

Abstract
AtNOS1 was previously identified as a potential nitric-oxide synthase (NOS) in Arabidopsis thaliana, despite lack of sequence similarity to animal NOSs. Although the dwarf and yellowish leaf phenotype of Atnos1 knock-out mutant plants can be rescued by treatment with exogenous NO, doubts have recently been raised as to whether AtNOS1 is a true NOS. Moreover, depending on the type of physiological responses studied, Atnos1 is not always deficient in NO induction and/or detection, as previously reported. Here, we present experimental evidence showing that AtNOS1 is unable to bind and oxidize arginine to NO. These results support the argument that AtNOS1 is not a NOS. We also show that the renamed NO-associated protein 1 (AtNOA1) is a member of the circularly permuted GTPase family (cGTPase). AtNOA1 specifically binds GTP and hydrolyzes it. Complementation experiments of Atnoa1 mutant plants with different constructs of AtNOA1 show that GTP hydrolysis is necessary but not sufficient for the physiological function of AtNOA1. Mutant AtNOA1 lacking the C-terminal domain, although retaining GTPase activity, failed to complement Atnoa1, suggesting that this domain plays a crucial role in planta. cGTPases appear to be RNA-binding proteins, and the closest homolog of AtNOA1, the Bacillus subtilis YqeH, has been shown to participate in ribosome assembly and stability. We propose a similar function for AtNOA1 and discuss it in the light of its potential role in NO accumulation and plant development.
Numerous studies have demonstrated that plants, like animals, generate nitric oxide (NO)3 to regulate a wide range of physiological processes. NO is involved in plant development; it represses flowering (1), reduces seed dormancy (2), and regulates germination (3). NO production has also been detected following different environmental stresses. For example, NO regulates stomata closure in response to abiotic stress (4), and in response to biotic stress, NO participates in induction of plant defenses (5-7).
Although NO plays a role as significant in plants as it does in animals, NO synthesis in planta is still a matter of debate (8). Two major routes have been proposed for NO formation in plants. The first one relies on the reduction of nitrite to NO. Several studies demonstrate that nitrate reductase, whose primary function is to catalyze the reduction of nitrate to nitrite, can convert nitrite to NO with low efficiency (9, 10). Nitrite can also be reduced to NO by a plasma membrane-bound nitrite:NOreductase (11), by a mitochondrial electron transport-dependent reductase (12), or nonenzymatically in acidic, reducing environments (13). The second probable NO biosynthetic pathway uses arginine as a substrate, following a reaction similar to that observed for the well characterized animal NOSs. Indeed, several lines of evidence suggest the existence of a mammalian NOS-like enzyme in plants. Application of arginine analogs, inhibitors of animal NOSs, results in a reduction of NO detected in plants (5, 6, 14-17). Arginine-dependent citrulline formation, a co-product of the NOS reaction, has also been observed in plant extracts (5, 6, 15).
Two potential plant NOSs have been reported thus far, but in both cases, further investigation failed to confirm NO biosynthesis activity. Data demonstrating NOS activity of a variant form of the P protein of the glycine decarboxylase complex (18) were found to be nonreproducible and unreliable and thus were retracted (19). Crawford and co-workers (20) identified the second potential NOS in A. thaliana, AtNOS1, based on homology to a hypothetical snail NOS or NOS partner that cross-reacted with mammalian NOS antibody (23% identity, 39.5% similarity, 30.1% gap between AtNOS1 and the snail protein using a local alignment) (20, 21). Interestingly, AtNOS1 T-DNA knock-out plants (Atnos1) have a growth phenotype that can be rescued by the application of NO donor compounds. Moreover, chemical probes sensitive to NO indicated reduced NO levels in Atnos1 compared with wild type plants (20, 22-24). However, several groups, including our laboratory and Crawford's, cannot reproduce the originally reported NOS activity with recombinant AtNOS1, calling into question the true function of this protein (25-27).
AtNOS1 is a 561-amino acid protein that has no sequence homology to the animal NOSs. It belongs to the circularly permuted GTPase (cGTPase) family (28). The central domain of AtNOS1-(176-350) contains guanine-binding motifs (G motifs) characteristic of small GTPases like Ras, Rho, and Cdc42 (29), but in an unusual arrangement; G4-G5 are N-terminal of G1-G2-G3. Among the four subfamilies of cGTPase represented by YlqF (in Bacillus subtilis), YjeQ (in Escherichia coli), and YawG (in Saccharomyces pombe), YqeH (in B. subtilis) is the closest homolog of AtNOS1 (30% identity, 43.6% similarity, 15.5% gap) (Fig. 2A). Both proteins contain four conserved cysteines in the N-terminal region (zinc-binding domain (ZBD)) that can form a zinc finger motif CXGCXnCXRC of the treble clef family (30, 31). They also possess a very similar C-terminal domain (CTD) of unknown function (40.8% similarity, 22.3% gap). AtNOS1 contains an additional 101 residues at the N terminus comprising a predicted mitochondria targeting sequence and a short stretch of basic lysine residues (KKKKK).
FIGURE 2.

Primary and quaternary structure comparison of AtNOA1 and gsYqeH. A, sequence alignment of AtNOA1 (A), gsYqeH (Y), and the murine homolog of AtNOA1 (M). Conserved residues are colored by Chroma (black background). The three protein domains comprising the ZBD, the CPG domain with guanine nucleotide-binding regions G1 to G5 (red), and the CTD are shown in blue. The insertions (I1-I4) present in AtNOA1 sequence relative to gsYqeH are shown in green. B, the GDP-bound form of gsYqeH (blue) and the modeled structure of Δ101 (magenta) are superimposed. The overall structures of the two proteins almost completely overlap (backbone root mean square deviation of 0.49 Å). The insertions I1-I4 (green) present in AtNOA1 do not interfere with the overall fold and are located in β-turns and loops.
Little is known concerning the function of cGTPases in eukaryotes that could shed light on the possible role of AtNOS1 in plants, particularly in regard to NO accumulation. Bacterial cGTPases are essential for cell growth (32-34). In bacteria and in some eukaryotes, this family of GTP-binding proteins is associated with RNA/ribosome binding function (28, 35-37). For example, YqeH has been shown to be essential for the viability of B. subtilis (33) and to participate in ribosome biogenesis and assembly (38, 39). Eukaryotic homologs are found in other plants, such as tomato (64.7% identity) and rice (Q6YPG5, 60.5% identity) as well as in mice (NP_062810) and humans (NP_115689). Mammalian homologs have less homology to AtNOS1 (22.4 and 23.2% identity and 35 and 34.5% similarity for the mouse and the human homologs, respectively) and contain longer amino acid insertions (34.6 and 31.4% gap for the mouse and human homologs, respectively). These proteins also contain a mitochondrial targeting peptide at their N termini and, like AtNOS1, they seem to localize in this organelle (22, 40).
The function of AtNOS1 as an authentic NOS has been recently questioned. This led to the renaming of AtNOS1 as NO-associated protein 1 (AtNOA1) (25). Nevertheless, publications still refer to AtNOS/A1 as a potential NOS (41, 42). Here, we examined the ability of AtNOS1 protein to bind and oxidize arginine into NO, using several independent assays. We demonstrate that AtNOS1 is not a NOS but a functional GTPase. We show that its GTPase activity is necessary but not sufficient for its function in planta. This new activity is discussed in the context of the defective NO accumulation phenotype of Atnos1.
EXPERIMENTAL PROCEDURES
GTP and NADPH were purchased from Roche Applied Science, and (6R)-5,6,7,8-tetrahydro-l-biopterin (BH4) came from Shircks Laboratory (Jona, Switzerland). All other chemicals were from Sigma, unless otherwise indicated.
Proteins Expression and Purification—Plasmids used for neuronal NOS (nNOS) expression, pCWori+ containing the rat nNOS cDNA and pGroESL for chaperone protein expression, were kindly provided by Dr. M. A. Sari (CNRS UMR 8601, Université René Descartes, Paris, France). The expression and purification of nNOS protein was conducted in the presence of arginine and BH4, as previously reported (43). The protein was buffer-exchanged using a Sephadex G25 column prior to use.
The AtNOA1 cDNA clone was obtained from Arabidopsis Biological Resource Center. The cDNA encoding AtNOA1 full-length, the N-terminal deletion of 101 amino acids (Δ101) and the T327A mutant, the N-terminal truncation containing only the circularly permuted G-motif domains (CPG domains) and the CTD (residue 351-561) and the C-terminal truncated proteins (residues 102-350) were all generated by PCR and cloned into pET28 expression vector (Novagen) as N-terminal His6 tag fusions. Plasmids were transformed in E. coli BL21 (DE3) cells. These cells were grown in LB media at 37 °C until A600 nm ∼ 0.6, and expression was induced by 100 μm isopropyl 1-thio-β-d-galactopyranoside. After 20 h at 18 °C, cells were collected and lysed in buffer A supplemented with 5 mm imidazole (buffer A: 25 mm HEPES, pH 7.5, 300 mm NaCl, 2 mm MgCl2, 10% glycerol, 2 mm dithiothreitol, 1 mm phenylmethylsulfonyl fluoride). Soluble protein supernatant was applied to a Ni2+-nitrilotriacetic acid-agarose column (Qiagen), washed with buffer A containing 20 mm imidazole, and eluted with 300 mm imidazole. Size exclusion chromatography (Superdex 200; Amersham Biosciences) in buffer B supplemented with 2 mm MgCl2 was performed to further purify the proteins (buffer B: 50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10% glycerol, and 2 mm dithiothreitol). Fractions of interest were pooled and concentrated before storage at -80 °C until further use. Protein concentrations were evaluated using the Bradford reagent (Bio-Rad) and bovine serum albumin as a standard.
NO Formation—Rates of NO synthesis were determined at room temperature on a Cary 50 spectrophotometer using the oxyhemoglobin assay for NO (44). A 150-μl reaction containing 20 μm oxyhemoglobin, 100 units/ml superoxide dismutase and catalase, 10 μm BH4, 10 mm CaCl2, 10 μg/ml calmodulin (CaM), 1 mm NADPH, and 100 μm arginine in 50 mm Hepes buffer (pH 7.5) and 5 mm dithiothreitol was prepared. The addition of protein (nNOS or AtNOA1) initiated the reaction. The NO-dependent conversion of oxyhemoglobin to methemoglobin was monitored by scanning every 0.5 min between 380 and 450 nm. An extinction coefficient of 77 mm-1 cm-1 between the peak at 401 nm and the valley at 420 nm was used to quantify NO. All other NOS assays were conducted as previously described (45).
Arginine Binding—Arginine-binding experiments were carried out with 100 nm [2,3,4-3H]arginine (41 Ci/mmol; PerkinElmer Life Sciences) with or without unlabeled arginine (100,000-fold molar excess) in 120 μl of buffer B for 15 min on ice. Unbound ligand was removed with a 1-ml G-25 Superfine (Amersham Biosciences) column by centrifugation for 2 min at 1,000 × g. Bound [2,3,4-3H]arginine was quantified by scintillation counting of 90 μl of filtrate.
Homology Modeling—Alignment of AtNOA1-(175-534) (containing CPG and CTD) and YqeH-(59-369) was performed using the alignment program Tcoffee (46). A three-dimensional model for AtNOA1-(175-534) was generated by comparative protein modeling through satisfaction of spatial restraints with the program MODELLER (47) using the x-ray structure for gsYqeH-(59-369) as a template (48).
Complementation—Atnoa1 seeds were obtained from Dr. Nigel Crawford (University of California at San Diego). For constitutive expression of AtNOA1 or AtNOA1D226N in plants, the open reading frame of the corresponding gene was amplified and cloned into plant transformation vector pF3PZPY122 (49) that encodes for the corresponding protein tagged by three tandem FLAG epitopes at the C terminus. For the expression of the CTD-truncated AtNOA1 in plants, AtNOA1-(1-386) cDNA was amplified and cloned into the plant transformation vector pBIN61HA that encodes AtNOA1-(1-386) protein tagged with an HA epitope at the C terminus (50). The different constructs were transformed into Agrobacterium medium strain GV3101, and recombinants were screened on LB medium containing chloramphenicol (50 μg/ml for pF3PZPY122) or kanamycin (30 μg/ml for pBIN61HA). Atnoa1 plants were transformed by the floral dip method (51). The transgenic plants were selected for gentamicin (50 μg/ml in Murashige-Skoog medium for pF3ZPY122) or kanamycin (50 μg/ml for pBIN61HA) resistance and allowed to set seeds. Although the Atnoa1 mutants were originally generated by an insertion of T-DNA conferring kanamycin resistance, they developed antibiotic sensitivity after inbreeding for several generations (data not shown) (20, 52). Seeds of a T2 family were planted on Murashige-Skoog medium containing the corresponding antibiotics, incubated at 4 °C for 3 days in darkness, and transferred at 22 °C for 2 weeks under the long day condition (18-h light/6-h darkness). For AtNOA1-(1-386), seedlings displaying vigorous root growth were transferred to soil for further growth under the same long day condition. As controls, wild-type and Atnoa1 seeds were treated similarly except for the germination on Murashige-Skoog medium without the antibiotics. The genotypes of T1 and T2 plants were confirmed by genomic DNA PCR for knock-out of wild type AtNOA. Constitutive expression of AtNOA1 and AtNOA1D226N was examined by Western blotting analysis with anti-FLAG antibody (Sigma). Anti-HA antibody (Roche Applied Science) was used to probe for AtNOA1-(1-386) expression.
GTP Binding—N-Methylanthraniloyl-labeled GDP (MantGDP) was provided by Dr. Richard Cerione (Cornell University). Fluorescence of MantGDP was measured on a Varian Eclipse spectrofluorimeter in Dr. Cerione's laboratory. Kd for MantGDP binding to AtNOA1 was determined with 1 μm protein at 25 °C in 50 mm Tris-HCl, pH 7.5, 150 mm NaCl supplemented with 200 mm KCl, and 5 mm MgCl2 when indicated. Increasing amounts of MantGDP (up to 2 μm final concentration) were added, and the fluorescence was measured (excitation, 356 nm; emission, 450 nm) and corrected with the corresponding signal in the absence of protein. The resulting relative fluorescence of MantGDP-Δ101 complex was plotted as a function of MantGDP concentration, and the curve was fitted to a one-site-binding hyperbolic function with Origin Pro 7.5 software (OriginLab Corp., Northampton, MA). For competition assays, proteins were mixed with 200 nm MantGDP until saturation of the fluorescence signal (∼1.5 μm Δ101). Increasing amounts of GTP, GDP, ATP, or CTP were added, and the fluorescence signal was recorded at equilibrium. The relative fluorescence was calculated using the following equation: relative fluorescence (%) = ((MantGDP fluorescence in the presence of competitor - basal MantGDP fluorescence)/(MantGDP fluorescence in the absence of competitor - basal MantGDP fluorescence)) × 100. The percentage of bound competitor was derived from the relative fluorescence and plotted as a function of the competitor concentration. IC50 values, corresponding to the amount of competitor necessary to observe a 50% decrease in fluorescence, were calculated.
GTPase Activity—To demonstrate that Δ101 was indeed a functional GTPase, 20 or 40 μm protein was incubated with 500 μm GTP, 2 mm MgCl2, 200 mm KCl in buffer B at 37 °C overnight. Samples were boiled for 5 min to stop the reaction and precipitate the proteins and then were centrifuged for 5 min. The supernatant was analyzed by reverse phase HPLC on a Waters Sunfire C18 5 μm (4.5 × 250-mm) column. Nucleotides were separated with an isocratic condition at 1 ml/min of 100 mm KH2PO4, pH 6.5, 10 mm tetrabutylammonium bromide, 0.2 mm NaN3, and 7.5% acetonitrile. Control reactions in the absence of protein were analyzed following the same procedure.
Rates of GTP hydrolysis were quantified by measuring [32P]phosphate release (53). Reactions containing 1 nm [γ-32P]GTP (2 μCi) and varying amounts of cold GTP were prepared in 300 μl of buffer B supplemented with 5 mm MgCl2 and 200 mm KCl. The reaction was started by the addition of the protein. At various times, as indicated in the figures, 50-μl aliquots were mixed with 1 ml of activated charcoal (5% in 50 mm NaH2PO4). After a 1-min centrifugation, [γ-32P]phosphates in the supernatant were counted on a liquid scintillation counter. Counts/min were plotted as a function of time for the different GTP concentrations. Reactions in the absence of protein were conducted to control for spontaneous hydrolysis. Km and Vmax values were determined by plotting the initial velocity of GTP hydrolysis (v0) as a function of the substrate concentration. Curves were fitted to the equation, v0 = (Vmax × [GTP])/(Km + [GTP]) using Origin Pro 7.5 software.
RESULTS
AtNOS1 Is Not a Nitric-oxide Synthase—The AtNOS1 full-length (fl) and a deletion variant with the first 101 amino acids removed (Δ101) were expressed in E. coli and tested for their ability to generate NO from arginine. The N-terminal deletion increases the solubility and stability of the protein by removing a putative mitochondrial targeting sequence as well as additional residues not found in the bacterial homolog YqeH (see Fig. 2A). Five different assays were used to assess the NO synthesis ability of AtNOS1, with the rat nNOS included in all assays as a positive control. First, the hemoglobin assay was used to follow the rapid conversion of oxyhemoglobin to methemoglobin by enzymatically generated NO. The reactions were performed with AtNOS1 fl and Δ101 in the presence of arginine and NADPH and with or without mammalian NOS cofactors, such as CaM, calcium, or BH4. Methemoglobin formation was not observed with AtNOS1 in any of the conditions tested (Fig. 1A). Second, a colorimetric Griess assay was used to detect the NO oxidation product nitrite in reactions including AtNOS1 fl or Δ101 and a combination of possible cofactors. Again, the recombinant proteins were unable to generate nitrite via NO from arginine (data not shown). Mammalian NOSs generate citrulline from arginine as a NO co-product; AtNOS1 failed to produce radiolabeled citrulline from [3H]arginine in this third assay (data not shown). All three NOS assays require electron transfer from NADPH for product formation. We were unable to detect NADPH oxidation or cytochrome c reduction by AtNOS1 (data not shown). To test the possibilities that AtNOS1 converts arginine to NO via a mechanism different from that of mammalian NOSs or that the lack of a suitable reductase component in our reconstituted system was preventing the detection of AtNOS1 activity, we assessed whether AtNOS1 is at least competent in binding its hypothetical substrate arginine. Neither Δ101 nor fl bound [3H]arginine in an exclusion chromatography assay (Fig. 1B). The addition of potential cofactors, such as NADPH, CaM, and Ca2+, as suggested by the original report (20) did not promote arginine binding to Δ101 (Fig. 1B). In this and all other assays, the mammalian nNOS control gave positive results, indicating that these assays were working. Together, these results demonstrate that AtNOS1 neither binds nor oxidizes arginine to NO via a mechanism analogous to that of the mammalian NOSs (Table 1). Therefore, from this point onward, we will follow the suggestion to use AtNOA1 (A. thaliana NO-associated protein 1) to refer to this protein (25).
FIGURE 1.

NO synthase properties of nNOS and AtNOA1. A, NO synthase activity of rat nNOS and Δ101. Differential spectra of oxyhemoglobin were recorded every 0.5 min for 5 min in a reaction containing either 3.9 μg of nNOS (•) or 155 μg of Δ101 (▪) and the necessary cofactors, as indicated under “Experimental Procedures.” Optical density differences between 401 and 420 nm are plotted as a function of time and shows the absence of methemoglobin formation with AtNOA1 in contrast to its production with nNOS. B, binding of [3H]arginine by nNOS, Δ101, and full-length AtNOA1 using exclusion chromatography. A 120-μl reaction containing 0.5 μCi of [3H]arginine (100 nm) in the absence of protein or in the presence of 2 μm nNOS, Δ101, or fl was equilibrated on ice for 15 min. The cofactors (100 μm), CaM (10 μg/ml), and CaCl2 (0.01 m) were also included when indicated (+cofactors). A 100-μl aliquot of each reaction was chromatographed on a 1-ml Sephadex G25 column, and the excluded [3H]arginine bound to protein was counted (dashed bars). The specificity of arginine binding to the protein was assessed in reactions containing 10 mm unlabeled arginine in addition to the [3H]arginine (open bars).
TABLE 1.
Lack of expected AtNOA1 NOS-like activities
| AtNOA1 | nNOS/inducible NOS | |
|---|---|---|
| Oxyhemoglobin assay | − | +a,b |
| [3H]Arginine → [3H]citrulline | − | +c |
| Nitrite formation (Griess assay) | − | +c |
| NADPH oxidation | − | +a |
| Cytochrome c reduction | − | +c |
| Arginine binding | − | +a,b |
Purified recombinant rat nNOS was used as a positive control.
See Fig. 1 for details.
E. coli lysate containing murine recombinant inducible NOS (Cayman Chemicals) was used in a parallel experiment as a positive control.
Structure-Function Analyses Reveal the Importance of the CPG and CTD Domains—Taking advantage of the close sequence similarity between Δ101 and YqeH (Fig. 2A), a model for the three-dimensional structure of Δ101 was built based on the x-ray crystal structure of the Geobacillus stearothermophilus YqeH (gsYqeH) (Fig. 2B). The model suggests that, like gsYqeH, the CPG domain of AtNOA1 displays a fold similar to that of canonical GTPases. In the GDP-bound form the nucleotide is probably exposed to the solvent. As observed with other small GTPases like Ras (29), the aspartate residue in the G4 motif (Asp-226; Fig. 2A) is positioned favorably to stabilize the guanine ring moiety. Introduction of fl wild type AtNOA1 into Atnoa1 mutant plants restored the wild type phenotype, including normal plant size and green coloration of leaves, as shown in Figs. 3, A and B (20, 48). In contrast, expression of a mutant AtNOA1D226N in Atnoa1 mutant plants failed to restore normal growth or leaf coloration to Atnoa1 (Figs. 3, A and B). This suggests that the disruption of GTP/GDP binding leads to loss of function of AtNOA1 in planta, thus highlighting the essential role of CPG domain in AtNOA1 physiological function.
FIGURE 3.

Complementation of Atnoa1 with mutants of AtNOA1. A (from left to right), 2-week-old seedlings of wild type Col-0, Atnoa1, and Atnoa1 transformed with either AtNOA1D226N or AtNOA1 fl. Seeds were planted on Murashige-Skoog medium, incubated at 4 °C for 3 days, and then transferred to 22 °C for germination and further growth. The size bars correspond to 3 mm. The arrowheads point to the emerging rosette leaves. B, Western blot of leaf extracts using anti-FLAG antibody to assess the expression of FLAG-tagged AtNOA1D226N and AtNOA1 in Atnoa1 plants. C shows that the C-terminal truncated AtNOA1 fails to complement Atnoa1 mutant plants and shows (from left to right) wild type, Atnoa1, and Atnoa1 transformed with Atnoa1-(1-386) (white bars, correspond to 2 cm). Seed germination was synchronized by cold treatment, and photographs were taken 4 weeks after germination. D, Western blot of leaf extracts of wild type, Atnoa1, and Atnoa1 plants expressing HA-tagged AtNOA1-(1-386) using anti-HA antibody. In both B and D, the Rubisco protein band stained with Coomassie Blue shows comparable loading of protein extracts.
Based on structural analysis of gsYqeH and other GTPases (48), GTP binding may trigger a conformational change in the connection between the CPG domain and the CTD. Repositioning of the CTD may modulate the potential GTPase activity of AtNOA1 or vice versa. In both cases, the spatial arrangement of the CTD in relation to the CPG domain suggests a possible important role of CTD in AtNOA1 function. The structure of AtNOA1 CTD is predicted to be very similar to that of gsYqeH. Insertions I2-I4 present in AtNOA1 are located in loops and β-turns (Fig. 2, A and B). Therefore, they are unlikely to change the overall fold of this domain from that found in gsYqeH. The arrangement of the CTD is interesting, since it displays a novel topology involving two pseudosymmetric β-sheet units. Structural similarity has been found between each of these subunits and the RNA-binding protein TRAP (Trp RNA-binding attenuating protein) (48). To assess the importance of this domain in planta, we conducted complementation experiments to evaluate if the CTD truncated AtNOA1 was able to function like fl AtNOA1 in plants. In contrast to what was observed with the introduction of the fl AtNOA1, introduction into Atnoa1 of AtNOA1-(1-386), which contains both the N-terminal zinc-binding and CPG domains but not the CTD, failed to rescue the yellowish color of the first true leaves and the dwarf phenotype of Atnoa1 mutant plants (Figs. 3, C and D). This result suggests that CTD is necessary for AtNOA1 function in planta.
AtNOA1 Binds GDP More Tightly than GTP—The AtNOA1 CPG domain displays a three-dimensional arrangement very similar to canonical GTPases, suggesting that AtNOA1 might be a functional GTPase. To assess this possibility, we first determined the ability of AtNOA1 to bind GDP and/or GTP using the fluorescent MantGDP (54). The addition of Δ101 to a solution of MantGDP led to a rapid increase in fluorescence, indicating the binding of MantGDP to the protein (Fig. 4A, 1). The further addition of 50 μm ATP or CTP did not change the fluorescence, suggesting that neither ATP nor CTP were able to compete with MantGDP for binding (Fig. 4A, 2 and 3). Even higher concentrations of ATP or CTP (up to 500 μm) did not lead to any significant change in fluorescence (Fig. S1). This rules out the possibility of a low affinity binding of those nucleotide to Δ101. However, the addition of 50 μm GTP decreased MantGDP fluorescence by over 50%. This result argues that binding of MantGDP to Δ101 is reversible and specific. Both GTP and GDP were able to displace MantGDP; together these data indicate that AtNOA1 specifically binds the guanidine nucleotide. Magnesium salt regulates nucleotide release in small GTPases (55); however, its presence did not modify the observed GTP/GDP binding of AtNOA1. Moreover, it did not modify the high affinity of MantGDP for Δ101 (∼560 nm; Table 2). A competition assay between MantGDP and either GDP or GTP was used to quantify GTP and GDP binding to AtNOA1 (Fig. 4B). Increasing amounts of GTP or GDP were added to the preformed MantGDP-Δ101 complex, and nucleotide exchange was monitored by following the decrease in fluorescence. The resulting IC50 for GTP and GDP is a measure of their relative binding affinities for Δ101. GDP consistently bound more tightly than GTP to Δ101 with an approximately 3-fold difference in IC50 that was little affected by the presence of MgCl2 and KCl (Table 2).
FIGURE 4.

GTP and GDP binding to Δ101. A, specific binding of MantGDP and GTP to Δ101. MantGDP fluorescence change (in arbitrary units (a.u.)) caused by the addition of Δ101 to 1.5 μm (1) was monitored before and after the successive addition to 50 μm final concentration of ATP (2), CTP (3), or GTP (4) in buffer B. Only GTP was able to compete with MantGDP for binding to Δ101. B, binding competition assay between MantGDP and either GTP (▪) or GDP (•). The data used for plotting were derived from the relative fluorescence percentage, as described under “Experimental Procedures,” and the IC50 values were calculated. Each experiment was carried out in triplicate.
TABLE 2.
Affinities of MantGDP, GDP, and GTP for AtNOA1Δ101
| Kd (MantGDP) | IC50 (GDP) | IC50 (GTP) | |
|---|---|---|---|
| nm | μm | μm | |
| Buffer B with 200 mm KCl, 5 mm MgCl2 | 546 ± 98 | 5.2 ± 0.9 | 18.9 ± 2.5 |
| Buffer B | 577 ± 237 | 6.0 ± 1.8 | 15.4 ± 1.8 |
AtNOA1 Is a Slow GTPase, Whose Activity Is Independent of Its ZBD and CTD—The ability of AtNOA1 to hydrolyze GTP was assessed by HPLC analysis of the reaction products of Δ101 with 500 μm GTP. After incubation at 37 °C overnight, in the absence of Δ101, very little GDP was produced with the majority of the guanine remaining as GTP (Fig. 5A, bottom). In the presence of Δ101, GTP was converted to GDP in a dose-dependent manner, with the majority of it hydrolyzed to GDP with the higher amount of Δ101 (Fig. 5A, top). The GTPase activity of Δ101 was quantified using an activated charcoal pull-down assay with radioactive [γ-32P] GTP (Fig. 5B). In this assay, Δ101 was incubated at 37 °C with [γ-32P]GTP and varying amounts of unlabeled GTP. The initial velocity of GTP hydrolysis was calculated from the counts/min data and used to determine the Km and Vmax values of the protein (64.5 ± 5.7 μm and 0.072 ± 0.01 min-1, respectively). The GTPase activity required MgCl2 (Fig. 6). The presence of either KCl or (NH4)2SO4 was needed for the activity as well (data not shown). The conserved threonine residue of the G2 motif, within the Switch I region, is usually involved in coordination of Mg2+ via its side chain hydroxyl and is in contact with the γ phosphate of GTP via its main chain NH. Mutation of this threonine residue to alanine has been shown to result in loss of GTPase activity of other GTPases (48, 56). Mutation of the corresponding threonine in Δ101 (T327A) similarly abolished its GTP hydrolysis activity (Fig. 6). This result demonstrates that, although Δ101 hydrolysis activity is low, this protein is an authentic GTPase. It also reveals the crucial role of the Switch I region for the GTPase activity of AtNOA1. Different constructs of AtNOA1 were also tested to determine whether the ZBD or CTD of AtNOA1 might alter its GTPase activity (Fig. 6). Removal of the ZBD or CTD did not significantly change its GTPase activity. AtNOA1-(102-350) and AtNOA1-(176-561) hydrolyzed GTP with activities corresponding to 80 ± 50 and 130 ± 50% of the GTPase activity of Δ101, respectively.
FIGURE 5.

GTPase activity of Δ101. A, HPLC chromatograms showing the elution profile of 500 μm GTP after an overnight incubation at 37 °C in the absence (bottom) or presence of 20 or 40 μm Δ101 (top). GTP and GDP elutes at 13 and 8 min, respectively. B, counts/min versus time of a reaction containing 5 μm Δ101, 2 μCi of [γ-32P]GTP, and increasing concentrations of GTP (10 μm (▪), 25 μm (□), 50 μm (▴), 100 μm (▾), 200 μm (•), 400 μm (○), and 800 μm (▸)) in buffer B supplemented with 200 mm KCl and 5 mm MgCl2 at 37 °C. Linear regression of the counts/min was mathematically converted into pmol of GTP hydrolyzed/min/μmol of protein. The Lineweaver-Burk reciprocal plot (inset) leads to the Km and Vmax for the protein.
FIGURE 6.

GTPase activity of different constructs of AtNOA1 and magnesium dependence of the reaction. The amounts of phosphate released, expressed in nmol of GTP hydrolyzed/nmol of protein, are presented for reactions containing MgCl2 plus no protein (○) or 3 μm Δ101 (▪), of Δ101T327A (♦), of AtNOA1-(176-561) (CPG-CTD domains (▴)), of AtNOA1-(102-350) (ZBD-CPG domains (♦)), or Δ101 in the absence of MgCl2 (□). All reactions were conducted at 37 °C in the presence of 4 μCi of [γ-32P]GTP and 500 μm GTP. Data points with slightly negative values were set to zero.
DISCUSSION
From AtNOS1 to AtNOA1—AtNOS1 was reported in 2003 to have NOS activity (20). However, using a range of assays with varying sensitivities to monitor different properties of NOSs, our results indicate that this protein does not have NOS-like activities (Table 1). The NO formation activity of AtNOS1 was reported to be regulated by Ca2+-CaM binding (20); thus, the rat nNOS isoform was chosen as a positive control in our experiments, since its activity is also regulated by Ca2+-CaM binding (43). Reactions containing AtNOS1 fl or Δ101, arginine, and NADPH, as an electron donor, failed to produce NO (oxyhemoglobin assay; Fig. 1A) or NO-derived nitrite (Griess assay) regardless of the presence of CaM, CaCl2, BH4, and flavines like FMN or FAD. Under the same conditions, nNOS produced 147 pmol of NO/min/mg of protein at room temperature as measured with the oxyhemoglobin assay and as previously published (43). The NOS activity reported for the recombinant AtNOS1 was ∼30 pmol of NO/min/mg of protein (20), which is in the same range and should have been detected by this assay. Indeed, this level of activity should have resulted in a detectable optical difference of 0.01 between 401 and 420 nm in our experiments (with 155 μg of protein in a 150-μl reaction for 5 min). This was not observed even at later time points and higher protein concentrations. A third assay, the very sensitive detection of radio-labeled citrulline from [3H]arginine, also failed to detect NOS activity of AtNOS1. In addition, AtNOS1 did not have electron transfer and arginine binding (Fig. 1B) activities, two properties of NOS-like enzymes. Moreover, the structure of the bacterial homolog gsYqeH and the modeling of AtNOS1 do not reveal any fold that might account for a NOS function or the binding of necessary cofactors. In summary, our data show that AtNOS1 does not possess any of the expected characteristics of a NOS or NOS-like enzyme (Table 1). These findings are consistent with the recent communications questioning the NOS activity of AtNOS1 (25, 27) and the renaming of AtNOS1 as AtNOA1.
Characterization of AtNOA1, a Plant cGTPase—The amino acid sequence of AtNOA1 reveals the presence of a circularly permuted GTP-binding domain (57). Unlike the classical small GTPases like Rho, Ras, and Ran that have been extensively studied, little is known about the cGTPase family and its GTPase activity, especially in eukaryotes. Moreover, the catalytic glutamine or histamine found in the G3 motif of small GTPases, which maintains the water molecule in an orientation necessary for hydrolysis, is replaced with a hydrophobic residue in AtNOA1 and its homologs (Val-349 in AtNOA1; see Fig. 2A). Although such a mutation in Ras disrupts GTP hydrolysis, many HAS GTPases (hydrophobic amino acid substituted for catalytic glutamine residue GTPases) retain their GTPase activity (58).
We demonstrated by HPLC analysis that AtNOA1 is able to hydrolyze GTP to GDP and showed the requirement for both MgCl2 and a monovalent salt, such as KCl or (NH4)2SO4 (Fig. 4A; data not shown for (NH4)2SO4 effect). The GTPase activity of AtNOA1 was higher and more reproducible in the presence of KCl than in the presence of (NH4)2SO4. This stimulating effect of potassium ions on GTP hydrolysis has been observed in another HAS GTPase, MnmE (59). In that case, the positive charge of the potassium ion stabilizes the transition state in MnmE. Further study of the AtNOA1 GTPase mechanism of catalysis is required to determine if this is also the case for AtNOA1. Examination of GTP binding by AtNOA1 revealed additional interesting characteristic of this cGTPase. Although magnesium plays an inhibitory role in guanine nucleotide exchange in small GTPases (55, 60), the presence of MgCl2 did not alter AtNOA1 affinities for GTP, GDP, or a fluorescent GDP analog (Table 2), as has been reported for the Rho GTPases (61). This characteristic might explain why magnesium ions were not detected in the crystal structure of the bacterial homolog gsYqeH and the other cGTPase YjeQ (48, 62). Nonetheless, magnesium ions are essential for GTPase activity and thus may be bound only transiently to assist catalysis. Second, neither ATP nor CTP could compete with the fluorescent GDP analog, even when present at high concentrations (2500-fold molar excess, Figs. 4A and S1). Thus, AtNOA1 appears to specifically bind the guanosine nucleotide, a characteristic shared with the bacterial cGTPases YqeH, YlqF, and YloQ (33, 63). Third, the higher affinity of AtNOA1 for GDP than for GTP (Fig. 4B and Table 2) might explain its slow steady-state GTP hydrolysis rate. Indeed, its Vmax of 0.07 ± 0.01 min-1 falls within the range of nonactivated Ras (0.028 min-1) or EF-Tu (0.036 min-1) GTPase activity (64, 65). It is also comparable with nonactivated cGTPase YjeQ (0.15 min-1) of E. coli (66, 67) and Yloq of B. subtilis (0.22 min-1) (63) but unexpectedly lower than YqeH high intrinsic GTPase activity (0.93 min-1) (39). Small GTPases like Ras or Rho interact with a guanine nucleotide exchange factor or GTPase-activating protein, which leads toa102- to 105-fold increase of the rate of GTP hydrolysis (68). Interestingly, for several bacterial cGTPases, interaction with ribosome subunits modulates their GTPase activity; YjeQ GTPase activity in vitro was enhanced 160-fold in the presence of the 30 S subunit (67, 69), whereas the 50 S subunit stimulates the activity of YlqF (35). The slow steady-state GTP hydrolysis rate of AtNOA1 suggests that it may require a guanine nucleotide exchange factor or a GTPase-activating protein to reach a physiologically relevant GTPase activity. It remains to be determined whether ribosome and/or RNA binding enhances AtNOA1 GTPase activity.
Despite the circular permutation of the G domain, the GTPase domain folding in Yloq and YjeQ, as well as in YqeH and the model of AtNOA1, is similar to the one observed with classical small GTPases (48, 62, 70). However, the rearrangement of the G subdomains leads to repositioning at the C-terminal end of the G3 region (Switch II; Fig. 2A). This region contains the traditional catalytic glutamine residue and is therefore essential for catalysis. In all members of the cGTPase family, a C-terminal domain connects directly to G3 due to the sequence permutation of the G protein homology regions (28). This suggests that the CTD may influence the GTPase activity or vice versa (i.e. GTP hydrolysis may modulate CTD function). In our study, removal of either the ZBD or the CTD did not significantly change the GTPase activity of AtNOA1. This is particularly interesting considering that not only the GTPase domain but also the CTD is important for the function of AtNOA1 in planta (Fig. 3). As G proteins cycle between an inactive GDP-bound and an active GTP-bound state, they undergo conformational changes that allow for interaction with effectors. We suspect that the CTD of AtNOA1 has a critical function in planta that is modulated by GTP hydrolysis.
What Function of AtNOA1 Could Account for the Impaired NO Accumulation in Atnoa1 Mutant Plants?—We suspect that AtNOA1 binds ribosomes and consequently plays a role in their proper assembly and/or stability, which leads to appropriate levels of protein synthesis. Although Zemojtel et al. (27) previously speculated that AtNOA1 is involved in mitochondrial ribosome biogenesis and/or translation, our suspicion is based on several observations. First, AtNOA1 belongs to the Era/Obg subfamily of small GTPases that have been predicted and/or shown to be associated with ribosomes (36, 69). Studies show that bacterial YlqF participates in the final steps of 50 S ribosomal subunit assembly (35, 37), evidence points toward a role of YjeQ in 30 S ribosome biogenesis and subunit association (69, 71), and preliminary results indicate that YloQ activity is enhanced by purified E. coli ribosomes (63). Second, in eukaryotes, the yeast cGTPase NUG1 associates with 60 S pre-ribosomal particles (72). Third and of particular relevance is the finding that YqeH, the closest bacterial homolog of AtNOA1, is involved in 30 S subunit biogenesis in B. subtilis (38, 39). Since YqeH complements the Atnoa1 mutant (48, 73), it is very likely that AtNOA1 plays a similar role in plants as YqeH does in bacteria. The ZBD, CTD, or both may play such a ribosome/RNA-binding role. Indeed, zinc finger motifs have nucleic acid-binding properties. The treble clef motif found in AtNOA1 is associated with many types of activities, from binding nucleic acids, proteins, or small molecules to phophodiester bond hydrolysis. Interestingly, the ribosomal proteins L24E and S14 contain such a domain (30). Also, the unique structure of the C-terminal domain of gsYqeH is found in the predicted structure of the modeled AtNOA1 and is similar to the TRAP protein, which has the ability to bind RNA (48). In particular, two triads of residues involved in RNA binding in the TRAP protein (74) are well conserved in AtNOA1 and its homologs (e.g. Asp-483, Trp-491, and Arg-530 and Phe-401, Arg-407, and Asp-409 in AtNOA1). The x-ray structure of gsYqeH and the predicted structure of the modeled AtNOA1 suggest that these residues are exposed and match the spatial arrangement observed in the RNA-binding site of TRAP (48). Whether the CTD binds RNA alone or in association with the N-terminal domain remains to be demonstrated, but the essential role of the CTD in AtNOA1 function (Fig. 3, C and D) suggests that it may fulfill the important role of RNA/ribosome binding.
Based on its sequence, AtNOA1 is predicted to be targeted to either
mitochondria (score of 77.9% on TargetP, 6.5 on Psort) or chloroplasts (6.5 on
Psort). According to the same localization programs, both the tomato (64.7%
identity) and rice (60.5%) homologs are predicted to be imported into
chloroplasts. Moreover, in contrast to evidence for mitochondrial localization
of AtNOA1 in Arabidopsis roots
(22), a recent publication has
shown that AtNOA1 co-localized with chloroplasts in leaves and is imported
into isolated leaf chloroplasts
(73). Regardless of whether
AtNOA1 is in mitochondria and/or chloroplasts, these are both sites of
electron transfer that can lead to reactive oxygen species (ROS) production
(75). We propose that the
defective ribosome/RNA assembly in Atnoa1 leads to increased
production of ROS, such as superoxide ion
(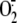 ), hydroxyl radical
(OH.), and hydrogen peroxide (H2O2) in either
or both of these organelles. Consistent with this proposition, Atnoa1
mutant plants exhibit a constitutively elevated level of ROS and oxidized
lipids and proteins (22,
76).
), hydroxyl radical
(OH.), and hydrogen peroxide (H2O2) in either
or both of these organelles. Consistent with this proposition, Atnoa1
mutant plants exhibit a constitutively elevated level of ROS and oxidized
lipids and proteins (22,
76).
We propose that the elevated amount of ROS observed in the Atnoa1
mutant is responsible for the reduced NO accumulation, since NO can react very
quickly with 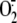 and lipid
radicals (77) and thus reduce
the amount of detectable NO. Indeed, peroxynitrite generated by the rapid
reaction between NO and
and lipid
radicals (77) and thus reduce
the amount of detectable NO. Indeed, peroxynitrite generated by the rapid
reaction between NO and 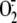 is unable to activate the extensively used fluorescent probe
diaminofluorescein (78).
Moreover, although there is an abundant literature documenting the lower
accumulation and/or detection of NO in Atnoa1
(20,
22,
24,
79,
80), there are also reports
showing that Atnoa1 is not always impaired in NO accumulation. For
example, in response to H2O2, to iron, to indole
3-butyric acid, to Verticillium dahliae toxins, or to zeatin, NO
production is as high in Atnoa1 as in wild type plants
(23,
41,
42,
81,
82). The presence of a
nitrite-dependent NO production pathway in Atnoa1 might account for
those conflicting observations. Consideration of subcellular localization of
ROS and NO may be another way to reconcile these differences. If both reactive
species are produced in the same subcellular location, NO detection may be
inefficient due to its rapid interaction with the elevated levels of ROS in
Atnoa1; in contrast, elevated ROS levels in Atnoa1 in a
subcellular localization different from that of NO production may not affect
the detected NO levels. Thus, it is likely that the association of AtNOA1 with
NO is the result of the pleiotropic effects of mal-functioning organelles that
overproduce ROS, which can rapidly react with NO, thereby reducing the amount
of NO free to react in the various NO detection assays. A very recent study
shows that a mutant of AtNOA1 (also called rif1 for
resistant to inhibition by FSM) has reduced
chloroplastic protein synthesis and elevated expression of methylerythritol
phosphate (MEP) pathway enzymes
(73). Although exogenous
application of NO partially rescued the pale yellowish leaf phenotype of
Atnoa1/rif1 mutant plants, the other physiological traits associated
with Atnoa1/rif1 were not rescued (e.g. chloroplastic
protein synthesis, MEP pathway regulation). The rescue of the morphological
phenotype of Atnoa1 by application of SNP was the central argument in
favor of a direct relation between AtNOA1 and NO production
(20). The recent result with
rif1 reinforces the possibility that the connection between NO and
AtNOA1 is indirect. The addition of exogenous NO might rescue the pale
phenotype via its antioxidant property that counteracts the high ROS
environment of Atnoa1. However, this antioxidant effect is not
sufficient to rescue the loss of function of AtNOA1 associated with
deregulation of the MEP pathway enzymes and protein synthesis in chloroplasts
(73).
is unable to activate the extensively used fluorescent probe
diaminofluorescein (78).
Moreover, although there is an abundant literature documenting the lower
accumulation and/or detection of NO in Atnoa1
(20,
22,
24,
79,
80), there are also reports
showing that Atnoa1 is not always impaired in NO accumulation. For
example, in response to H2O2, to iron, to indole
3-butyric acid, to Verticillium dahliae toxins, or to zeatin, NO
production is as high in Atnoa1 as in wild type plants
(23,
41,
42,
81,
82). The presence of a
nitrite-dependent NO production pathway in Atnoa1 might account for
those conflicting observations. Consideration of subcellular localization of
ROS and NO may be another way to reconcile these differences. If both reactive
species are produced in the same subcellular location, NO detection may be
inefficient due to its rapid interaction with the elevated levels of ROS in
Atnoa1; in contrast, elevated ROS levels in Atnoa1 in a
subcellular localization different from that of NO production may not affect
the detected NO levels. Thus, it is likely that the association of AtNOA1 with
NO is the result of the pleiotropic effects of mal-functioning organelles that
overproduce ROS, which can rapidly react with NO, thereby reducing the amount
of NO free to react in the various NO detection assays. A very recent study
shows that a mutant of AtNOA1 (also called rif1 for
resistant to inhibition by FSM) has reduced
chloroplastic protein synthesis and elevated expression of methylerythritol
phosphate (MEP) pathway enzymes
(73). Although exogenous
application of NO partially rescued the pale yellowish leaf phenotype of
Atnoa1/rif1 mutant plants, the other physiological traits associated
with Atnoa1/rif1 were not rescued (e.g. chloroplastic
protein synthesis, MEP pathway regulation). The rescue of the morphological
phenotype of Atnoa1 by application of SNP was the central argument in
favor of a direct relation between AtNOA1 and NO production
(20). The recent result with
rif1 reinforces the possibility that the connection between NO and
AtNOA1 is indirect. The addition of exogenous NO might rescue the pale
phenotype via its antioxidant property that counteracts the high ROS
environment of Atnoa1. However, this antioxidant effect is not
sufficient to rescue the loss of function of AtNOA1 associated with
deregulation of the MEP pathway enzymes and protein synthesis in chloroplasts
(73).
In conclusion, our study provides strong evidence that AtNOA1 is not an NOS, but a cGTPase, whose enzyme activity is necessary but not sufficient for its function in planta. Characterization of this new class of plant GTPases demonstrates the importance of a previously uncharacterized protein domain, the CTD. Additional studies are needed to elucidate the role of AtNOA1 in plants, in particular its possible function in mitochondrial and/or chloroplastic ribosome biogenesis and maintenance.
Supplementary Material
Acknowledgments
We thank Dr. Nigel Crawford for providing Atnoa1 seeds and Jawahar Sudhamsu for useful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant 5R01GM067011 (to D. F. K.). This work was also supported by National Science Foundation Grant (to B. R. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: NO, nitric oxide; NOS, nitric-oxide synthase; CTD, C-terminal domain; CPG domain, circularly permuted G-motif domain; ZBD, zinc-binding domain; CaM, calmodulin; cGTPase, circularly permuted GTPase; BH4, tetrahydro-l-biopterin; nNOS, neuronal NOS; HA, hemagglutinin; MantGDP, N-methylanthraniloyl-labeled GDP; fl, full-length; gsYqeH, G. stearothermophilus YqeH; ROS, reactive oxygen species.
References
- 1.He, Y., Tang, R. H., Hao, Y., Stevens, R. D., Cook, C. W., Ahn, S. M., Jing, L., Yang, Z., Chen, L., Guo, F., Fiorani, F., Jackson, R. B., Crawford, N. M., and Pei, Z. M. (2004) Science 305 1968-1971 [DOI] [PubMed] [Google Scholar]
- 2.Bethke, P. C., Libourel, I. G., and Jones, R. L. (2006) J. Exp. Bot. 57 517-526 [DOI] [PubMed] [Google Scholar]
- 3.Beligni, M. V., and Lamattina, L. (2000) Planta 210 215-221 [DOI] [PubMed] [Google Scholar]
- 4.Neill, S. J., Desikan, R., Clarke, A., and Hancock, J. T. (2002) Plant Physiol. 128 13-16 [PMC free article] [PubMed] [Google Scholar]
- 5.Durner, J., Wendehenne, D., and Klessig, D. F. (1998) Proc. Natl. Acad. Sci. U. S. A 95 10328-10333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Delledonne, M., Xia, Y., Dixon, R. A., and Lamb, C. (1998) Nature 394 585-588 [DOI] [PubMed] [Google Scholar]
- 7.Wendehenne, D., Durner, J., and Klessig, D. F. (2004) Curr. Opin. Plant Biol. 7 449-455 [DOI] [PubMed] [Google Scholar]
- 8.Wilson, I. D., Neill, S. J., and Hancock, J. T. (2008) Plant Cell Environ. 31 622-631 [DOI] [PubMed] [Google Scholar]
- 9.Yamasaki, H., and Sakihama, Y. (2000) FEBS Lett. 468 89-92 [DOI] [PubMed] [Google Scholar]
- 10.Rockel, P., Strube, F., Rockel, A., Wildt, J., and Kaiser, W. M. (2002) J. Exp. Bot. 53 103-110 [PubMed] [Google Scholar]
- 11.Stohr, C., and Stremlau, S. (2006) J. Exp. Bot. 57 463-470 [DOI] [PubMed] [Google Scholar]
- 12.Planchet, E., Gupta, K. J., Sonoda, M., and Kaiser, W. M. (2005) Plant J. 41 732-743 [DOI] [PubMed] [Google Scholar]
- 13.Bethke, P. C., Badger, M. R., and Jones, R. L. (2004) Plant Cell 16 332-341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ninnemann, H., and Maier, J. (1996) Photochem. Photobiol. 64 393-398 [DOI] [PubMed] [Google Scholar]
- 15.Cueto, M., Hernandez-Perera, O., Martin, R., Bentura, M. L., Rodrigo, J., Lamas, S., and Golvano, M. P. (1996) FEBS Lett. 398 159-164 [DOI] [PubMed] [Google Scholar]
- 16.Mur, L. A., Carver, T. L., and Prats, E. (2006) J. Exp. Bot. 57 489-505 [DOI] [PubMed] [Google Scholar]
- 17.Lamotte, O., Gould, K., Lecourieux, D., Sequeira-Legrand, A., Lebrun-Garcia, A., Durner, J., Pugin, A., and Wendehenne, D. (2004) Plant Physiol. 135 516-529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandok, M. R., Ytterberg, A. J., van Wijk, K. J., and Klessig, D. F. (2003) Cell 113 469-482 [DOI] [PubMed] [Google Scholar]
- 19.Travis, J. (2004) Science 306 960. [DOI] [PubMed] [Google Scholar]
- 20.Guo, F. Q., Okamoto, M., and Crawford, N. M. (2003) Science 302 100-103 [DOI] [PubMed] [Google Scholar]
- 21.Huang, S., Kerschbaum, H. H., Engel, E., and Hermann, A. (1997) J. Neurochem. 69 2516-2528 [DOI] [PubMed] [Google Scholar]
- 22.Guo, F. Q., and Crawford, N. M. (2005) Plant Cell 17 3436-3450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bright, J., Desikan, R., Hancock, J. T., Weir, I. S., and Neill, S. J. (2006) Plant J. 45 113-122 [DOI] [PubMed] [Google Scholar]
- 24.Zeidler, D., Zahringer, U., Gerber, I., Dubery, I., Hartung, T., Bors, W., Hutzler, P., and Durner, J. (2004) Proc. Natl. Acad. Sci. U. S. A 101 15811-15816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crawford, N. M., Galli, M., Tischner, R., Heimer, Y. M., Okamoto, M., and Mack, A. (2006) Trends Plant Sci. 11 526-527 [Google Scholar]
- 26.Guo, F. G. (2006) Trends Plant Sci. 11 527-528 [Google Scholar]
- 27.Zemojtel, T., Frohlich, A., Palmieri, M. C., Kolanczyk, M., Mikula, I., Wyrwicz, L. S., Wanker, E. E., Mundlos, S., Vingron, M., Martasek, P., and Durner, J. (2006) Trends Plant Sci. 11 524-525 [DOI] [PubMed] [Google Scholar]
- 28.Anand, B., Verma, S. K., and Prakash, B. (2006) Nucleic Acids Res. 34 2196-2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bourne, H. R., Sanders, D. A., and McCormick, F. (1991) Nature 349 117-127 [DOI] [PubMed] [Google Scholar]
- 30.Grishin, N. V. (2001) Nucleic Acids Res. 29 1703-1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krishna, S. S., Majumdar, I., and Grishin, N. V. (2003) Nucleic Acids Res. 31 532-550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arigoni, F., Talabot, F., Peitsch, M., Edgerton, M. D., Meldrum, E., Allet, E., Fish, R., Jamotte, T., Curchod, M. L., and Loferer, H. (1998) Nat. Biotechnol. 16 851-856 [DOI] [PubMed] [Google Scholar]
- 33.Morimoto, T., Loh, P. C., Hirai, T., Asai, K., Kobayashi, K., Moriya, S., and Ogasawara, N. (2002) Microbiology 148 3539-3552 [DOI] [PubMed] [Google Scholar]
- 34.Zalacain, M., Biswas, S., Ingraham, K. A., Ambrad, J., Bryant, A., Chalker, A. F., Iordanescu, S., Fan, J., Fan, F., Lunsford, R. D., O'Dwyer, K., Palmer, L. M., So, C., Sylvester, D., Volker, C., Warren, P., McDevitt, D., Brown, J. R., Holmes, D. J., and Burnham, M. K. (2003) J. Mol. Microbiol. Biotechnol. 6 109-126 [DOI] [PubMed] [Google Scholar]
- 35.Matsuo, Y., Morimoto, T., Kuwano, M., Loh, P. C., Oshima, T., and Ogasawara, N. (2006) J. Biol. Chem. 281 8110-8117 [DOI] [PubMed] [Google Scholar]
- 36.Leipe, D. D., Wolf, Y. I., Koonin, E. V., and Aravind, L. (2002) J. Mol. Biol. 317 41-72 [DOI] [PubMed] [Google Scholar]
- 37.Uicker, W. C., Schaefer, L., and Britton, R. A. (2006) Mol. Microbiol. 59 528-540 [DOI] [PubMed] [Google Scholar]
- 38.Uicker, W. C., Schaefer, L., Koenigsknecht, M., and Britton, R. A. (2007) J. Bacteriol. 189 2926-2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Loh, P. C., Morimoto, T., Matsuo, Y., Oshima, T., and Ogasawara, N. (2007) Genes Genet. Syst. 82 281-289 [DOI] [PubMed] [Google Scholar]
- 40.Zemojtel, T., Kolanczyk, M., Kossler, N., Stricker, S., Lurz, R., Mikula, I., Duchniewicz, M., Schuelke, M., Ghafourifar, P., Martasek, P., Vingron, M., and Mundlos, S. (2006) FEBS Lett. 580 455-462 [DOI] [PubMed] [Google Scholar]
- 41.Shi, F. M., and Li, Y. Z. (2008) BMB Rep. 41 79-85 [DOI] [PubMed] [Google Scholar]
- 42.Tun, N. N., Livaja, M., Kieber, J. J., and Scherer, G. F. (2008) New Phytol. 178 515-531 [DOI] [PubMed] [Google Scholar]
- 43.Moreau, M., Takahashi, H., Sari, M. A., Boucher, J. L., Sagami, I., Shimizu, T., and Mansuy, D. (2004) J. Inorg. Biochem. 98 1200-1209 [DOI] [PubMed] [Google Scholar]
- 44.Murphy, M. E., and Noack, E. (1994) Methods Enzymol. 233 240-250 [DOI] [PubMed] [Google Scholar]
- 45.Hevel, J. M., and Marletta, M. A. (1994) Methods Enzymol. 233 250-258 [DOI] [PubMed] [Google Scholar]
- 46.Poirot, O., O'Toole, E., and Notredame, C. (2003) Nucleic Acids Res. 31 3503-3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sali, A., and Blundell, T. L. (1993) J. Mol. Biol. 234 779-815 [DOI] [PubMed] [Google Scholar]
- 48.Sudhamsu, J., Lee, G. I., Klessig, D. F., and Crane, B. R. (2008) J. Biol. Chem. 283 32968-32976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Feng, S. H., Ma, L. G., Wang, X. P., Xie, D. X., Dinesh-Kumar, S. P., Wei, N., and Deng, X. W. (2003) Plant Cell 15 1083-1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bendahmane, A., Farnham, G., Moffett, P., and Baulcombe, D. C. (2002) Plant J. 32 195-204 [DOI] [PubMed] [Google Scholar]
- 51.Zhang, X., Henriques, R., Lin, S. S., Niu, Q. W., and Chua, N. H. (2006) Nat. Prot. 1 641-646 [DOI] [PubMed] [Google Scholar]
- 52.Kilby, N. J., Leyser, H. M. O., and Furner, I. J. (1992) Plant Mol. Biol. 20 103-112 [DOI] [PubMed] [Google Scholar]
- 53.Majumdar, S., Ramachandran, S., and Cerione, R. A. (2004) J. Biol. Chem. 279 40137-40145 [DOI] [PubMed] [Google Scholar]
- 54.Remmers, A. E., Posner, R., and Neubig, R. R. (1994) J. Biol. Chem. 269 13771-13778 [PubMed] [Google Scholar]
- 55.Hall, A., and Self, A. J. (1986) J. Biol. Chem. 261 10963-10965 [PubMed] [Google Scholar]
- 56.Martinez-Vicente, M., Yim, L., Villarroya, M., Mellado, M., Perez-Paya, E., Bjork, G. R., and Armengod, M. E. (2005) J. Biol. Chem. 280 30660-30670 [DOI] [PubMed] [Google Scholar]
- 57.Zemojtel, T., Penzkofer, T., Dandekar, T., and Schultz, J. (2004) Trends Biochem. Sci. 29 224-226 [DOI] [PubMed] [Google Scholar]
- 58.Mishra, R., Gara, S. K., Mishra, S., and Prakash, B. (2005) Proteins Struct. Funct. Bioinf. 59 332-338 [DOI] [PubMed] [Google Scholar]
- 59.Scrima, A., and Wittinghofer, A. (2006) EMBO J. 25 2940-2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan, J. Y., and Wessling-Resnick, M. (1998) BioEssays 20 516-521 [DOI] [PubMed] [Google Scholar]
- 61.Zhang, B., Zhang, Y., Wang, Z., and Zheng, Y. (2000) J. Biol. Chem. 275 25299-25307 [DOI] [PubMed] [Google Scholar]
- 62.Shin, D. H., Lou, Y., Jancarik, J., Yokota, H., Kim, R., and Kim, S. H. (2004) Proc. Natl. Acad. Sci. U. S. A 101 13198-13203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cladiere, L., Hamze, K., Madec, E., Levdikov, V. M., Wilkinson, A. J., Holland, I. B., and Seror, S. J. (2006) Mol. Genet. Genomics 275 409-420 [DOI] [PubMed] [Google Scholar]
- 64.Frech, M., Darden, T. A., Pedersen, L. G., Foley, C. K., Charifson, P. S., Anderson, M. W., and Wittinghofer, A. (1994) Biochemistry 33 3237-3244 [DOI] [PubMed] [Google Scholar]
- 65.Rutthard, H., Banerjee, A., and Makinen, M. W. (2001) J. Biol. Chem. 276 18728-18733 [DOI] [PubMed] [Google Scholar]
- 66.Daigle, D. M., Rossi, L., Berghuis, A. M., Aravind, L., Koonin, E. V., and Brown, E. D. (2002) Biochemistry 41 11109-11117 [DOI] [PubMed] [Google Scholar]
- 67.Himeno, H., Hanawa-Suetsugu, K., Kimura, T., Takagi, K., Sugiyama, W., Shirata, S., Mikami, T., Odagiri, F., Osanai, Y., Watanabe, D., Goto, S., Kalachnyuk, L., Ushida, C., and Muto, A. (2004) Nucleic Acids Res. 32 5303-5309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bos, J. L., Rehmann, H., and Wittinghofer, A. (2007) Cell 129 865-877 [DOI] [PubMed] [Google Scholar]
- 69.Daigle, D. M., and Brown, E. D. (2004) J. Bacteriol. 186 1381-1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Levdikov, V. M., Blagova, E. V., Brannigan, J. A., Cladiere, L., Antson, A. A., Isupov, M. N., Seror, S. J., and Wilkinson, A. J. (2004) J. Mol. Biol. 340 767-782 [DOI] [PubMed] [Google Scholar]
- 71.Campbell, T. L., and Brown, E. D. (2008) J. Bacteriol. 190 2537-2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bassler, J., Kallas, M., and Hurt, E. (2006) J. Biol. Chem. 281 24737-24744 [DOI] [PubMed] [Google Scholar]
- 73.Flores-Perez, U., Sauret-Gueto, S., Gas, E., Jarvis, P., and Rodriguez-Concepcion, M. (2008) Plant Cell 20 1303-1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang, M., Chen, X., Militello, K., Hoffman, R., Fernandez, B., Baumann, C., and Gollnick, P. (1997) J. Mol. Biol. 270 696-710 [DOI] [PubMed] [Google Scholar]
- 75.Gechev, T. S., Van Breusegem, F., Stone, J. M., Denev, I., and Laloi, C. (2006) BioEssays 28 1091-1101 [DOI] [PubMed] [Google Scholar]
- 76.Zhao, M., Zhao, X., Wu, Y., and Zhang, L. (2007) J. Plant Physiol. 164 737-745 [DOI] [PubMed] [Google Scholar]
- 77.Wink, D. A., Miranda, K. M., Espey, M. G., Pluta, R. M., Hewett, S. J., Colton, C., Vitek, M., Feelisch, M., and Grisham, M. B. (2001) Antioxid. Redox Signal. 3 203-213 [DOI] [PubMed] [Google Scholar]
- 78.Kojima, H., Nakatsubo, N., Kikuchi, K., Kawahara, S., Kirino, Y., Nagoshi, H., Hirata, Y., and Nagano, T. (1998) Anal. Chem. 70 2446-2453 [DOI] [PubMed] [Google Scholar]
- 79.Zhao, M. G., Tian, Q. Y., and Zhang, W. H. (2007) Plant Physiol. 144 206-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zottini, M., Costa, A., De Michele, R., Ruzzene, M., Carimi, F., and Lo Schiavo, F. (2007) J. Exp. Bot. 58 1397-1405 [DOI] [PubMed] [Google Scholar]
- 81.Arnaud, N., Murgia, I., Boucherez, J., Briat, J. F., Cellier, F., and Gaymard, F. (2006) J. Biol. Chem. 281 23579-23588 [DOI] [PubMed] [Google Scholar]
- 82.Kolbert, Z., Bartha, B., and Erdei, L. (2008) J. Plant Physiol. 165 967-675 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.