

Abstract
Several bacterial solute transport mechanisms involve members of the periplasmic binding protein (PBP) superfamily that bind and deliver ligand to integral membrane transport proteins in the ATP-binding cassette, tripartite tricarboxylate transporter, or tripartite ATP-independent (TRAP) families. PBPs involved in ATP-binding cassette transport systems have been well characterized, but only a few PBPs involved in TRAP transport have been studied. We have measured the thermal stability, determined the oligomerization state by small angle x-ray scattering, and solved the x-ray crystal structure to 1.9 Å resolution of a TRAP-PBP (open reading frame tm0322) from the hyperthermophilic bacterium Thermotoga maritima (TM0322). The overall fold of TM0322 is similar to other TRAP transport related PBPs, although the structural similarity of backbone atoms (2.5-3.1 Å root mean square deviation) is unusually low for PBPs within the same group. Individual monomers within the tetrameric asymmetric unit of TM0322 exhibit high root mean square deviation (0.9 Å) to each other as a consequence of conformational heterogeneity in their binding pockets. The gel filtration elution profile and the small angle x-ray scattering analysis indicate that TM0322 assembles as dimers in solution that in turn assemble into a dimer of dimers in the crystallographic asymmetric unit. Tetramerization has been previously observed in another TRAP-PBP (the Rhodobacter sphaeroides α-keto acid-binding protein) where quaternary structure formation is postulated to be an important requisite for the transmembrane transport process.
Bacterial periplasmic binding proteins (PBPs)2 frequently are associated with one of three active transport systems: the ATP-binding cassette transport systems (1) that couple ATP hydrolysis to transport (2, 3) and the tripartite ATP-independent periplasmic (TRAP) (4, 5) and tripartite tricarboxylate transporter families (6, 7) that utilize the H+/M+ motive force to translocate ligands. Only a small number of TRAP transport systems have been characterized in detail (4, 8). TRAP transport systems, such as that from Rhodobacter capsulatus (4), are comprised of three proteins: a PBP, a small integral membrane protein, and a large integral membrane protein that shows sequence similarity to ion symporters. Arrangements of these components can also be fused into multi-domain polypeptides (9). Ligands that are known to be transported in TRAP systems, such as ascorbate (10), sialic acid (8), pyroglutamic acid (11), and C4-dicarboxylates (12) are usually not transported by ATP-binding cassette transporters.
PBPs form a protein superfamily characterized by two α/β domains with a ligand-binding site located at the domain interface (13). The two domains typically are connected by two to three β-strands that function as a hinge in ligand-mediated changes in the relative orientation of the two domains (14). The ligand is completely enveloped within the interdomain interface of the closed state. Three groups (I, II, and III) can be distinguished within the PBP superfamily categorized upon the topology of β-strands in each the two domains (15). Tam and Saier (13) originally identified TRAP-PBPs as members of the PBP superfamily based on sequence similarity. In the absence of a three-dimensional structure, it was not possible to further assign TRAP-PBPs to one of the three structural groups. The crystal structures of two (the sialic acid-binding protein from Haemophilus influenzae (SaBP) (16) and the α-keto acid-binding protein from Rhodobacter sphaeroides (αKaBP) (17)) TRAP-PBPs revealed they belong to the PBP group II. Unlike previously studied PBPs, the αKaBP was shown to be an obligate dimer that forms tetramers in solution (17).
Elucidation of the complete genomic sequence of the hyperthermophilic bacterium Thermotoga maritima has revealed that this organism is rich in metabolite uptake systems (18). To further understand this important aspect of microbial biochemistry, it is necessary to supplement sequence-based information with functional and structural studies. A survey of the genomic sequence of T. maritima (18) identified a large number of potential PBP open reading frames (ORFs) in metabolite transport operons. Among these, ORF tm0322 exhibits high amino acid and operon similarity to TRAP-associated PBPs but not to ATP-binding cassette transport-associated PBPs. Immediately adjacent to tm0322 are two ORFs (tm0323 and tm0324) that show significant sequence similarity to the R. capsulatus TRAP integral membrane transporters (4). This gene cluster is therefore postulated to correspond to a T. maritima TRAP transport system. Here we present the initial biochemical characterization, small angle x-ray scattering (SAXS) analysis, and x-ray crystal structure of the PBP component of this putative TRAP transport system.
EXPERIMENTAL PROCEDURES
Cloning, Overexpression, and Purification—The tm0322 gene was amplified from T. maritima genomic DNA by sticky end PCR (19) using the following primers: PO4-TATGTCAGCGGTATTTGGCGCGAAGTACACACTGAG, and TCAGCGGTATTTGGCGCGAAGTACACACTGAG for the 5′ half of the gene; PO4-AATTCTAATGGTGATGGTGATGGTGCCCAGACTCTCCCTTCACTTCCTTGATCAGCTGG, and CTAATGGTGATGGTGATGGTGCCCAGACTCTCCCTTCACTTCCTTGATCAGCTGG for the 3′ half of the gene. The resulting fragment, lacking the putative periplasmic signal sequence, was cloned into the NdeI/EcoRI sites of a pET21a (Novagen) plasmid for overexpression in Escherichia coli. A hexahistidine affinity tag was fused in-frame at the C terminus to facilitate purification by immobilized metal affinity chromatography (20) followed by gel filtration chromatography (Superdex S75 26/60). TM0322 elutes from the gel filtration column as multiple peaks, which based on calculated hydrodynamic radius are monomers and dimers (see Fig. 1). Protein concentration was determined spectrophotometrically using an extinction coefficient (ε = 52,000 m-1 cm-1) (21) calculated from the number of tryptophan and tyrosine residues in the protein sequence.
FIGURE 1.

Elution profiles of TM0322. A, gel filtration chromatogram of TM0322. Putative monomer and dimer peaks are indicated. B, overlaid native (top) and SDS-PAGE gels (bottom) of Superdex S75 fractions (dashed line indicates the division of the two gels); the bottom of lane 15 contains SDS-PAGE molecular mass standards.
Selenomethionine-substituted TM0322 was grown in the E. coli methionine auxotroph B834 (Novagen). A total of 100 ml of an overnight culture grown in TB was pelleted and resuspended in 4 liters of M9 minimal medium supplemented with 100 mg/liter selenomethionine. This was grown to an A600 of 0.7 at 37 °C and induced by the addition of isopropyl β-d-thiogalactopyranoside (final concentration, 1 mm), followed by further growth at 37 °C for 3-4 h. Protein was purified as described above for the native protein except that 5 mm dithiothreitol was added to all buffers.
SAXS Data Acquisition and Processing—SAXS data were collected at beam line X21 at the National Synchrotron Light Source (Brookhaven National Laboratory). The wavelength of the beam was 1.24 Å. Monomeric (see Fig. 1B, lane 7) and dimeric (see Fig. 1B, lane 14) fractions from the gel filtration column were concentrated to 3.0 and 1.7 mg/ml, respectively, in a 10 mm Tris, 300 mm NaCl, pH 7.8, buffer for SAXS analysis. Monomer, dimer, and buffer samples were exposed for 45 s at 10 °C in a quartz flow cell at a flow rate of 28 μl/min. SAXS data on a standard protein, hen egg white lysozyme (14.2 kDa; Acros Organics, Morris Plains, NJ), were collected over a concentration series in the same capillary. For the monomer and dimer, the recorded intensities of three independent runs/sample were averaged together after they were circularly averaged and scaled to obtain a relative scattering intensity (I) as a function of momentum transfer vector, q (q = [4πsinθ]/λ), after subtraction of buffer scattering contributions. I0 values, determined for the monomeric and dimeric fractions relative to that of lysozyme (22), were as expected (see Table 1).
TABLE 1.
Summary of P(r) analysis using GNOM
|
Sample
|
Molecular mass
|
Io/c
|
Protein
|
Rg
|
Dmax
|
|
|---|---|---|---|---|---|---|
| Observed | Expected | |||||
| kDa | mg/ml | Å | Å | |||
| Lysozyme | 14.2 | 35.0 | 1.0 | 14.6 ± 0.06 | 45 | |
| Monomeric TM0322 | 37.2 | 90.1 | 91.7 | 3.0 | 22.10 ± 0.03 | 80 |
| Dimeric TM0322 | 74.4 | 188.5 | 183.4 | 1.7 | 31.47 ± 0.45 | 115 |
SAXS Data Analysis—For monodisperse globular proteins, a
plot of ln(I(q)) versus q2, where
q·Rg ≤ 1, was linear and was fit to
Equation 1
to provide an estimation of the scattering particle radius of gyration
(Rg) and forward or zero angle scatter
(I0) from the slope
(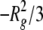 ) and the y-intercept
I0 (Guinier analysis
(23)). All of the scattering
data were analyzed in this manner using the Primus software package
(24). Indirect Fourier
transformation of the scattering data over the measured q range gives
a pairwise distribution function of interatomic vectors,
P(r). The inverse Fourier transform of
I(q) yields P(r), which represents the
distribution of vector lengths connecting small volume elements within the
entire volume of the scattering particle (Equation
2).
) and the y-intercept
I0 (Guinier analysis
(23)). All of the scattering
data were analyzed in this manner using the Primus software package
(24). Indirect Fourier
transformation of the scattering data over the measured q range gives
a pairwise distribution function of interatomic vectors,
P(r). The inverse Fourier transform of
I(q) yields P(r), which represents the
distribution of vector lengths connecting small volume elements within the
entire volume of the scattering particle (Equation
2).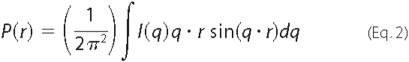
P(r) = 0 at r = 0 Å and approaches 0 at the maximum linear dimension of the particle, Dmax. The radius of gyration (Rg) and forward scattering (I0) were calculated from the second moment and the start of P(r) (25), respectively, where Rg is the root mean square of all elemental volumes from the center of mass of the particle, weighted by their scattering densities, and I0 is directly proportional to the molar particle concentration multiplied by the square of the scattering particle molecular weight for particles with the same mean scattering density (see Table 1) (25). The GNOM45 software package (26) was used for all P(r) analyses.
Ab Initio Model Construction—Three-dimensional shapes of the monomeric and dimeric proteins were constructed from their respective SAXS data using the GASBOR22IQW program (q range input for each analysis was from 0.01 to 0.04 Å-1) (27), by calculating the distribution of linearly connected 1.9 Å spheres (the number of spheres is set to the number of residues in the protein) that best fit the scattering data. Each calculation was repeated five times with different random starting points for the Monte Carlo optimization algorithm; no predefined shape or symmetry constraints were used. From these runs, the predicted structure with the lowest deviation of the calculated scattering profile from experimental data was used for interpretation. To compare the SAXS-based models of monomeric and dimeric TM0322, with the atomic structure obtained from x-ray crystallographic data, the structures were superimposed using the SUPCOMB13 (28) program.
Thermal Denaturation—Thermal denaturations were determined by measuring the circular dichroism signal at 222 nm (1-cm path length) as a function of temperature, using an Aviv model 202 circular dichroism spectrophotometer with 2 μm protein (12.5 mm sodium phosphate, pH 7.7, 5 mm NaCl) and guanidinium chloride (GdmCl) at various concentrations (4.0-6.5 m). Protein samples were equilibrated for 15 min prior to collecting data. Each measurement includes a 3-s averaging time for data collection and a 60-s equilibration period at each temperature. The data were fit to a two-state model to determine the appTm values (29, 30). The appTm values in the absence of denaturant were determined by linear extrapolation (31). A total of 10 ligands (succinate, fumarate, maleate, ectoine, sialic acid, xylulose, α-ketoglutarate, tartrate, glutamate, and oxalate) were screened by monitoring ligand-induced changes in the thermal melting transition; none of these ligands induced changes in the appTm value.
Crystallization and X-ray Data Collection—TM0322 was crystallized by hanging drop vapor diffusion in drops containing 2 μl of 10 mg/ml protein solution mixed with 2 μl of 1.9-2.3 m ammonium sulfate, 0.1 m phosphate/citrate buffer, pH 4.2, and equilibrated against 900 μl of 1.9-2.3 m ammonium sulfate, 0.1 m phosphate/citrate buffer, pH 4.2. Large selenomethionine-substituted crystals in the P6522 space group (a = 119 Å, b = 119 Å, c = 427.3 Å) typically grew within 3-4 days at 17 °C. The crystals were transferred stepwise to 2.2 m ammonium sulfate, 0.1 m phosphate/citrate buffer, pH 4.2, incrementally supplemented with glycerol in 5% steps up to 25% final concentration for cryoprotection. The crystals were mounted in a nylon loop and flash frozen in liquid nitrogen. All of the data were collected at 100 K at the SER-CAT 22ID and 22BM beam lines at the Advanced Photon Source. The diffraction data were scaled and indexed using XDS (32).
Structure Determination Methods—The structure was determined by the multiwavelength anomalous diffraction method (33) using a three-wavelength dataset collected for a selenomethionine TM0322 crystal at the selenium K/α absorption edge. The SHELXD (34) program was used to identify 36 of a possible 44 selenium sites, corresponding to four monomers in the asymmetric unit. The top substructure solution from SHELXD yielded a pseudo-free correlation coefficient of 74.35% and contrast and connectivity parameters of 0.831 and 0.944, respectively. Initial phases calculated after density modification in SHELXE produced an interpretable electron density map. The use of noncrystallographic symmetry aided further phase improvement and phase extension in RESOLVE (35). The improved phases were input into ARP/wARP (36), which was able to trace 1214 of the total 1316 residues in the asymmetric unit. Further manual model building was carried out in O (37) or COOT (38), and the model was refined in REFMAC5 (39). The final model includes four monomers and 529 water molecules. The model exhibits good stereochemistry as determined by PROCHECK (40); the final refinement statistics are listed in Table 2. The atomic coordinates and diffraction data for the selenomethionine TM0322 structure were deposited in the Protein Data Bank with accession code 2HPG.
TABLE 2.
Data collection and refinement statistics
| λ1 | λ2 | λ3 | |
|---|---|---|---|
| Data collection | |||
| Detector type | Mar 300 | Mar 300 | Mar 300 |
| Wavelength (Å) | 0.97167 | 0.97934 | 0.97920 |
| Resolution (Å) | 30.0-1.9 | 30.0-1.9 | 30.0-1.9 |
| Measured reflections | 1365842 | 1406722 | 1365936 |
| Unique reflections | 142555 | 142631 | 142987 |
| Mean I/σ(I)a | 14.7 (4.3) | 13.0 (4.1) | 14.1 (3.7) |
| Completeness (%)a | 100 (100) | 100 (100) | 100 (100) |
| Rsym (%)a | 9.4 (40.5) | 10.2 (41.5) | 10.5 (47.2) |
| Redundancya | 9.6 (8.4) | 9.9 (8.6) | 9.6 (8.3) |
| Phasing (20-1.9) Å | |||
| SHELXE phasing statistics | |||
| Contrast/connectivity | 0.831/0.944 | ||
| Pseudo-free CC (%) | 74.35 | ||
| Refinement | |||
| Resolution (Å) | 30-1.9 | ||
| Number of reflections (working set/test set) | 135259/7152 | ||
| Rcryst (%) | 21.3 (31.4) | ||
| Rfree (%)b | 23.9 (35.3) | ||
| Number of atoms | |||
| Protein | 10351 | ||
| Water | 529 | ||
| Root mean square deviation | |||
| Bond lengths (Å) | 0.011 | ||
| Bond angles (°) | 1.25 | ||
| Average B factor (Å2) | |||
| Main chain | 27.9 | ||
| Side chain | 29.1 | ||
| Solvent | 32.0 | ||
| Protein geometry | |||
| Ramachandran outliers (%) | 0.24 | ||
| Ramachandran favored (%) | 97.8 | ||
| Rotamer outliers (%) | 2.6 |
The numbers in parentheses represent values in the highest resolution shell.
Rfree is the R factor based on 5% of the data excluded from refinement.
RESULTS
Cloning, Expression, and Protein Purification—ORF tm0322 in the T. maritima genome sequence (18) was postulated to encode a TRAP-PBP of unknown function based on its sequence similarity to the known R. capsulatus C4-dicarboxylate-binding protein and its position within a putative operon containing ORFs homologous to other TRAP transporters. The DNA for ORF tm0322, lacking a putative periplasmic signal sequence (41) (residues 1-16), was amplified from T. maritima genomic DNA by the polymerase chain reaction. The resulting DNA fragment was cloned into a pET21a vector in-frame with a C-terminal hexahistidine tag. Overexpression of native (in BL21 cells) and selenomethionine-substituted protein (in B834 cells) yielded ∼70 mg of pure protein/liter of culture.
C-terminally hexahistidine-tagged TM0322 was purified by immobilized metal affinity chromatography (20) followed by gel filtration chromatography (Superdex S75 26/60). TM0322 elutes from the gel filtration column in multiple peaks that are assigned to monomers, dimers, and higher order assemblies based on the calculated hydrodynamic radius (Fig. 1). Quaternary assemblies exceeding dimers cannot be resolved because they elute in the void volume of the gel filtration column (>75 kDa). Comparison of native and denaturing PAGE analysis of the eluate indicates there are at least three noncovalent populations of protein assemblies (Fig. 1). Both the monomeric (lanes 11-15) and dimeric (lanes 7-10) fractions are homogeneous in the native PAGE gel; mobility shifts in the native gel correlate with the peaks observed in the gel filtration chromatogram (Fig. 1).
Thermal Stability—The stability of TM0322 was determined by thermal denaturation using circular dichroism (30). In the absence of GdmCl, no significant change in the circular dichroism signal could be observed as a function of temperature (data not shown). All of the subsequent measurements were carried out in the presence of GdmCl (Fig. 2). Thermal denaturations fit a two-state model (29, 30). The apo-protein has an apparent thermal transition midpoint (appTm) of 130 °C in the absence of GdmCl, as determined by linear extrapolation of thermal denaturations measured at different GdmCl concentrations (Fig. 2) (31).
FIGURE 2.

Thermal stability of TM0322. A, thermal denaturation of TM0322 at various guanidinium chloride concentrations (▪, 4.0 m; •, 4.5 m; ▴, 5.0 m; ▾, 5.5 m; ♦, 6.0 m; +, 6.5 m). The solid lines in A are fit to a two-state model that accounts for the native and denatured baseline slopes (29, 30). B, extrapolated appTm values of TM0322 obtained from the series of thermal melting curves at different concentrations of GdmCl. The solid line represents a linear fit to the observations.
Small Angle X-ray Scattering—The putative monomeric (Fig. 1B, lane 7) and dimeric (Fig. 1B, lane 14) fractions were concentrated and analyzed by small angle x-ray scattering (Fig. 3). Their Guinier plots are linear over a q range of 0.012-0.058 Å-1 for the monomeric and 0.012-0.040 Å-1 for the dimeric fractions, which suggests that both samples are monodisperse and furthermore indicates Rg values of 22.1 ± 0.03 and 31.5 ± 0.45 Å for the monomeric and dimeric fractions, respectively (Fig. 3 and Table 1). The P(r) data for the monomeric protein indicates particles with a Dmax of ∼80 Å, whereas the P(r) for the dimeric protein has particles with a Dmax of ∼115 Å (Fig. 3 and Table 1).
FIGURE 3.

SAXS intensity data and P(r) distribution for monomeric and dimeric fractions. A, SAXS intensity I(q) data for monomeric (black squares) and dimeric TM0322 (gray squares). Inset, Guinier region for monomer (black squares) and dimer (gray squares). B, pairwise vector length distribution curve (Fourier transform of I(q) data) for monomeric (black squares) and dimeric (gray squares) protein.
The GASBOR22IQW (27) program was used for ab initio shape construction based on the best fit of a modeled set of spheres to the I(q) scattering data. The models of monomeric and dimeric TM0322 used 342 and 684 1.9 Å radius spheres (corresponding to the expected number of residues in each of the assemblies) and 234 and 611 water molecules, respectively, based on a Fibonacci grid of 12 and 14, respectively (27); no shape or symmetry constraints were imposed. The resulting models are shown in Fig. 4. The monomer model has an overall shape (∼65 Å ×∼40 Å ×∼20 Å) with dimensions consistent with other TRAP-PBPs (11, 16, 17) (Fig. 4). The dimer model has an overall shape of a three-dimensional triangle (∼70 Å × ∼100 Å ×∼110 Å ×∼25 Å) (Fig. 4). It shares little resemblance to αKaBP, which is an obligate dimer. The model can be bisected into two fragments equal to those of the monomeric SAXS model, suggesting that TM0322 assembles into a dimer arranged differently from αKaBP (Fig. 4).
FIGURE 4.

Ab initio models derived from SAXS. A, orthogonal views of the monomer SAXS model. B, orthogonal views of the dimer SAXS model. The dimer can be divided into two fragments (dashed line) each with dimensions equal to that of the SAXS monomer. The amino acid spheres and water molecules (gray) are shown in a surface representation.
Structure Determination by X-ray Diffraction—Both monomeric and dimeric fractions of TM0322 were used in crystallization experiments; no crystals were obtained for the latter. The crystal structure of TM0322 was solved by a three-wave-length anomalous diffraction experiment (33) to a resolution of 1.90 Å using crystals of selenomethionine-substituted protein (Fig. 5). Experimental phases were determined with SHELX (34) using 36 of 44 selenium atoms from four molecules in the crystallographic asymmetric unit. The final model has an R of 21.3% and an Rfree of 23.9%. Data collection, phasing, refinement, and stereochemistry statistics are summarized in Table 2. The program ARP/wARP (36) was able to automatically trace 1214 of the total 1316 residues in the asymmetric unit. Further manual model building was carried out in O (37) or COOT (38), and the model was refined in REFMAC5 (39).
FIGURE 5.

Overall structure of TM0322. A, stereo diagram of TM0322 colored by secondary structure (green, β-strands; red, α-helix; blue, 310-helix;). Secondary structure elements that connect the two domains are indicated. B, close-up view of the binding pocket region overlaid with Fo - Fc electron density (blue) contoured at 2.5 σ. The unknown, putative α-keto acid, ligand is shown in gray ball and stick representation. Potential hydrogen bonds (black dashed line) are indicated.
Overall Structure—TM0322 contains four monomers (residues 17-336; numbering according to NCBI NP_228134) in the crystallographic asymmetric unit and 529 water molecules. Electron density for an unknown ligand is present in the binding pocket of molecule A (see below). In each monomer residues 18-20 and the hexahistidine tag were disordered and thus not included in the model; in molecule C residues 77-79 were omitted because of disorder. 97.8% of residues have conformations that are in the favored Ramachandran regions, whereas 99.8% are in conformations in the allowed regions. The three Ramachandran outliers (Leu67 and Glu70 from molecule D, and Ala33 from molecule D) are found in the disordered loops that construct the binding pocket region.
TM0322 adopts the canonical two-domain α/β fold characteristic of the PBP superfamily. Based on the ordering of its β-strands, we assign it to be a group II PBP (Fig. 5) (15). Unlike other group II PBPs, TM0322 has several regions of 310 helix, the largest being a seven-residue helix (Lys97-Asn103) located adjacent to the interdomain hinge. 310 helices have previously been observed only in group III PBPs (42). Both the N- and C-terminal domains are α/β/α sandwiches with significantly twisted β-sheet cores. The central β-sheets of the two domains are similar in strand order; the C-terminal domain has an additional β-strand. The β-strands are connected by either α- or 310-helices (Fig. 5). The two domains are joined by a hinge consisting of two β-strands (β5 and β10) and an α-helix (α11) (Fig. 5). Besides forming part of the hinge, α11 is a major structural component in both the N- and C-terminal domains and spans the entire length protein molecule (almost 55 Å). An equivalent, long α-helix has been observed in other TRAP-PBPs (11, 16, 17) and in the group III PBPs (42, 43). With the exception of TRAP-PBPs, no other group II PBPs contain an α-helix with a similar structural role.
Each of the four molecules in the asymmetric unit adopts a similar overall conformation with respect to the degree of closing and twisting of their two domains. Nevertheless, the monomers have a relatively high root mean square deviation (0.9 Å average for backbone atoms) with respect to each other. The observed differences are confined mainly to the loops that form the ligand-binding pocket of the N-terminal domain (residues 30-37 and 61-70) and the α-helix (α2) and β-strand (β2) that respectively precede and follow the loop formed by residues 61-70 (Fig. 6). These regions are buried in the interior of the tetramer and exhibit high B factors; the presence of ligand in molecule A has little effect on the temperature factors. The observed differences in conformation in each of the four molecules suggest that the binding pocket region of TM0322 is flexible (Fig. 6).
FIGURE 6.

Conformational heterogeneity of the TM0322 binding pocket region. A, ribbon representation colored by chain of the structural alignment of the four monomers found in the crystallographic asymmetric unit of TM0322. The region exhibiting large conformational heterogeneity is highlighted with a dashed line. B, surface representation of the crystallographic asymmetric unit of TM0322. The two noncrystallographic dimers A/D and B/C are colored in magenta and cyan, respectively. The two noncrystallographic symmetry axes that relate the symmetry of the dimer of dimers are indicated. Axis 1, A/B and C/D; axis 2, A/D and B/C. A cut away from the interior of the tetramer highlighting the region exhibiting large conformational heterogeneity is shown.
The crystallographic asymmetric unit of TM0322 contains two noncrystallographic symmetry axes that divide the unit cell into dimers (Fig. 6). The interface within the A/D and B/C dimers buries ∼2,500 Å2 of solvent-accessible surface area where within the A/B and C/D dimers ∼1,200 Å2 is buried (44) This suggests that dimers A/D and B/C are more stable and likely to correspond to the dimers observed in solution.
Comparison of SAXS and Crystallographic Models—The program SUPCOMB13 was used to superimpose the SAXS monomer and dimer models onto the TM0322 crystallographic x-ray structure (Fig. 7). The overlay of an individual monomer from the x-ray structure correlates well with the SAXS monomer envelope shape (final SUPCOMB distance of 1.8 Å using all protein atoms in crystal structure). The model for the SAXS dimer correlates better with the crystallographic A/D or B/C dimers (final SUPCOMB distance of 2.5 Å, using all protein atoms in crystal structure) rather than the A/B or C/D dimers (final SUPCOMB distance of 2.8 Å, using all protein atoms in crystal structure). This suggests that the solution dimer is similar to that observed in the crystal. Furthermore, the SAXS dimer can be assembled into a tetramer that recapitulates the crystallographic asymmetric unit, by fitting together the concave interfaces of the dimers (Fig. 7).
FIGURE 7.

Superposition of SAXS and x-ray crystallographic models. A, surface representation of the monomeric TM0322 SAXS model overlaid with a ribbon model of the monomeric x-ray crystal structure. B, surface representation of the dimeric TM0322 SAXS model overlaid with a ribbon model of the dimeric (molecules A/D) x-ray crystal structure. C, the surface complementarity of the SAXS dimer model and the crystal dimer recapitulates the crystallographic tetramer. Top, crystallographic dimer of molecules B/C; middle, crystallographic dimer of molecules A/D superimposed with the SAXS dimer model; bottom, SAXS dimer model superimposed on the tetrameric crystal structure. SAXS water molecules are represented as gray spheres.
Comparison with Other TRAP-PBP Structures—Currently, the Protein Data Bank contains the x-ray crystal structures of four TRAP-associated PBPs (45): the monomeric SaBP (16), the two pyroglutamic acid-binding proteins from Bordetella pertussis (11), and dimeric αKaBP (17). PBPs from the same group have high structural similarity even though the amino acid similarity may be low. The amino acid sequence (∼50% similarity of TRAP-PBPs in Protein Data Bank), overall topology, and ordering of secondary structure elements of TM0322 are similar with other TRAP PBPs; TM0322, however, has low structural similarity with these group members (2.5-3.1 Å root mean square deviation of backbone atoms), using either the apo- or ligand-bound forms of the other TRAP-PBPs. Molecule A of TM0322 does share the most structural similarity (2.5 Å root mean square deviation of backbone atoms) to the ligand-bound form of the SaBP, where ligand is also present in one of the four molecules in the asymmetric unit (16).
Ligand-Binding Site—C4-dicarboxylates (46), ectoine (47), α-ketoglutarate (10), and sialic acid (16) are known to be transported by TRAP transport systems. Binding of these and five other ligands (see “Experimental Procedures”) to TM0322 was examined by monitoring changes in appTm in their presence (48). None of the ligands tested were found to bind to TM0322 by this criterion. Consequently no exogenous ligands were added in the crystallization trials. Nevertheless, we observed electron density in the region where the ligand is expected to bind based on similarity with other TRAP-PBPs. This electron density was present in only one (molecule A) of the four monomers in the asymmetric unit. We attribute the observed density to a serendipitously bound ligand present in E. coli or the fermentation broth that persists through purification (Fig. 5). Occupancy of only one in four ligand-binding sites has also been observed in SaBP (16).
Based on the pattern of hydrogen bond donors and acceptors of the amino acid side chains surrounding the observed density, we conclude that the ligand is consistent with an α-keto acid with an unknown substituent group. We postulate that the unknown substituent is a polar substituted seven-membered nonplanar cyclic structure preceded by a smaller polar R group (Fig. 5). The putative α-keto acid group is surrounded by main chain atoms (Gly180 and Val198) that can potentially form two hydrogen bonds with the oxygens of the carboxylate group. An additional hydrogen bond can potentially be formed by a specifically bound water molecule (Wat331) that is coordinated by three hydrogen bonds with main chain polar atoms (Fig. 5). Adjacent to the putative α-keto acid is electron density that is postulated to be a seven-membered nonplanar ring structure (Fig. 5). The identity of polar amino acids surrounding the electron density of the ring indicates that the ring (or its R groups) is polar; only one nonpolar amino acid (Trp184) forms potential van der Waals interactions with the ring. Four amino acids (Asp88, Gln154, Arg157, and Asn244) form an annulus of polar hydrogen bond donors and acceptors, which, based on distance from the ring (3.2-4.5 Å), may potentially form hydrogen bonds with the ring and its substituents. Next to the putative ring there is additional electron density that could account for an unknown polar R group coordinated by the guanidino nitrogens of Arg157 and Arg177. Because the identity of the ligand could not be conclusively determined, it was not included in the final model of TM0322.
DISCUSSION
We have carried out initial biochemical characterization, SAXS analysis, and x-ray crystal structure determination of ORF TM0322, the putative PBP component of an uncharacterized TRAP transport system from T. maritima. TM0322 adopts the two-domain fold that is characteristic of PBPs. It shares the highest structural similarity with PBPs that are associated with TRAP transport, of which TM0322 is the only thermophilic member whose structure has been determined. Although electron density is observed for a serendipitously bound ligand in one of the monomers in the asymmetric unit, we have not been able to identify this ligand. This ligand originates from the cytoplasm of E. coli or the growth medium (yeast extract) and cannot be guaranteed to correspond to the natural cognate ligand in T. maritima. The density and the putative hydrogen-bonding pattern of the surrounding residues are consistent with an α-keto acid with a large R group.
The gel filtration elution profile and the SAXS analysis indicate that TM0322 assembles into a dimer in solution. The dimer shape differs from that of the R. sphaeroides α-keto acid-binding protein, which forms stable obligate dimers through a helix swapping mechanism (17). The mechanism of dimer assembly in TM0322 is unknown. The dimers are stable and not in obvious equilibrium with the monomeric form (the monomeric gel filtration fraction does not form dimers over time). Also absent is a clear conformational change (such as helix swapping). This suggests that an unknown mechanism shifts the equilibrium toward dimer formation. We postulate that this is accomplished via a ligand-induced conformational change, in this case the serendipitously bound unknown ligand.
It has been suggested elsewhere that quaternary structure formation is a prerequisite for the ligand co-transport process in PBP-mediated TRAP transport; oligomerization is postulated to create a channel that forms a solvent-excluded ligand-binding pocket important for subsequent transfer to transmembrane H+/M+ transporters (17). Based on the crystal structure of TM0322, the SAXS model, and the elution profile, we postulate that the tetrameric assembly observed in the crystallographic asymmetric unit could correspond to such a complex. This dimer of dimers results in the positioning of each of the four binding pockets into a solvent-excluded chamber in the center of the tetramer (Fig. 6). The large tetrameric complex may also serve as a plug to prevent ligand escape (17, 49) and to prevent uncoupled H+/M+ transport. Future work will address the role of TM0322 quaternary assembly in its interactions with the membrane transport machinery to gain a better understanding of PBP-mediated transport in the TRAP system.
Acknowledgments
We thank G. Shirman for protein expression and purification and the laboratories of G. Thomas and D. Kelly for release of their H. influenzae SaBP coordinates and manuscript prior to publication.
This work was supported, in whole or in part, by National Institutes of Health Grant 5 DP1 OD000122-02 (to H. W. H.). This work was also supported by Homeland Security Advanced Research Projects Agency Grant W81XWH-05-C-0161 (to H. W. H.), a National Institutes of Health sponsored biological chemistry training grant (to M. J. C.), Bonnie Cone Fellowship National Science Foundation ADVANCE Grant SBE-0548401 (to J. K. K.), and United States Department of Energy, Office of Science, Office of Basic Energy Sciences Contract DE-AC02-98CH10886. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: PBP, periplasmic binding protein; TRAP, tripartite ATP-independent; SAXS, small angle x-ray scattering; SaBP, sialic acid-binding protein from H. influenzae; αKaBP, α-keto acid-binding protein from R. sphaeroides; ORF, open reading frame; GdmCl, guanidinium chloride.
References
- 1.Nikaido, H. (1994) FEBS Lett. 346 55-58 [DOI] [PubMed] [Google Scholar]
- 2.Davidson, A. L., Shuman, H. A., and Nikaido, H. (1992) Proc. Natl. Acad. Sci. U. S. A. 89 2360-2364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hor, L. I., and Shuman, H. A. (1993) J. Mol. Biol. 233 659-670 [DOI] [PubMed] [Google Scholar]
- 4.Forward, J. A., Behrendt, M. C., Wyborn, N. R., Cross, R., and Kelly, D. J. (1997) J. Bacteriol. 179 5482-5493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rabus, R., Jack, D. L., Kelly, D. J., and Saier, M. H., Jr. (1999) Microbiology 145 3431-3445 [DOI] [PubMed] [Google Scholar]
- 6.Widenhorn, K. A., Boos, W., Somers, J. M., and Kay, W. W. (1988) J. Bacteriol. 170 883-888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winnen, B., Hvorup, R. N., and Saier, M. H., Jr. (2003) Res. Microbiol. 154 457-465 [DOI] [PubMed] [Google Scholar]
- 8.Severi, E., Randle, G., Kivlin, P., Whitfield, K., Young, R., Moxon, R., Kelly, D., Hood, D., and Thomas, G. H. (2005) Mol. Microbiol. 58 1173-1185 [DOI] [PubMed] [Google Scholar]
- 9.Kelly, D. J., and Thomas, G. H. (2001) FEMS Microbiol Rev. 25 405-424 [DOI] [PubMed] [Google Scholar]
- 10.Thomas, G. H., Southworth, T., Leon-Kempis, M. R., Leech, A., and Kelly, D. J. (2006) Microbiology 152 187-198 [DOI] [PubMed] [Google Scholar]
- 11.Rucktooa, P., Antoine, R., Herrou, J., Huvent, I., Locht, C., Jacob-Dubuisson, F., Villeret, V., and Bompard, C. (2007) J. Mol. Biol. 370 93-106 [DOI] [PubMed] [Google Scholar]
- 12.Walmsley, A. R., Shaw, J. G., and Kelly, D. J. (1992) J. Biol. Chem. 267 8064-8072 [PubMed] [Google Scholar]
- 13.Tam, R., and Saier, M. H., Jr. (1993) Microbiol. Rev. 57 320-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjorkman, A. J., and Mowbray, S. L. (1998) J. Mol. Biol. 279 651-664 [DOI] [PubMed] [Google Scholar]
- 15.Fukami-Kobayashi, K., Tateno, Y., and Nishikawa, K. (1999) J. Mol. Biol. 286 279-290 [DOI] [PubMed] [Google Scholar]
- 16.Muller, A., Severi, E., Mulligan, C., Watts, A. G., Kelly, D. J., Wilson, K. S., Wilkinson, A. J., and Thomas, G. H. (2006) J. Biol. Chem. 281 22212-22222 [DOI] [PubMed] [Google Scholar]
- 17.Gonin, S., Arnoux, P., Pierru, B., Lavergne, J., Alonso, B., Sabaty, M., and Pignol, D. (2007) BMC Struct. Biol. 7 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nelson, K. E., Clayton, R. A., Gill, S. R., Gwinn, M. L., Dodson, R. J., Haft, D. H., Hickey, E. K., Peterson, J. D., Nelson, W. C., Ketchum, K. A., McDonald, L., Utterback, T. R., Malek, J. A., Linher, K. D., Garrett, M. M., Stewart, A. M., Cotton, M. D., Pratt, M. S., Phillips, C. A., Richardson, D., Heidelberg, J., Sutton, G. G., Fleischmann, R. D., Eisen, J. A., White, O., Salzberg, S. L., Smith, H. O., Venter, J. C., and Fraser, C. M. (1999) Nature 399 323-329 [DOI] [PubMed] [Google Scholar]
- 19.Zeng, G. (1998) BioTechniques 25 206-208 [DOI] [PubMed] [Google Scholar]
- 20.Yip, T. T., and Hutchens, T. W. (2004) Methods Mol. Biol. 244 179-190 [DOI] [PubMed] [Google Scholar]
- 21.Gill, S. C., and von Hippel, P. H. (1989) Anal. Biochem. 182 319-326 [DOI] [PubMed] [Google Scholar]
- 22.Krigbaum, W. R., and Kugler, F. R. (1970) Biochemistry 9 1216-1223 [DOI] [PubMed] [Google Scholar]
- 23.Glatter, O., and Kratky, O. (1982) Small Angle X-ray Scattering, pp. 18-37, Acdemic Press, New York
- 24.Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003) J. Appl. Crystallogr. 36 1277-1282 [Google Scholar]
- 25.Ashish, Juncadella, I. J., Garg, R., Boone, C. D., Anguita, J., and Krueger, J. K. (2008) J. Biol. Chem. 283 2761-2772 [DOI] [PubMed] [Google Scholar]
- 26.Svergun, D. I. (1992) J. Appl. Crystallogr. 25 495-503 [Google Scholar]
- 27.Svergun, D. I., Petoukhov, M. V., and Koch, M. H. (2001) Biophys. J. 80 2946-2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kozin, M. B., and Svergun, D. I. (2000) J. Appl. Crystallogr. 34 33-41 [Google Scholar]
- 29.Schellman, J. A. (1987) Annu. Rev. Biophys. Biophys. Chem. 16 115-137 [DOI] [PubMed] [Google Scholar]
- 30.Cohen, D. S., and Pielak, G. J. (1994) Protein Sci. 3 1253-1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cuneo, M. J., Changela, A., Warren, J. J., Beese, L. S., and Hellinga, H. W. (2006) J. Mol. Biol. 362 259-270 [DOI] [PubMed] [Google Scholar]
- 32.Kabsch, W. (1993) J. Appl. Crystallogr. 26 795-800 [Google Scholar]
- 33.Hendrickson, W. A., Smith, J. L., and Sheriff, S. (1985) Methods Enzymol. 115 41-55 [DOI] [PubMed] [Google Scholar]
- 34.Schneider, T. R., and Sheldrick, G. M. (2002) Acta Crystallogr. Sect. D Biol. Crystallogr. 58 1772-1779 [DOI] [PubMed] [Google Scholar]
- 35.Terwilliger, T. C. (2000) Acta Crystallogr. Sect. D Biol. Crystallogr. 56 965-972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perrakis, A., Harkiolaki, M., Wilson, K. S., and Lamzin, V. S. (2001) Acta Crystallogr. Sect. D Biol. Crystallogr. 57 1445-1450 [DOI] [PubMed] [Google Scholar]
- 37.Jones, T. A., Zou, J. Y., Cowan, S. W., and Kjeldgaard. (1991) Acta Crystallogr A 47 110-119 [DOI] [PubMed] [Google Scholar]
- 38.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 39.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 40.Laskowski, R. A., MacArthur, M.W., Moss, D.S., Thornton, J.M. (1993) J. Appl. Crystallogr. 26 283-291 [Google Scholar]
- 41.Nakai, K., and Horton, P. (1999) Trends Biochem. Sci 24 34-36 [DOI] [PubMed] [Google Scholar]
- 42.Karpowich, N. K., Huang, H. H., Smith, P. C., and Hunt, J. F. (2003) J. Biol. Chem. 278 8429-8434 [DOI] [PubMed] [Google Scholar]
- 43.Clarke, T. E., Braun, V., Winkelmann, G., Tari, L. W., and Vogel, H. J. (2002) J. Biol. Chem. 277 13966-13972 [DOI] [PubMed] [Google Scholar]
- 44.Krissinel, E., and Henrick, K. (2007) J. Mol. Biol. 372 774-797 [DOI] [PubMed] [Google Scholar]
- 45.Holm, L., and Sander, C. (1993) J. Mol. Biol. 233 123-138 [DOI] [PubMed] [Google Scholar]
- 46.Shaw, J. G., Hamblin, M. J., and Kelly, D. J. (1991) Mol Microbiol 5 3055-3062 [DOI] [PubMed] [Google Scholar]
- 47.Tetsch, L., and Kunte, H. J. (2002) FEMS Microbiol Lett 211 213-218 [DOI] [PubMed] [Google Scholar]
- 48.Matulis, D., Kranz, J. K., Salemme, F. R., and Todd, M. J. (2005) Biochemistry 44 5258-5266 [DOI] [PubMed] [Google Scholar]
- 49.Chen, J., Sharma, S., Quiocho, F. A., and Davidson, A. L. (2001) Proc. Natl. Acad. Sci. U. S. A. 98 1525-1530 [DOI] [PMC free article] [PubMed] [Google Scholar]