

Abstract
In addition to repressing ERBB2 promoter function, histone deacetylase (HDAC) inhibitors induce accelerated decay of mature ERBB2 transcripts; the mechanism mediating this transcript destabilization is unknown but depends on the 3′ untranslated region (UTR) of ERBB2 mRNA. Using ERBB2 overexpressing human breast cancer cells (SKBR3), the mRNA stability factor HuR was shown to support ERBB2 transcript integrity, bind and endogenously associate with a conserved U-rich element within the ERBB2 transcript 3′ UTR, co-immunoprecipitate with RNA-associated HDAC activity, and co-localize with HDAC6. HDAC6 also co-immunoprecipitates with HuR in an RNA-dependent manner; and within 6 h exposure to a pan-HDAC inhibitor dose that does not significantly alter cytosolic HuR levels or HuR binding to ERBB2 mRNA, cellular ERBB2 transcript levels decline while remaining physically associated with HDAC6. Knockdown of HDAC6 protein by siRNA partially suppressed the ERBB2 transcript decay induced by either pan-HDAC or HDAC6-selective enzymatic inhibitors. Three novel hydroxamates, ST71, ST17 and ST80, were synthesized and shown to inhibit HDAC6 with 14 to 31-fold greater selectivity over their binding and inhibition of HDAC1. Unlike more potent pan-HDAC inhibitors, these HDAC6-selective inhibitors produced dose-dependent growth arrest of ERBB2 overexpressing breast cancer cells by accelerating decay of mature ERBB2 mRNA without repressing ERBB2 promoter function. In sum, these findings point to the therapeutic potential of HuR and HDAC6-selective inhibitors, contrasting ERBB2 stability effects induced by HDAC6 enzymatic inhibition and HDAC6 protein knockdown, and demonstrate that ERBB2 transcript stability mechanisms include exploitable targets for development of novel anticancer therapies.
Keywords: ERBB2 mRNA, HDAC6, HuR, HDAC inhibitors, breast cancer
Introduction
Although the overexpressed receptor tyrosine kinase encoded by the amplified ERBB2 oncogene is a well validated therapeutic target, there remains a clinical need for newer anti-ERBB2 therapeutic strategies. Following a high-throughput screen of cell permeable drug-like small molecules, nonspecific histone deacetylase (HDAC) inhibitors emerged as a promising drug class capable of selectively repressing endogenous ERBB2 promoter function; curiously, such pan-HDAC inhibitors were subsequently found capable of rapidly destabilizing mature ERBB2 transcripts (1–3). The antitumor and anti-ERBB2 properties of pan-HDAC inhibiting hydroxamates like trichostatin A (TSA), vorinostat (suberoylanilide hydroxamic acid/SAHA, Merck & Co., Whitehouse Station, NJ) and LAQ824 (Novartis Pharmaceuticals, East Hanover, NJ) were validated against different ERBB2 overexpressing breast cancer cell lines (e.g. SKBR3, BT474, MDA-453), a trastuzumab-sensitive ERBB2-positive breast cancer xenograft model (e.g. BT474) and a trastuzumab-resistant ERBB2-positive breast cancer xenograft model (B585); in all these studies tumor growth inhibition correlated with selective downregulation of ERBB2 transcript and protein levels (1–4).
We initially explored how pan-HDAC inhibitors capable of repressing ERBB2 gene transcription might also accelerate decay of mature cytoplasmic ERBB2 transcripts. An ERBB2-independent breast cancer cell line (MCF7) was transfected by paired luciferase expressing constructs, one with and one without downstream insertion of the ERBB2 3′ untranslated region (UTR) which contains an evolutionarily conserved U-rich regulatory element putatively capable of binding transcript stability factors like HuR or AUF1 (5, 6; Figure 1A). After either transient or stable introduction of the reporter genes, cells treated with a pan-HDAC inhibitor (TSA, 5–10 h) showed substantially reduced mRNA and protein expression of the ERBB2 3′ UTR-regulated reporter, suggesting that endogenous ERBB2 transcripts might be similarly regulated (7). To date, neither AUF1 nor HuR have been shown to bind the ERBB2 3′ UTR, although high cytoplasmic levels of the shuttling factor, HuR, have recently been associated with poorer prognosis breast cancers (8).
Figure 1.

ERBB2 expression is regulated by the transcript stability factor, HuR, which binds a conserved U-rich element in the 3′UTR of ERBB2 mRNA. A. Near the poly-A tail of the ERBB2 3′ UTR is an evolutionarily conserved U-rich region (nt 465–505). B. In vitro binding assay using SKBR3 cytosol showing that HuR, but not AUF1, is capable of binding to a radiolabelled probe consisting of the conserved U-rich element of the ERBB2 3′ UTR mRNA. HuR antibody induces a supershift in the HuR-probe complex with loss of the higher mobility probe-bound HuR band. C. Immunoblots showing downregulation of cytosolic AUF1 and HuR levels after siRNA treatment (48 h) of SKBR3 cells, with resultant effects on ERBB2 levels normalized to β-actin.
Cytosolic 3′ UTR binding proteins like AUF1 and HuR, or other regulators of mRNA translation and stability, have not previously been identified as HDAC substrates. In some cell systems, pan-HDAC inhibitors have been shown to significantly reduce cytosolic (but not total) HuR levels by acetylation of importin-α1, resulting in reduced stability of HuR target transcripts (9). Unlike HuR, HDAC6 is known to be primarily cytoplasmic in location where it functions to deacetylate such substrates as the chaperoning protein HSP90, the microtubule component α-tubulin, and the actin cytoskeleton protein cortactin (3, 10–13). The unique protein structure of HDAC6, with its tandem catalytic deacetylase domains and carboxy(C)-terminal binding-of-ubiquitin zinc (BUZ) finger domain found almost exclusively in deubiquitinating enzymes, also suggests that HDAC6 serves largely nongenomic cell functions (13). In fact, HDAC6 has been shown to play essential microtubule, cytoskeleton and dynein motor associated roles in regulating cell migration, macropinocytosis, protein trafficking and accumulation of misfolded proteins into the aggresome (10–13). Some of these nongenomic functions may not require HDAC6 catalytic activity, yet all appear to be dependent on the non-catalytic ubiquitin binding BUZ finger domain (13, 14). Although HDAC6 is generally acknowledged to be extranuclear in both its location and function, nuclear translocation of HDAC6 by chromatin associated proteins has also been described (12, 13). As well, a recent immunohistochemical study of normal and malignant breast tissues noted that HDAC6 is localized primarily in the nucleus of normal mammary epithelial cells but largely in the cytoplasm of most breast cancer cells (15). Thus, while HDAC6 is a logical HDAC candidate to be involved in the maintenance of ERBB2 cytoplasmic mRNA stability in human breast cancer cells, to date there have been no reports identifying any specific HDAC with eukaryotic regulation of mRNA translation or stability.
The present study was undertaken to further investigate HDAC regulation of ERBB2 transcript stability in an ERBB2-positive human breast cancer cell line, SKBR3. We observed that ERBB2 transcript levels are dependent on cytosolic HuR, which binds to the evolutionarily conserved U-rich region in the 3′ UTR of ERBB2 mRNA, co-precipitates with HDAC enzymatic activity, and co-localizes with cytosolic HDAC6. In turn, HDAC6 immunoprecipitates are enriched with HuR. A series of novel HDAC6-selective inhibitors were synthesized and shown to produce antiproliferative effects at culture concentrations that fail to repress nuclear ERBB2 promoter activity but induce decay of mature ERBB2 mRNA and reduce ERBB2 protein levels. In contrast to the ERBB2 inhibiting effects of catalytic pan-HDAC and HDAC6-selective inhibitors, knockdown of HDAC6 protein in SKBR3 by siRNA produced a paradoxical increase in ERBB2 protein expression with no effect on transcript levels, demonstrating that HDAC6 enzymatic inhibition and protein downregulation are not functionally equivalent. HDAC6 knockdown, however, offset ERBB2 mRNA decay induced by either pan-HDAC or HDAC6-selective inhibitors.
Materials and Methods
Cells, culture growth and luciferase expression assays
The human breast cancer cell line SKBR3 was originally obtained from the American Type Culture Collection (ATCC; Manassas, VA) and is grown in McCoys medium (Cellgro, Herndon, VA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. HCT116 human carcinoma cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech, Hemdon, VA) supplemented with 10% FBS, 1% penicillin/streptomycin and 2 mM L-glutamine (GIBCO, Invitrogen, Carlsbad, CA). The luciferase expressing ERBB2 promoter-reporting subline, MCF7/R067pGL-4, was constructed as previously described (1, 2) and was grown in DME H-16 medium supplemented with 10% FBS, 10 μg/ml insulin, 1% penicillin/streptomycin, and 500 μg/ml neomycin (G418, Mediatech). Culture growth was measured using an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma-Aldrich, St. Louis, MO) assay after plating cells into 96-well, 12-well or 10 cm culture dishes, treating with drug or vehicle (0.5% DMSO) at the indicated concentrations and times. MTT was added to washed monolayers at a final concentration of 0.5 mg/ml (4 h), and absorbance of the colored soluble formazan product was quantitated at 570 nm by spectrophotometric miocroplate reader (Molecular Devices, Sunnyvale, CA), as previously described (1, 2). Luciferase expession was measured from plated, treated and washed cells after lysis (Passive Lysis Buffer E194A, Promega, Madison, WI) using a commercial assay kit (Promega) and microplate luminometer (LabSystems Fluoroskan Ascent FL, Thermo Electron Corp., Milfort, MA), as previously described (1, 2).
Hydroxamates and HDAC assays
Trichostatin A (TSA) was commercially obtained (Sigma-Aldrich) and LAQ824 was a kind gift from Novartis Pharmaceuticals, Inc. (East Hanover, NJ); these were prepared in 0.1 M stock solutions kept in DMSO at −20°C and protected from light. Three new hydroxamates (Figure 4), ST17, ST80 and ST71, were synthesized by our previously established route using tritylhydroxylamine, for ST17, and benzylhydroxylamine, for ST80 and ST71, in the synthetic sequence (16). Identity and purity of each compound were confirmed by IR, NMR and elemental analysis (data not shown). The relative HDAC inhibiting potential of these compounds was first compared against an HDAC-enriched rat liver extract, as previously described (17). The relative HDAC6 binding and inhibitory specificity of the novel hydroxamates was tested using immunoprecipitated FLAG-tagged HDAC1 and HDAC6 subtypes (18, 19), and were scored as IC50 (50% inhibitory concentration) values calculated for each compound based on inhibition of the enzymatic reactivity of a fluorescent small molecule substrate (20), as previously described (17). A commercially obtained fluorogenic deacetylation assay kit was also used to evaluate HDAC enzymatic activity in various cellular immunoprecipitates (Upstate, Lake Placid, NY). To confirm the intracellular specificity of the HDAC6-specific inhibitors, their ability to induce acetylation of α-tubulin (α-AcTubulin), an HDAC6 specific substrate, relative to total acetylation of histone H4 (AcH4) or K12-acetylation of H4, substrates for nuclear associated HDACs, was assessed by immunoblotting extracts from control and treated cells.
Figure 4.

Structure and HDAC6-selective activity of three newly synthesized hydroxamic acids. A. Chemical structures of ST17, ST80, and ST71. B. Selective in vitro micromolar activity (IC50, 50% μM inhibitory concentration) of inhibitors against deacetylase activity of affinity purified FLAG-tagged HDAC1 and HDAC6 subtypes, measured as described in Materials and Methods. C. Differential levels of acetylated α-tubulin (α-AcTubulin) and Histone H4 K12 (AcH4K12) in whole cell lystes from SKBR3 cells treated for 24 h with 0.5 μM LAQ824 or 50 μM of ST71, ST80 and ST17. Control (C) lysate from vehicle treated SKBR3 cells as shown. Densitometry determined ratios for α-AcTubulin/AcH4K12 were normalized by the value from LAQ824 treatment.
ERBB2 3′ UTR mRNA binding assays
The binding of intracellular HuR and AUF1 proteins to the ERBB2 3′ UTR U-rich mRNA element was measured by electrophoretic mobility shift assay, using an RNA probe spanning the conserved terminal U-rich mRNA element (nt 465–505). Cytosolic extracts were incubated with 1–2 mg/ml poly[d(I-C)] in 100 mM KCl, 10 mM Tris-HCl, ph 7.5, 2 mM DTT, and 20% vol/vol glycerol for 20 min at 23°C. The binding reaction was then initiated by adding a P32 labeled RNA probe generated from a PCR fragment spanning the UTR U-rich region and incorporating a T3 RNA polymerase binding site for the production of sense strand transcripts, using P32 UTP as the labeled nucleotide in the T3 RNA polymerase reaction with the labeled RNA probe purified by Nuc Spin columns (Ambion, Austin, TX). Some binding reactions were co-incubated with antibodies to HuR (Santa Cruz Biotechnology, Santa Cruz, CA) or AUF1 (Upstate). Following treatment of the RNA binding reaction with RNase T1 for 10 minutes at 23°C to eliminate unprotected RNA, reaction products were electrophoretically separated on 4.2% acrylamide (29:1 acrylamide:bisacrylamide) gels in 0.5x TBE (50 mM Tris, pH 7.5, 50 mM boric acid, and 1 mM EDTA) running buffer at 150 V. Gels were vacuum-dried and gel shifted bands were visualized by autoradiography.
Transient transfection and siRNA reagents
Validated siRNA reagents (and associated control siRNAs) specific for HuR and AUF1 (Qiagen, Valencia, CA) or HDAC6 (Dharmacon, Lafayette, CO) were commercially obtained. Cells were transfected with the various siRNAs at a final concentration of 100 nM using Oligofectamine (Invitrogen) in serum-free medium for 7 hours. Following transfection, cells were incubated for an additional 48 hours (HuR, AUF1) or 72 hours (HDAC6) in media supplemented with 10% serum (+/− additional treatments as indicated), prior to RNA and/or protein extraction. Transfections were repeated three times to assure consistency of results.
Immunoblotting and immunoprecipitation
Cytosolic lysates used for immunoprecipitation were prepared from cells dounced in a hypotonic buffer containing 50 mM Hepes (pH 7.4), 10 mM KCL, 0.3% Nonidet P-40, 10 mM MgCl2, 100 U/ml SUPERasein (Ambion), and mini-complete protease inhibitors (Roche Diagnostics, Mannheim, Germany). Following removal of nuclei by centrifugation, the cytoplasmic lysate was brought to 50 mM KCl and incubated at 4°C for 5 hours with either 0.2 μg of HuR, HDAC6 or control antibodies together with 12 μl of protein G Sepharose beads (GE Healthcare, Uppsala, Sweeden) per ml of lysate. The protein G beads from the immunoprecipitations were washed once at 4°C in wash buffer (20 mM Tris pH 7.4, 50 mM KCL, 10 mM MgCl2 0.3% Nonidet P-40), resuspended in 1X loading buffer (20 mM Tris pH 6.9, 2% SDS, 1% 2-mercaptoethanol, 50 mM NaCl and 10% glycerol), heated to 95°C for 10 minutes. Equal aliquots of immunoprecipitated material were electrophoresed using 4–12% gradient gels (Invitrogen), transferred to nitrocellulose (Amersham Bioscience) and probed for HuR and HDAC6 in hybridization buffers of 20 mM Tris pH7.5, 130 mM NaCl, 0.05% Tween-20 with 5% non-fat milk. Horseradish peroxidase (HRP) coupled goat anti-mouse (BioRad, Hercules, CA) and native IgG HRP Detection Reagent (Pierce, Rockford, IL) were used for detection of bound antibody by the manufacturer’s chemiluminescence enhancement procedure. Western analysis of lysates prepared from whole cells solubilized in loading buffer using brief sonication was preformed as described above for immunoprecipitated samples. Antibodies used included ERBB2 (Calbiochem, San Diego, CA), β-actin (Abcam, Cambridge, MA), α-tubulin (Sigma), acetylated α-tubulin (Sigma), AUF1 (Upstate), HuR (mouse 3A2, Santa Cruz Biotechnology), Lex A (mouse control Ab for HuR IPs, Santa Cruz Biotechnology), HDAC6 (sc-11420, Santa Cruz Biotechnology), ERα (rabbit control Ab for HDAC6 IPs, Santa Cruz Biotechnology) acetylated H4 (Upstate) or K12-acetylated H4 (Cell Signaling Technology, Danvers, MA).
Northern blots and RT-PCR detection of specific transcripts
RNA from control or treated cells was isolated using TRIzol (Invitrogen) according to the manufacturer’s specifications. For Northern blotting,15 μg of total RNA per lane was electrophoresed into 1% agarose-formaldehyde gels, then transferred onto Hybond Plus membranes (Amersham, Piscataway, NJ) that were UV cross-linked, hybridized with P32 labeled cDNA probes for ERBB2 or GAPDH, and washed as previously described (1, 2). Hybridizing bands were visualized by autoradiography and quantified by densitometry (GS-710 Calibrated Imaging Densitometer, BioRad, Hercules, CA), and transcript ratios (ERBB2/GAPDH) calculated. RNA isolated using TRIzol (Invitrogen) from cellular immunoprecipitates prepared as described above was reverse transcribed by oligo dT priming using SuperScript II reverse transcriptase (Invitrogen) in 20 μl volumes, according to manufacturer’s instructions, then subjected to PCR amplification using ERBB2 or GAPDH specific primers (forward, reverse). GAPDH detection served as a control for nonspecific RNA binding to cellular immunoprecipitates. ERBB2 specific primers were designed to amplify a 137 nt sequence within the ERBB2 3′ UTR, just upstream of the evolutionarily conserved terminal U-rich sequence (nt 465–505). PCR reactions were performed with Phusion (New England BioLabs, Ipswich, MA) polymerase using 2 μl aliquots from the RT reactions with primers at 1μM final concentration. Linearity of PCR reactions were confirmed by examination of identically prepared reactions following 23–26 cycles. Reaction products were examined on precast 8% polyacrylaminde TBE gels (Invitrogen) using HaeIII cut PhiX DNA markers (New England BioLabs) and visualized by ethidium bromide staining. Primers pairs used were as follows: ERBB2 (137 bp amplimer) forward: 5′ GGTACTGAAAGCCTTAGGGAAGC 3′ reverse: 5′ ACACCATTGCTGTTCCTTCCTC 3′ GAPDH (234 bp amplimer) forward: 5′ CGAATTGGCTACAGCAACAGG 3′ reverse: 5′ GTACATGACAAGGTGCGGCTC 3′
Immunofluorescence imaging
SKBr3 cells were fixed and incubated with rabbit polyclonal anti-HDAC6 and mouse monoclonal anti-HuR primary antibodies (Santa Cruz Biotechnology), followed by donkey anti-rabbit Alexa 555 and anti-mouse Alexa 488 secondary antibodies (Invitrogen). Cells were visualized using an LSM 510 NLO Confocal Scanning System (mounted on an Axiovert 200 Inverted Microscope; Carl Zeiss Ltd., Cambridge, UK) equipped with a two-photon Chameleon laser (Coherent Inc. Santa Clara, CA). Confocal images were generated using a 40x objective with 4x digital zoom, processed by Imaris software including analysis by the co-localization module (Bitplane AG., Zurich, Switzerland).
Results
As shown in Figure 1A, embedded near the poly-A tail of the ERBB2 3′ UTR sequence is an evolutionarily conserved U-rich region (nt 465–505) putatively capable of binding factors like HuR or AUF1 that regulate mRNA stability (5, 6). While both AUF1 and HuR are abundantly expressed in SKBR3 cells, Figure 1B shows that only HuR is capable of in vitro binding to the conserved terminal U-rich element (nt 465–505) of the ERBB2 3′ UTR mRNA. Treatment of SKBR3 cells with a pan-HDAC inhibitor (TSA, 0.4 μM × 20 h) capable of destabilizing ERBB2 mRNA neither induced AUF1 nor inhibited HuR binding to this ERBB2 3′ UTR probe (data not shown). Consistent with the in vitro observation of HuR binding to ERBB2 mRNA, siRNA-mediated downregulation of AUF and HuR protein levels in SKBR3 cells demonstrated that after 48 h only HuR downregulation is associated with altered intracellular expression of ERBB2 (Figure 1C).
HuR immunoprecipitated from SKBR3 cells demonstrated endogenous binding to ERBB2 transcripts and associated RNA-dependent HDAC activity. As shown in Figure 2A, immunoprecipitated HuR contained specifically increased levels of ERBB2 mRNA, detected by RT-PCR using ERBB2 specific primers chosen to amplify a 137 nt sequence within the ERBB2 3′ UTR just upstream of the conserved U-rich region (nt 465–505). In contrast, control immunoprecipitates from SKBR3 contained almost undetectable levels of ERBB2 mRNA, and both control and HuR immunoprecipitates showed comparable levels of nonspecific association with GAPDH mRNA. Using a commercial fluorogenic deacetylation assay to monitor total HDAC enzymatic activity, these same ERBB2 mRNA-associated HuR immunoprecipitates were found to contain 3-fold higher HDAC activity than control immunoprecipitates, and this increased HDAC activity was shown to be dependent on HuR-associated mRNA since it was eliminated by concomitant RNase treatment (Figure 2B). Since the majority of cytosolic HDAC activity is thought to be due to HDAC6, SKBR3 cytosols were subjected to IP-immunoblotting to detect any physical association between HuR and HDAC6. As shown in Figure 2C, HuR immunoprecipitates were enriched in HDAC6 and HDAC6 immunoprecipitates were enriched in HuR. Interestingly, 3 h pretreatment of SKBR3 with a pan-HDAC inhibitor (0.4 μM TSA) did not significantly alter these IP-immunoblot results; however, RNase pretreatment of the SKBR3 cytosols abolished the co-association of HuR with HDAC6 (data not shown). Microscopic imaging studies were undertaken to further address the apparent physical association between HuR and HDAC6. As seen in Figure 2D, confocal immunofluorescence imaging confirmed the major nuclear and minor cytoplasmic abundance of HuR (green image) and the major cytoplasmic and minor nuclear abundance of HDAC6 (red image) in normally growing SKBR3 cells. Planar merging of these digitized confocal images readily demonstrated the nuclear and cytoplasmic co-localization of HuR with HDAC6 (yellow image).
Figure 2.

Association of cytosolic HuR with ERBB2 mRNA and HDAC enzymatic activity, and the co-precipitation and co-localization of HuR with HDAC6 in SKBR3 cells. A. Immunoprecipitation of cytoplasmic HuR by specific (or isotype control) antibody followed by RT-PCR assessment of co-precipitated ERBB2 and GAPDH transcripts. PCR primer pairs were chosen to amplify a 137 nt sequence within the ERBB2 3′ UTR or a 183 nt sequence within GAPDH transcripts. B. Following control or HuR immunoprecipitations from SKBR3 cytosols +/− RNase treatment, a fluorogenic deacetylation assay was used to monitor co-precipitating total HDAC enzymatic activity. C. SKBR3 cytosolic lysates subjected to immunoprecipitation (IP) followed by Western blotting (WB), demonstrating HuR IP enriched with HDAC6 and HDAC6 IP enriched with HuR (also compared to β-actin co-precipitation). Longer WB exposures (not shown) confirmed equivalent lane loading by IgG heavy and light chain band intensities. D. Immunofluorescence microscopic imaging (40x objective with 4x digital zoom) showing major nuclear and minor cytoplasmic abundance of HuR (green) and major cytoplasmic and minor nuclear abundance of HDAC6 (red), and their nuclear and cytoplasmic co-localization (yellow), in cultured SKBR3 cells.
Functional relationships between ERBB2, HuR and HDAC6 in SKBR3 cells were explored using catalytic inhibitors of pan-HDAC enzymatic activity (HDACi: TSA or LAQ824) and siRNA induced downregulation of HuR (or AUF1) and HDAC6 protein levels. As shown in Figure 3A, even after 20 h of SKBR3 treatment with TSA (0.4 μM), cellular levels of HuR remained unchanged while ERBB2 protein levels showed the expected level of decline as previously reported (1). While total HuR levels remained unchanged, 3–6 h of TSA treatment produced ≤30% reduction in SKBR3 cytosolic HuR levels (data not shown), a much less pronounced reduction in cytosolic HuR levels as reported in other TSA treated cell systems (9). To determine how much the destabilization of ERBB2 transcripts by HDACi depended on intracellular HuR or AUF1, 48 h of siRNA pretreatment was employed to first reduce HuR or AUF1 levels and cells were then treated with LAQ824 (10 μM × 6 h) or drug vehicle. As shown in the Northern blots of Figure 3B, pan-HDAC inhibition substantially reduced ERBB2 mRNA levels following AUF1 downregulation and had a slightly attenuated ERBB2 mRNA reducing effect following HuR downregulation. Note that in the absence of HDACi, HuR downregulation alone reduced ERBB2 mRNA levels relative to AUF1 downregulation, consistent with earlier described results (Figure 1C).
Figure 3.

ERBB2 effects of pan-HDAC enzymatic inhibitors (HDACi) are independent of HuR levels and associated with transcript binding to HDAC6, but dissimilar to that of HDAC6 protein downregulation by siRNA. A. ERBB2 protein inhibition by 16–20 h of SKBR3 treatment with TSA (0.4 μM), with no effect on total HuR expression. B. Effect of AUF1 or HuR downregulation by siRNA treatment (48 h, as described in Figure 1) followed by 6 h treatment with LAQ824 (10 μM) on SKBR3 ERBB2 mRNA levels (normalized to 28S and 18S rRNA). C. Immunoprecipitation of cytoplasmic HDAC6 followed by RT-PCR assessment of co-precipitated ERBB2 and GAPDH transcripts (as described in Figure 2) from control (untreated) and 3 hr LAQ824 (HDACi) treated SKBR3 cells. Untreated and HDACi RT samples were PCR amplified for 24 and 26 cycles respectively. D. HDAC6 knockdown by siRNA (72 h) in SKBR3 cells and resultant effects on ERBB2 and acetylated α-tubulin levels, normalized to total α-tubulin.
Analogous to experiments measuring ERBB2 mRNA associated with cytosolic HuR immunoprecipitates (Figure 2A), HDAC6 cytosolic immunoprecipitates were prepared in the presence or absence of SKBR3 exposure to HDACi (3 h), and co-precipitating ERBB2 transcripts were measured by RT-PCR. Figure 3C indicates that SKBR3 cytosolic HDAC6 is enriched for ERBB2 3′ UTR mRNA, and this enrichment was not diminished even in the presence of LAQ824 (10 μM × 3 h). Unlike HDACi treatments that reduced ERBB2 mRNA and protein levels, siRNA induced HDAC6 knockdown sufficient to allow for α-tubulin acetylation resulted in slightly increased levels of SKBR3 ERBB2 protein by 72 h (Figure 3D). Consistent with these Western blots, Northern blots from the same cell treatments showed no significant change in ERBB2 mRNA levels (data not shown). These latter results indicate that HDAC6 protein knockdown is not functionally equivalent to catalytic inhibition of its deacetylase activity, and that α-tubulin acetylation does not functionally correlate with ERBB2 mRNA destabilization.
A series of novel HDAC6-selective inhibitors were synthesized to evaluate their effects on SKBR3 ERBB2 expression and growth. Figure 4 shows the structural differences between the hydroxamates ST17, ST80 and ST71 (panel A) with in vitro assays demonstrating their 14 to 31-fold greater catalytic inhibition (IC50 values) of purified HDAC6 activity relative to HDAC1 activity, with ST80 showing the greatest selectivity (panel B). The intracellular functionality of the HDAC6-selective inhibitors relative to the pan-HDAC inhibitor LAQ824 was determined by assaying the relative levels of the HDAC6-specific target, acetylated α-tubulin, to histone H4 acetylated at K12. As shown in Figure 4C, SKBR3 cells treated with the HDAC6-selective inhibitors displayed significantly greater α-tubulin acetylation relative to histone H4 K12 acetylation when compared to the level in LAQ824 treated cells, with ST80 displaying an 80-fold enhancement in the acetylated α-tubulin to acetylated K12 histone H4 ratio. The inability of any of these three HDAC6-selective hydroxamate inhibitors to repress endogenous ERBB2 promoter activity after 24 h exposure to doses exceeding 50 μM, as compared to the well described ERBB2 promoter repressing effects of sub-micromolar doses of other pan-HDAC inhibiting hydroxamates (1–3), was demonstrated using the same ERBB2 promoter-reporting (luciferase expressing) MCF7/R06pGL-4 cell line formerly used to screen chemical libraries for cell permeable drugs with specific ERBB2 promoter repressing function (2). Figure 5A shows a representative dose-response comparison of ST71 with LAQ824 (24 h treatment) against MCF7/R06pGL-4 cells; while neither drug altered the viability of this ERBB2 independent subline (measured by MTT assay) over this 24 h treatment interval, the HDAC6-selective inhibitor was unable to repress luciferase expression at any drug dose in contrast to the pan-HDAC inhibitor which exhibited the expected dose-dependent repression of ERBB2 promoter-driven luciferase activity. When ST71 and LAQ824 were compared for their ability to inhibit SKBR3 cell growth after 72 h continuous exposure, both showed dose-dependent antiproliferative activities, but LAQ824 exhibited 3-log greater potency (lower IC50) over ST71 (Figure 5B).
Figure 5.

HDAC6-selective inhibitor ST71 fails to repress intracellular ERBB2 promoter activity but inhibits culture growth of ERBB2-positive SKBR3 cells. A. Dose-response comparison against ERBB2 promoter-reporting (luciferase expressing) MCF7/R06pGL-4 cells following exposure to HDAC6-specific inhibitor ST71 or pan-HDAC inhibitor LAQ824, showing 24 h effects (% control) on endogenous luciferase expression (blue, solid circles) and MTT cell viability (red, solid squares). B. Dose-response comparison against ERBB2-positive SKBR3 cells after exposure to ST71 or LAQ824, with loss of MTT cell viability as a measure of growth inhibition following 72 h treatment (% control).
To confirm the impact of HDAC6-selective inhibitors on ERBB2 transcript decay, experiments were designed using SKBR3 cells pre-transfected for 72 h with HDAC6 siRNA. As shown in Figure 6A, the HDAC6 siRNA transfected SKBR3 cells were significantly less susceptible to LAQ824 or ST80 induced ERBB2 mRNA decay as control siRNA treated cells. To assess the ability of antiproliferative doses of the HDAC6-selective inhibitors to promote ERBB2 protein downregulation, Figure 6B demonstrates that following a 24 h treatment of SKBR3 cells with 50 μM of ST71, ST80 or ST17, suppression of ERBB2 protein levels were relatively equivalent to that achieved by 0.5 μM LAQ824.
Figure 6.
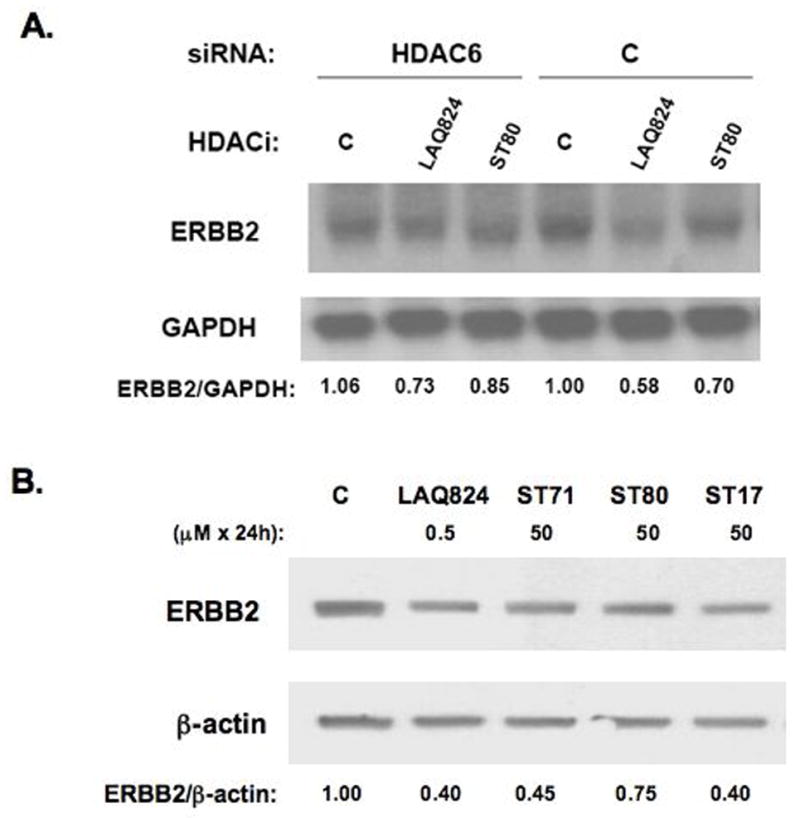
HDAC6 knockdown offsets ERBB2 mRNA decay induced by pan-HDACi and HDAC6-selective inhibitors that comparably reduce SKBR3 ERBB2 mRNA and protein levels. A. Northern blot showing the influence of 3 h treatment with 0.5 μM LAQ824, 50 μM ST80 or vehicle control (C) on ERBB2 mRNA levels in SKBR3 cells pre-transfected for 72 h with HDAC6 or control (C) siRNA. Densitometry determined ratios of ERBB2/GAPDH band intensities were normalized by the value obtained from vehicle treated control siRNA transfectants (C). B. Western blots showing 24 h treatment effects of indicated drug and concentration on SKBR3 ERBB2 and β-actin protein levels; densitometry measured band intensities were used to calculate ERBB2/β-actin protein ratios, normalized such that untreated control (C) ratio = 1.
Discussion
We initially reported that pan-HDAC inhibitors, while capable of repressing ERBB2 promoter function and accelerating the decay of mature ERBB2 transcripts, are unable to affect ERBB2 mRNA expressed off an ectopic cDNA construct lacking both 5′ and 3′ ERBB2 UTR sequences (1). By fusing the ERBB2 3′ UTR onto the terminus of a luciferase cDNA reporter and transfecting this into human breast cancer cells, we subsequently confirmed that the ERBB2 3′ UTR sequence is capable of accelerating transcript decay upon exposure (≥5 h) to pan-HDAC inhibitors (7). As well, by employing an expression array strategy to identify and quantitate all U-rich UTR-bearing genes endogenously expressed in SKBR3 cells, we demonstrated that ERBB2 is among more than 300 different U-rich transcripts (including cyclins D1 and D2, and the importin β family member exportin 4) rapidly destabilized within 6 h of treatment with such pan-HDAC inhibiting hydroxamates as TSA and LAQ824 (21). By sequence inspection the conserved U-rich element (nt 465–505; Figure 1A) within the 3′ UTR of ERBB2 mRNA could putatively bind either the destabilizer, AUF1, or the stabilizer, HuR (5, 6). However, the present study demonstrates that while both AUF1 and HuR are abundantly expressed in SKBR3 cells, only HuR appears capable of binding to the ERBB2 U-rich element (Figure 1B) and regulating intracellular levels of ERBB2 mRNA and protein (Figures 1C and 3B).
As cytosolic HuR immunoprecipitates were found to possess increased HDAC enzymatic activity (Figure 2B) and numerous previous studies have shown that HDAC6 is the primary source of most cytoplasmic HDAC enzymatic activity (10–13), Western analysis of HuR immunoprecipitates confirmed the presence of HDAC6, and the reciprocal experiment confirmed the association of HuR with HDAC6 immunoprecipitates (Figure 2C). This co-association of HuR with HDAC6 was RNA-dependent; furthermore, confocal imaging in SKBR3 showed cytoplasmic and nuclear co-localization of HDAC6 with HuR (Figure 2D). Like HuR, HDAC6 immunoprecipitates appeared to be enriched with ERBB2 transcripts, and this association was maintained for at least for three hours following treatment with an ERBB2 transcript destabilizing dose of HDACi (Figure 3C). It remains unclear how HDAC6 physically associates with ERBB2 mRNA. Based on evidence provided here it may be suggested that HDAC6, which has no structural means of directly binding to nucleic acids, associates with HuR in an RNA-dependent interaction (Figure 2B); alternatively, HDAC6 may be tethered by an unknown RNA-binding protein to the 3′UTR of ERBB2 transcripts.
HDAC6, while not essential for normal mammalian development (11, 12), has been shown to regulate cell shape and motility in human breast cancer cells (22), growth factor induced endocytosis and nuclear receptor translocation in fibroblasts (11), and T-cell dependent antibody response (12) and chemotaxis in lymphocytes (14). Curiously, lymphocyte chemotaxis does not require the deacetylase activity but does require the protein scaffolding and BUZ finger domain functions of HDAC6 (14). In contrast, nuclear receptor translocation in fibroblasts is significantly more impaired by loss of HDAC6 catalytic function than by total absence of HDAC6 protein (12). This complexity involving HDAC6 catalytic and non-catalytic functions likely explains the paradoxical observation that knockdown of SKBR3 HDAC6 protein levels by siRNA did not destabilize ERBB2 mRNA as did inhibition of HDAC6 catalytic activity by either selective or non-selective HDAC6 binding hydroxamates. ERBB2 transcript destabilization may require not only the absence of HDAC6 deacetylase activity but also the presence of HDAC6 protein to provide a scaffolding or shuttling function for transport of destabilized mRNA from polyribosomes into decapping and degradation compartments, such as stress granules and the processing (P) body (5). In contrast to mRNA decay, ERBB2 protein decay is known to be mediated by cytoplasmic proteasome and aggresome systems (23), both of which can be inhibited by HDACi (13, 24). Thus, the modest increase in SKBR3 ERBB2 protein levels observed following siRNA knockdown of HDAC6 possibly reflects impairment in both these protein degradation systems induced by the HDAC6 knockdown. The availability of functionally active HDAC6-specific hydroxamates that either inhibit (e.g. Tubacin) or fail to inhibit (e.g. Niltubacin) its deacetylase activity might help to discern the relative importance of HDAC6 catalytic and non-catalytic functions in maintaining ERBB2 transcript stability. As well, further comparison of HDAC6 protein downregulation by siRNA with HDAC6 enzymatic inhibition may delineate those cytoplasmic compartments involved in ERBB2 mRNA decay from those involved in ERBB2 protein degradation.
Tubacin was the first HDAC6-selective hydroxamate inhibitor identified, and was shown to bind and inhibit one of the two deacetylase domains in HDAC6 (25); it typically inhibits intracellular HDAC6 tubulin deacetylase activity at culture concentrations exceeding 10 μM (14). The presently synthesized HDAC6-selective hydroxamate inhibitors, ST17, ST80 and ST71, show comparable specificity relative to Tubacin and modestly enhanced potency. They induce intracellular α-tubulin acetylation at drug concentrations ≥1 μM; even after 24 h of exposure to a 50 μM concentration sufficient to induce almost complete growth arrest of SKBR3 cells, these HDAC6-selective inhibitors predominantly induce α-tubulin acetylation relative to H4 acetylation (Figure 4C). Like Tubacin, these novel hydroxamates are useful chemical tools and prototypes for the development of more potent clinical drugs. As drug prototypes, these HDAC6-selective inhibitors demonstrate antiproliferative activity against SKBR3 cells at 3-log higher concentrations than that of one of the most potent pan-HDAC inhibiting drugs to enter clinical development, LAQ824 (Figure 5B). Following exposure to antiproliferative doses of these HDAC6-selective inhibitors, ERBB2 transcript and protein levels in SKBR3 cells declined just as rapidly as when these cells were treated with micromolar doses of LAQ824 (Figure 6). The explicit involvement of HDAC6 in mediating ERBB2 mRNA decay was shown using SKBR3 cells pre-transfected with siRNA that produced ~70% knockdown of HDAC6 protein levels (Figure 3D); this partial HDAC6 knockdown was sufficient to reduce the ERBB2 mRNA destabilizing ability of both a pan-HDACi (LAQ824) and an HDAC6-selective (ST80) inhibitor (Figure 6A).
While LAQ824 and other previously evaluated pan-HDAC inhibitors repressed endogenous ERBB2 promoter activity and accelerated decay of mature ERBB2 transcripts (1), none of the HDAC6-selective inhibitors (ST17, ST80, ST71) were able to affect endogenous ERBB2 promoter function (Figure 5A). These observations are consistent with the hypothesis that ERBB2 promoter function is regulated by one or more class I nuclear HDACs (e.g. HDAC1, HDAC2, HDAC3, and/or HDAC8) while a major component of ERBB2 transcript stability appears to be regulated by the class II cytoplasmic deacetylase, HDAC6. Altogether, these findings point to the therapeutic potential of HDAC6-selective inhibitors, and serve to demonstrate that ERBB2 transcript stability mechanisms employ targets like HuR and HDAC6 that are readily exploitable for the design of new anticancer therapies. Future studies aimed at dissecting the ERBB2 transcript stability mechanism are clearly needed, as it seems particularly important to better understand the structural and functional interactions between HDAC6 and HuR, as well as their potential involvement with another 3′ UTR mRNA associated stability mechanism, microRNA (26).
Acknowledgments
Financial support: National Institutes of Health sponsored grants R01-CA36773 and P50-CA58207 (UCSF Breast SPORE), and Hazel P. Munroe memorial funding to the Buck Institute. The Hans and Gertie Fischer-Foundation sponsored S.S. and M.J. who also thank Sanofi-Aventis for an [i]-travel award.
LAQ824 was generously provided by Novartis Pharmaceuticals, Inc. (East Hanover, NJ).
Abbreviations list
- UTR
untranslated region
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- TSA
trichostatin A
- siRNA
small inhibitory RNA
- RT-PCR
reverse transcription-polymerase chain reaction
- α-AcTubulin
acetylated α-tubulin
- AcH4
acetylated histone H4
References
- 1.Scott GK, Marden C, Xu F, Kirk L, Benz CC. Transcriptional repression of ErbB2 by histone deacetylase inhibitors detected by a genomically integrated ErbB2 promoter-reporting cell screen. Mol Cancer Therapeutics. 2002;1:385–392. [PubMed] [Google Scholar]
- 2.Marx C, Berger C, Xu F, Amend C, Scott GK, Hann B, Park JW, Benz CC. Validated high-throughput screening of drug-like small molecules for inhibitors of ErbB2 transcription. ASSAY and Drug Development Technologies. 2006;4:273–284. doi: 10.1089/adt.2006.4.273. [DOI] [PubMed] [Google Scholar]
- 3.Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Ann Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 4.Drummond DC, Marx C, Guo Z, Scott G, Noble C, Wang D, Pallavicini M, Park JW, Kirpotin DB, Benz CC. Enhanced pharmacodynamic and antitumor properties of a histone deacetylase inhibitor encapsulated in liposomes or ErbB2-targeted immunoliposomes. Clin Cancer Res. 2005;11:3392–3401. doi: 10.1158/1078-0432.CCR-04-2445. [DOI] [PubMed] [Google Scholar]
- 5.Barreau C, Paillard L, Osborne B. AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–7150. doi: 10.1093/nar/gki1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lopez de Silanes I, Zhan M, Lal A, Yang X, Gorospe M. Identification of a target RNA motif for RNA-binding protein HuR. Proc Natl Acad Sci (USA) 2004;101:2987–2992. doi: 10.1073/pnas.0306453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marx C, Berger C, Benz S, Mattie M, Benz C, Scott G. Therapeutic destabilization of ErbB2 transcripts mediated by U-rich mRNA binding proteins and microRNAs. Proc Amer Assoc Cancer Res. 2006;47:a5616. [Google Scholar]
- 8.Heinonen M, Bono P, Narko K, Chang S-H, Lundin J, Joensuu H, Furneaux H, Hla T, Haglund C, Ristimaki A. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 2005;65:2157–2161. doi: 10.1158/0008-5472.CAN-04-3765. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Yang X, Kawai T, Lopez de Silanes I, Mazan-Mamczarz K, Chen P, Chook YM, Quensel C, Köhler M, Gorospe M. AMP-activated protein kinase-regulated phosphorylation and acetylation of importin α1. J Biol Chem. 2004;279:48376–48388. doi: 10.1074/jbc.M409014200. [DOI] [PubMed] [Google Scholar]
- 10.Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 11.Gao Y, Hubbert CC, Lu J, Lee Y-S, Lee J-Y, Yau T-P. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol Cell Biol. 2007;27:8637–8647. doi: 10.1128/MCB.00393-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M, Cao C, Cheng H-L, Chua K, Lombard D, Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell, Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthias P, Yoshida M, Khochbin S. HDAC6, a new cellular stress surveillance factor. Cell Cycle. 2008;7:7–10. doi: 10.4161/cc.7.1.5186. [DOI] [PubMed] [Google Scholar]
- 14.Cabrero JR, Serrador JM, Barreiro O, Mittelbrunn M, Naranjo-Suarez S, Martin-Cofreces N, Vicente-Manzanares M, Mazitschek R, Bradner JE, Avila J, Valenzuela-Fernandez A, Sanchez-Madrid F. Lymphocyte chemotaxis is regulated by histone deacetylase 6 independently of its deacetylase activity. Mol Biol Cell. 2006;17:3435–3445. doi: 10.1091/mbc.E06-01-0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z, Yamashita H, Toyama T, Sugiura H, Omoto Y, Ando Y, Mita K, Hamaguchi M, Hayashi S, Iwase H. HDAC6 expression is correlated with better survival in breast cancer. Clin Cancer Res. 2004;10:6962–6968. doi: 10.1158/1078-0432.CCR-04-0455. [DOI] [PubMed] [Google Scholar]
- 16.Jung M, Hoffmann K, Brosch G, Loidl P. Analogues of trichostatin A and trapoxin B as histone deacetylase inhibitors. Bioorg Med Chem Lett. 1997;7:1655–1658. [Google Scholar]
- 17.Heltweg B, Jung M. A homogeneous non-isotopic assay for histone deacetylase activity. J Biomol Screen. 2003;8:89–95. doi: 10.1177/1087057102239644. [DOI] [PubMed] [Google Scholar]
- 18.Fischle W, Emiliani S, Hendzel MJ, Nagase T, Nomura N, Voelter W, Verdin E. A new family of human histone deacetylases related to Saccharomyces cerevisiae HDA1p. J Biol Chem. 1999;274:11713–11720. doi: 10.1074/jbc.274.17.11713. [DOI] [PubMed] [Google Scholar]
- 19.Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell. 2002;9:45–57. doi: 10.1016/s1097-2765(01)00429-4. [DOI] [PubMed] [Google Scholar]
- 20.Heltweg B, Dequiedt F, Verdin E, Jung M. A non isotopic substrate for assaying both human zinc and NAD+-dependent histone deacetylases. Anal. Biochem. 2003;319:42–48. doi: 10.1016/s0003-2697(03)00276-8. [DOI] [PubMed] [Google Scholar]
- 21.Benz SC, Scott GK, Marx C, Melov S, Benz CC. Histone deacetylase inhibitors accelerate decay of unique subsets of breast cancer transcripts that include ErbB2. Proc Amer Assoc Cancer Res. 2005;46:a5. [Google Scholar]
- 22.Saji S, Kawakami M, Hayashi S, Yoshida N, Hirose M, Horiguchi S, Itoh A, Funata N, Schreiber SL, Yoshida M, Toi M. Significance of HDAC6 regulation via estrogen signaling for cell motility and prognosis in estrogen receptor-positive breast cancer. Oncogene. 2005;24:4531–4539. doi: 10.1038/sj.onc.1208646. [DOI] [PubMed] [Google Scholar]
- 23.Marx C, Yau C, Banwait S, Zhou Y, Scott GK, Hann B, Park JW, Benz CC. Proteasome-regulated ERBB2 and estrogen receptor pathways in breast cancer. Mol Pharmacol. 2007;71:1525–1534. doi: 10.1124/mol.107.034090. [DOI] [PubMed] [Google Scholar]
- 24.Place RF, Noonan EJ, Giardina C. HDAC inhibition prevents NF-κB activation by suppressing proteasome activity: down-regulation of proteasome subunit expression stabilizes IκBα. Biochem Pharmacol. 2005;70:394–406. doi: 10.1016/j.bcp.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 25.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci (USA) 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scott GK, Goga A, Bhaumik D, Berger CE, Sullivan CS, Benz CC. Coordinate suppression of ERBB2 and ERBB3 by enforced expression of microRNA miR-125a or miR-125b. J. Biol. Chem. 2007;282:1479–1486. doi: 10.1074/jbc.M609383200. [DOI] [PubMed] [Google Scholar]