-fold
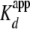 ) with refolded (H3/H4)2
tetramer. The
) with refolded (H3/H4)2
tetramer. The 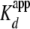 was 20.0 ±
0.7 nm with a nH of 1.5 ± 0.1 when the
concentration of H3/H4 was calculated as a H3/H4 heterodimer, and half that
when the concentration was calculated assuming a (H3/H4)2 tetramer
(Table 2). This measurement is
in excellent agreement with the value obtained from Alexa-546-H3/H4(E63C)
(Table 2), further validating
our assay. As with H2A/H2B, the removal of the histone tails of H3 and H4
resulted in an increased affinity for yNap1
(
was 20.0 ±
0.7 nm with a nH of 1.5 ± 0.1 when the
concentration of H3/H4 was calculated as a H3/H4 heterodimer, and half that
when the concentration was calculated assuming a (H3/H4)2 tetramer
(Table 2). This measurement is
in excellent agreement with the value obtained from Alexa-546-H3/H4(E63C)
(Table 2), further validating
our assay. As with H2A/H2B, the removal of the histone tails of H3 and H4
resulted in an increased affinity for yNap1
(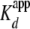 of 4.0 ± 0.2 ×
10–9 m and
Table 2).
of 4.0 ± 0.2 ×
10–9 m and
Table 2).
The stoichiometry of the H3/H4-yNap1 complex was measured as described above (Fig. 4A, inset). This experiment clearly shows that one H3/H4 histone-fold dimer binds one yNap1 monomer. If the predominant form of yNap1 under these conditions is assumed to be a dimer (as we believe is likely), this translates to two H3/H4 histone fold dimers (or one (H3/H4)2 tetramer) interacting with one yNap1 dimer (Fig. 3A).
FIGURE 4.

The interaction of yNap1 and DNA with histone tetramer. A, normalized fluorescence change as a function of histone tetramer binding to (yNap1m*)-Alexa-546 (closed circles), tail-less (H3/H4)2 (open squares), and H3(H113A)/H4 (plotted as tetramer) (open squares). Inset of A is the stoichiometry of (H3/H4)2 to yNAP dimer. (H3/H4)2 was titrated against 1×10–7 m (yNap1m*)-Alexa-546 to result in a stoichiometry of 1 histone tetramer to 1 yNap1 dimer. B, normalized fluorescence change as a function of DNA binding to (H3/H4(E63C))2-Alexa-488 (10–10 m). Buffer conditions were 20–50 mm Tris, pH 7.5, 1 mm dithiothreitol, 0.1 mg/ml bovine serum albumin, and 300 mm NaCl.
Our finding that H3/H4 and H2A/H2B bind yNap1 with comparable affinities and stoichiometries suggests that it is primarily the histone fold that is recognized by yNap1, and not any features that are specific to the H3-H3 four-helix bundle. To test this hypothesis, we prepared a mutant version of H3 that has been designed to abolish the H3-H3 interaction that is exclusively responsible for the formation of the (H3/H4)2 tetramer from two histone fold dimers H3(H113A). Indeed, when refolded with H4, the complex elutes from the size exclusion column as a dimer.5 The affinity of this H3(H113A)/H4 heterodimer for yNap1 was 16 ± 0.7 × 10–9 m with a nH of 1.3 ± 0.1, and thus is almost identical to the affinity of a wild-type H3/H4 complex (20.2 ± 0.7; Table 2).
The (H3/H4)2 Tetramer Binds DNA with a 10-Fold Higher
Affinity Than It Binds yNap1—We next wanted to test the hypothesis
that DNA competes with yNap1 for histones, as expected in light of the
assembly function of yNap1. To measure the affinity of (H3/H4)2
tetramer for DNA, we used Alexa-546-(H3/H4(E63C))2 (<5-fold the
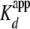 ) and titrated in a 146-base pair
DNA fragment (Fig.
4B). These data were fit to a simple binding isotherm
with a
) and titrated in a 146-base pair
DNA fragment (Fig.
4B). These data were fit to a simple binding isotherm
with a 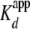 of 1.6 ± 0.2 ×
10–9 m. This is ∼5-fold tighter than the
interaction of (H3/H4)2 tetramer with yNap1, and translates into a
ΔΔG of –1.9 kcal/mol. While we cannot say at this
point if the (H3/H4)2-DNA interaction is a biologically relevant
tetrasome, our results clearly indicate that (H3/H4)2 interactions
with DNA are more favorable than the interaction with the chaperone.
of 1.6 ± 0.2 ×
10–9 m. This is ∼5-fold tighter than the
interaction of (H3/H4)2 tetramer with yNap1, and translates into a
ΔΔG of –1.9 kcal/mol. While we cannot say at this
point if the (H3/H4)2-DNA interaction is a biologically relevant
tetrasome, our results clearly indicate that (H3/H4)2 interactions
with DNA are more favorable than the interaction with the chaperone.
yNap1 Does Not Distinguish between Major Type Histones and Their
Variants—We measured the affinities of complexes containing
variants of H2A to yNap1 (Table
2). A dimer of H2A.Z-H2B or H2A. Bbd/H2B has an almost identical
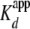 compared with major type H2A/H2B
dimer. Together with the similarities in yNap1 affinity for H2A/H2B and H3/H4,
our results lead us to conclude that yNap1 recognizes the architecture of the
basic histone fold that is common to H3/H4 and H2A/H2B (major type or variant
H2A) rather than specific amino acids.
compared with major type H2A/H2B
dimer. Together with the similarities in yNap1 affinity for H2A/H2B and H3/H4,
our results lead us to conclude that yNap1 recognizes the architecture of the
basic histone fold that is common to H3/H4 and H2A/H2B (major type or variant
H2A) rather than specific amino acids.
This interpretation is supported by our finding that yNap1 interacts with H2A (refolded in the absence of H2B) with high affinity (Table 2), while H3 binds with very low affinity. H2A is able to form a homo-dimer, most likely through a histone fold dimer-like assembly, whereas H3 alone forms aggregates (as shown by sedimentation equilibrium; data not shown). We were unable to detect any change in signal of (yNap1m)-Alexa-546 upon addition of homodimeric hMfB, an archaeal histone that binds and compacts DNA (19), indicating that no complex is formed under these conditions.
At Least Two Molecules of Linker Histone H1 Bind yNap1 with High
Affinity—yNap1 removes linker histone from chromatin
(21), but the affinity of H1
to nucleosomes has been estimated to be at least low nm
(22,
23). We measured the affinity
of H1 to yNap1 by titrating H1 against yNap1 (yNap1m*)-Alexa-546
(<5-fold the 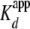 ). These data were
best described by a cooperative model with a
). These data were
best described by a cooperative model with a
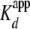 of 3.0 ± 0.1 × 10
–9 m and a nH of 1.4 ±
0.1 (Fig. 5). Under
stoichiometric conditions ((yNap1m*)-Alexa-546 > 10-fold the
of 3.0 ± 0.1 × 10
–9 m and a nH of 1.4 ±
0.1 (Fig. 5). Under
stoichiometric conditions ((yNap1m*)-Alexa-546 > 10-fold the
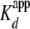 ) we observed the typical linear
increase, but it did not level out completely at higher ratios
(Fig. 5, inset). The
intersection of the two slopes occurs at two H1 histones per yNap1 dimer, but
the fact that a second less steep slope is observed suggests an additional
mode of H1 interaction with yNap1.
) we observed the typical linear
increase, but it did not level out completely at higher ratios
(Fig. 5, inset). The
intersection of the two slopes occurs at two H1 histones per yNap1 dimer, but
the fact that a second less steep slope is observed suggests an additional
mode of H1 interaction with yNap1.
FIGURE 5.

Binding and stoichiometry of H1 to yNap1. The normalized fluorescence change as a function of linker histone H1 binding to (yNap1m*)-Alexa-546. Inset is the stoichiometry of H1 to yNap1. While two phases were easily discernible the second did not level out as expected. These data suggest specific binding of two H1 linker histones to yNap1 and a possible third nonspecific H1-yNap1 interaction.
DISCUSSION
It has been suggested that yNap1 assembles and maintains chromatin in a thermodynamic manner (8). This model postulates that histones have a higher affinity for yNap1 than for all non-canonical (i.e. non-nucleosomal) association states with DNA. In order to fully explore this model we need to quantitatively evaluate the affinity of histone-yNap1 interactions. Here we have demonstrated that yNap1 indeed binds histones and their variants with very high affinity, and that each yNap1 monomer binds one histone fold dimer. The tails of both yNap1 and histones contribute to histone binding in unexpected ways. Our finding that H3/H4 binds to DNA more tightly than to yNap1 is consistent with the role of yNap1 in chromatin assembly.
Previously published assays to describe the interaction of yNap1 with histones are based on gel-shift and pull-down assays (9, 10). Both methods are limited due to the high concentrations of protein required for visualization and are difficult to quantify. In addition, they also suffer from changes in volume during gel loading or washing steps. This can be problematic if the system undergoes rapid equilibrium (koff faster than kon). The method described here uses fluorescently labeled yNap1 or histones. This provides us with the ability to titrate either protein at low concentrations and allows us to rule out the effect of fluorescent labels on binding.
The affinity measured for the titration of H2A/H2B into yNap1 is the same as that measured for yNap1 titrated into H2A/H2B when the concentration of yNap1 is assumed as a monomer. This, together with independent measurements of histone-yNap1 stoichiometries, suggests that each yNap1 monomer has an independent histone binding site that interacts with either one histone fold dimer or with one molecule of H1. Our previously published structural and biophysical studies strongly suggest that yNap1 exists as a homodimer (3, 5, 20), consistent with earlier biophysical analyses (6), implying that one yNap1 dimer binds two H2A/H2B dimers or one (H3/H4)2 tetramer (or two H3/H4 heterodimers), respectively.
Earlier in vitro GST pull-down and gel-shift assays concluded that yNap1 has a slight preference for H3/H4; however, this interaction was not observed in in vivo pull-down experiments (9, 10). Our quantitative analysis shows that yNap1 binds H3/H4 with slightly lower affinity than H2A/H2B, irrespective of the ability of H3 to form a heterotetramer via a four-helix bundle structure. In both cases, the basic histone tails had a negative effect on the interaction with yNap1. This counterintuitive result may be explained by an entropic penalty upon binding. While we do not know what effect, if any, post-translational modifications of histones have on the interaction with yNap1, it is intriguing to speculate that the affinity for yNap1 may be regulated by modification of histone tails (21). In light of our finding that complexes of H3/H4 and H2A/H2B bind with very similar affinities, it is not surprising that complexes refolded with histone H2A variants are not distinguished through differences in binding affinities. In contrast, the archaeal histone Hmfb does not interact with yNap1. This histone-fold dimer resembles eukaryotic histone fold dimers in that it has a basic pI and binds DNA, but it lacks the histone tails and secondary structure elements outside of the histone fold (22, 23).
yNap1 acts as a chaperone for the linker histone H1 in vitro and is capable of removing histone H1 from nucleosomal arrays (24). The affinity of the linker histone H1 to DNA has been estimated to be in the low nm range (25, 26). We find that one yNap1 dimer binds two molecules of linker histone H1 with high affinity; however, additional H1 molecules appear to bind with high affinity at higher H1:yNap1 ratios. Saccharomyces cerevisiae does not have stoichiometric amounts of linker histone; however, we find that metazoan Nap1 binds histones with comparable affinities to yNap1, and we expect the same to be true for linker histones (data not shown).
The binding of all histones to yNap1 is cooperative in nature with the exception of full-length histone dimers and their variants at high ionic strength. We consider at least three possible explanations for this observation. (i) yNap1 or histones could have a monomer to dimer Kd (or dimer to tetramer Kd for H3/H4) close to the concentrations we are working at; (ii) yNap1 undergoes a conformational change after the initial histone binding event; or (iii) the histone fold dimers interact with each other upon interaction with a yNap1 dimer. We should be able to observe differences in the binding of yNap1 to histones in scenarios (i) and (iii) by changing the protein being titrated or by studying mutations in histones that interfere with histone-histone interaction. To consider the effects of a yNap1 monomer-dimer transition on the affinities, we worked with several models; however, none were able to adequately explain our data. The possibility that the interaction between the histone fold dimers is responsible for the observed cooperativity is intriguing given that the H2A/H2B dimer binds with a Hill coefficient of 1, whereas the (H3/H4)2 tetramer binds with a Hill coefficient of 2 at 300 mm NaCl. A mutant H3(H113A)/H4 complex which remains in the dimeric state had little effect on the cooperativity but did alter the affinity. However, yNap1 titration into labeled H3/H4 is not cooperative, while the titration of H3/H4 is cooperative (see Reaction Scheme 2). While the fact that changing the protein being titrated changes the nH is consistent with the hypothesis that histone-histone interaction is responsible for the observed cooperativity; this hypothesis is not supported by the H3(H113A) mutation. It is possible that yNap1 facilitates the formation of (H3-H4)2 tetramer and that H3(H113A) does not alter the thermodynamics for tetramer formation enough to change the apparent cooperativity of binding. It is also possible that other histone-histone interactions exist. It is likely that the observed variability in cooperativity cannot be explained by one single the mechanisms presented here. Therefore, histone identity, histone tails, and ionic strength not only change the observed cooperative nature of binding but potentially the mechanism of cooperativity.
It has been proposed that the acidic C-terminal domain (CTAD) of yNap1 helps neutralize the charge of the histones (9). Consistent with this, it has been previously shown that a version of yNap1 lacking the CTAD fails to disassemble nucleosomes in vitro (10). We find that while the effects of removing either the CTAD or the less basic N-terminal tail have only moderate effects on the affinity, the removal of both affects histone binding synergistically. This makes sense in light of the structure of yNap1, where both tails emerge at the concave underside of the dome-shaped molecule (3).
The mechanism by which histone chaperones assemble and disassemble chromatin is largely unknown. The first assumption in a completely thermodynamic mechanism of Nap1-mediated chromatin assembly is that the (H3/H4)2 tetramer must have a higher affinity for DNA than for yNap1, as shown here. While this does not rule out other more complex mechanisms for chromatin assembly by yNap1, this is consistent with a thermodynamic model. Our data suggest that very small changes in binding energy may have large effects on chromatin/nucleosome structure.
Our working hypothesis is that yNap1 serves to prevent non-correct histone interaction and that only correct biologically relevant structures can compete with yNap1 for histones. Consistent with this we have measured the interaction of H2A/H2B dimer with DNA as ∼40 nm.5 The working model stipulates that yNap1 maintains a very low free histone pool to prevent incorrect binding. This model also suggests that the localization of yNap1 or other chaperones to either complexes such as p300 (21) or to the membrane as observed with Arabidopsis thaliana tNAP1 (27) could alter the saturation of nucleosomes on DNA. Alternatively, the possibility of a more direct interaction between yNap1 and the nucleosome, such as the tails of yNap1 interacting with an exposed surface of histone to promote histone release, cannot be excluded.
Acknowledgments
We thank Vidya Subramanian for critical reading of the manuscript, and Teri McLain at the W. M. Keck Protein Expression and Purification Facility for providing recombinant histones.
Author's Choice—Final version full access.
This work was supported, in whole or in part, by National Institutes of Health Grant GM067777 and by the Howard Hughes Medical Institute. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement”in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Nap1, nucleosome assembly protein 1; yNap1, yeast Nap1; yNap1m*, yNap1 C200A/C249A/C272A; CTAD, C-terminal acidic domain; f.c., fluorescence change.
M. Resch and K. Luger, unpublished results.
References
- 1.De Koning, L., Corpet, A., Haber, J. E., and Almouzni, G. (2007) Nat. Struct. Mol. Biol. 14 997–1007 [DOI] [PubMed] [Google Scholar]
- 2.Eitoku, M., Sato, L., Senda, T., and Horikoshi, M. (2008) Cell Mol. Life Sci. 65 414–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park, Y. J., and Luger, K. (2006) Biochem. Cell Biol. 84 549–558 [DOI] [PubMed] [Google Scholar]
- 4.Zlatanova, J., Seebart, C., and Tomschik, M. (2007) Faseb J. 21 1294–1310 [DOI] [PubMed] [Google Scholar]
- 5.Park, Y. J., and Luger, K. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McBryant, S. J., and Peersen, O. B. (2004) Biochemistry 43 10592–10599 [DOI] [PubMed] [Google Scholar]
- 7.Toth, K. F., Mazurkiewicz, J., and Rippe, K. (2005) J. Biol. Chem. 280 15690–15699 [DOI] [PubMed] [Google Scholar]
- 8.Mazurkiewicz, J., Kepert, J. F., and Rippe, K. (2006) J. Biol. Chem. 281 16462–16472 [DOI] [PubMed] [Google Scholar]
- 9.McBryant, S. J., Park, Y. J., Abernathy, S. M., Laybourn, P. J., Nyborg, J. K., and Luger, K. (2003) J. Biol. Chem. 278 44574–44583 [DOI] [PubMed] [Google Scholar]
- 10.Park, Y. J., Chodaparambil, J. V., Bao, Y., McBryant, S. J., and Luger, K. (2005) J. Biol. Chem. 280 1817–1825 [DOI] [PubMed] [Google Scholar]
- 11.Nakagawa, T., Bulger, M., Muramatsu, M., and Ito, T. (2001) J. Biol. Chem. 276 27384–27391 [DOI] [PubMed] [Google Scholar]
- 12.Luger, K., Rechsteiner, T. J., and Richmond, T. J. (1999) Methods Mol. Biol. 119 1–16 [DOI] [PubMed] [Google Scholar]
- 13.Dyer, P. N., Edayathumangalam, R. S., White, C. L., Bao, Y., Chakravarthy, S., Muthurajan, U. M., and Luger, K. (2004) Methods Enzymol. 375 23–44 [DOI] [PubMed] [Google Scholar]
- 14.Park, Y. J., Dyer, P. N., Tremethick, D. J., and Luger, K. (2004) J. Biol. Chem. 279 24274–24282 [DOI] [PubMed] [Google Scholar]
- 15.Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F., and Richmond, T. J. (1997) Nature 389 251–260 [DOI] [PubMed] [Google Scholar]
- 16.Karantza, V., Baxevanis, A. D., Freire, E., and Moudrianakis, E. N. (1995) Biochemistry 34 5988–5996 [DOI] [PubMed] [Google Scholar]
- 17.Karantza, V., Freire, E., and Moudrianakis, E. N. (1996) Biochemistry 35 2037–2046 [DOI] [PubMed] [Google Scholar]
- 18.Banks, D. D., and Gloss, L. M. (2004) Protein Sci. 13 1304–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Starich, M. R., Sandman, K., Reeve, J. N., and Summers, M. F. (1996) J. Mol. Biol. 255 187–203 [DOI] [PubMed] [Google Scholar]
- 20.Park, Y. J., McBryant, S. J., and Luger, K. (2008) J. Mol. Biol. 375 1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sharma, N., and Nyborg, J. K. (2008) Proc. Natl. Acad. Sci. U. S. A. 105 7959–7963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luger, K. (2000) Encyclopedia of Life Sciences
- 23.Luger, K. (2002) Nat. Genet. 16 16. [DOI] [PubMed] [Google Scholar]
- 24.Kepert, J. F., Mazurkiewicz, J., Heuvelman, G. L., Toth, K. F., and Rippe, K. (2005) J. Biol. Chem. 280 34063–34072 [DOI] [PubMed] [Google Scholar]
- 25.Varga-Weisz, P., van Holde, K., and Zlatanova, J. (1994) Biochem. Biophys. Res. Commun. 203 1904–1911 [DOI] [PubMed] [Google Scholar]
- 26.Varga-Weisz, P., Zlatanova, J., Leuba, S. H., Schroth, G. P., and van Holde, K. (1994) Proc. Natl. Acad. Sci. U. S. A. 91 3525–3529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galichet, A., and Gruissem, W. (2006) Plant Physiol. 142 1412–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]