

Abstract
Chimeric RNA/DNA oligonucleotides (“chimeraplasts”) have been shown to induce single base alterations in genomic DNA both in vitro and in vivo. The mdx mouse strain has a point mutation in the dystrophin gene, the consequence of which is a muscular dystrophy resulting from deficiency of the dystrophin protein in skeletal muscle. To test the feasibility of chimeraplast-mediated gene therapy for muscular dystrophies, we used a chimeraplast (designated “MDX1”) designed to correct the point mutation in the dystrophin gene in mdx mice. After direct injection of MDX1 into muscles of mdx mice, immunohistochemical analysis revealed dystrophin-positive fibers clustered around the injection site. Two weeks after single injections into tibialis anterior muscles, the maximum number of dystrophin-positive fibers (approximately 30) in any muscle represented 1–2% of the total number of fibers in that muscle. Ten weeks after single injections, the range of the number of dystrophin-positive fibers was similar to that seen after 2 wk, suggesting that the expression was stable, as would be predicted for a gene-conversion event. Staining with exon-specific antibodies showed that none of these were “revertant fibers.” Furthermore, dystrophin from MDX1-injected muscles was full length by immunoblot analysis. No dystrophin was detectable by immunohistochemical or immunoblot analysis after control chimeraplast injections. Finally, reverse transcription–PCR analysis demonstrated the presence of transcripts with the wild-type dystrophin sequence only in mdx muscles injected with MDX1 chimeraplasts. These results provide the foundation for further studies of chimeraplast-mediated gene therapy as a therapeutic approach to muscular dystrophies and other genetic disorders of muscle.
Muscular dystrophies are hereditary, degenerative disorders of muscle that result from defects in genes that encode a diverse group of proteins (1). The most common form of muscular dystrophy in humans is Duchenne muscular dystrophy (DMD), which occurs when a defect in the dystrophin gene results in a deficiency of dystrophin protein in skeletal muscle (2). The absence of dystrophin leads to muscle cell death and progressive muscle degeneration, although the pathogenetic mechanisms remain a mystery (3) and are the subject of active study (4, 5). As there are no effective long-term treatments for the muscular dystrophies, there has been much interest in gene therapy approaches to these disorders. Currently, the most actively studied methods involve viral-mediated delivery of normal genes to skeletal muscle, although all current viral vectors have limitations and/or adverse effects.
Recently, several groups have used a strategy to induce single base pair changes in genomic DNA in both mammalian cells (6–11) and plant cells (12, 13). The strategy involves the use of chimeric RNA/DNA oligonucleotides (“chimeraplasts”), each of which contains a stretch of oligonucleotides homologous to a sequence in the genome except for a single mismatched base. When the sequence in the chimeraplast aligns with that in the genomic DNA, the single base mismatch induces endogenous repair mechanisms to “correct” the base in the targeted gene (14–16). Chimeric oligonucleotides have been used to induce single nucleotide changes in different mammalian cell types both in vitro (6–10) and in vivo (9, 11). Whereas the efficiency of chimeraplast-mediated gene conversion has varied widely depending on the particular cell type and the experimental conditions (17), conversion rates as high as 40% have been reported (7, 9). Recently, Kren et al. (9) demonstrated alteration of a single base pair in the rat factor IX gene by using chimeric RNA/DNA oligonucleotides targeted to hepatocytes and delivered by i.v. injection, providing evidence that chimeraplast-mediated gene repair may be a powerful strategy for hepatic gene therapy without the use of viral vectors.
We report here the use of RNA/DNA oligonucleotides to direct the correction of a point mutation in the dystrophin gene in the mdx mouse. The mdx mouse has a point mutation at nucleotide position 3185 in the dystrophin gene that produces a stop codon in exon 23 (18). As a result, there is no dystrophin produced in skeletal muscle of these mice, and the muscle fibers undergo necrotic degeneration as in DMD. We found that injection of chimeraplasts designed to correct the point mutation resulted in the expression of dystrophin in muscle fibers locally around the site of injection. These results suggest that this approach might be useful for the treatment of genetic disorders of muscle.
Materials and Methods
Mice.
Mice of the mdx strain (C57BL/10ScSn-mdx) and the control (“C57”) strain (C57BL/10SnJ) were obtained from The Jackson Laboratory and were handled in accordance with guidelines of the Administrative Panel on Laboratory Animal Care of Stanford University. All injections were done in mice that were between 2 and 4 wk of age.
Chimeraplast Synthesis.
Chimeric RNA/DNA oligonucleotides were synthesized as described (19) and were provided by Kimeragen (Newtown, PA). The oligonucleotides were prepared with DNA and 2′-O-methyl RNA phosphoramidite nucleoside monomers on a PerSeptive Biosystems (Framingham, MA) Expedite Nucleic Acid Synthesizer, purified by HPLC, and quantified by UV absorbance. The Cy3-MDX1 chimeraplasts were purified by using PE Biosystems reverse-phase oligonucleotide purification cartridges and ethanol precipitated twice. More than 95% of the purified oligonucleotides were determined to be of full length.
Intramuscular Injections of Chimeraplasts.
Chimeraplasts were dissolved in PBS at a concentration of 4 μg/μl, and 5 μl was injected into tibialis anterior muscles of anesthetized mice (125 mg/kg ketamine; 25 mg/kg xylazine). In earlier pilot studies, different chimeraplast concentrations and injection volumes were tested.
Histological and Immunohistochemical Analysis.
Muscles were prepared for histological analysis as described (20). For dystrophin immunostaining, primary antibodies directed against the rod domain (MANDYS-8, 1:400 dilution; Sigma) and against the exon 26 segment of the dystrophin protein (MANDYS-18, 1:3 dilution; a kind gift from Glenn Morris, The North East Wales Institute, Wrexham, U.K.) were used. Specific antibody binding was detected with an Alexa-coupled, goat-anti-mouse secondary antibody (1:1,000 dilution; Molecular Probes). The number of dystrophin-positive fibers in a given muscle was determined in the serial section containing the greatest number of fibers. In some of the recent studies, we used a modification of a technique recently reported to reduce the interstitial staining with mouse mAbs in mdx muscle sections (21). The modification involved the dilution of the papain digest 1:10 before applying to the sections during the blocking step.
Immunoprecipitation and Immunoblot Analysis.
Immunoprecipitation and immunoblot procedures were as described (22). For dystrophin immunoprecipitation, equal amounts of protein (6 mg) from precleared extract were immunoprecipitated by using the MANDYS-8 antidystrophin antibody (1:100) for 3 h on ice, followed by protein G-agarose for 1 h. For detection of dystrophin, blots were probed with mouse mAbs to dystrophin (MANDYS-8, 1:400 dilution, or MANDYS-18, 1:100 dilution) followed by a horseradish peroxidase-coupled sheep-anti-mouse secondary antibody. Specific antibody binding was detected by an enhanced chemiluminescence system (Amersham Pharmacia).
Detection of Gene Conversion by mdx-Amplification-Resistant Mutation System (ARMS) Assay.
The technique of mdx-ARMS assay (23) was used with reverse transcriptase–PCR (RT-PCR) to test for conversion from the mdx to the wild-type (C57) dystrophin sequence. From each muscle, 5 μg of RNA was treated with 1 unit of DNase 1 (GIBCO/BRL) at room temperature to remove contaminating genomic DNA. First-strand cDNA was then synthesized by using 5 μg of DNase-treated RNA, oligo(dT), and avian myoblastosis virus (AMV) reverse transcriptase (GIBCO/BRL) at 42°C for 50 min. The product of this reaction was amplified by PCR using primers described below and AmpliTag polymerase (Perkin–Elmer). The 5′ primer (5′-GACACTTTACCACCAATGCG-3′) was designed to be complementary to a sequence located within exon 22 of the dystrophin message and was thus common to both the C57 and mdx sequences. The 3′ primers that were used to distinguish between the C57 and the mdx sequences were designated “C” for the C57-specific 3′ primer (5′-CTCAGATAGTTGAAGCCATTTTG-3′) and “M” for the mdx-specific 3′ primer (5′-CTCAGATAGTTGAAGCCATTTTA-3′). The DNA products of the PCR reactions were resolved on 1% agarose gels.
Results
Chimeraplast Design.
The primary sequence of the chimeraplast, termed MDX1, designed to correct the point mutation in the mdx dystrophin gene is shown in Fig. 1. Also shown is the presumed secondary hairpin loop structure and the homologous pairing/gene correction process that is postulated to occur in the endogenous dystrophin gene. Two chimeraplasts were used as controls with identical results—one has a sequence homologous to a region of the dog dystrophin gene (a 28-bp region spanning intron 6 and exon 7); the other is the chimeraplast used in the correction of the sickle cell mutation in the β-globin gene (designated SC1; ref. 6). The flanking sequences for both were the same as the flanking sequences in MDX1.
Figure 1.

Dystrophin and chimeraplast sequences. (A) The sequences of the dystrophin genes (nucleotide position 3164–3208) from normal C57 mice (Upper) and mdx mice (Lower) are shown. The nucleotides underlined in the normal sequence correspond to the sequence used to design the MDX1 chimeraplast. The mdx point mutation (C→T) is underlined. (B) The linear sequence of the MDX1 chimeraplast (uppercase, DNA residues; lowercase, 2′-O-methyl RNA residues) is shown (Upper). The underlined bases correspond to the sequence of the normal dystrophin gene underlined in A, and the “overlined” base (“G”) indicates the position of the base pair that is mutated in the mdx mouse. (Lower) The presumed secondary structure of the chimeraplast. The O-methylated RNA residues, poly(T) hairpin loops, and a 3′ G:C clamp provide chemical and thermal stability as well as resistance to helicases and RNA- and DNA-nucleases. (C) The postulated homologous pairing by which chimeraplasts are presumed to effect base pair alterations. The pairings between the chimeraplast residues and the endogenous gene nucleotides are shown by vertical lines; no pairing occurs at the mismatch. The mismatch is recognized by endogenous DNA repair mechanisms, resulting in a “correction” of the mdx sequence.
Distribution of Injected Chimeraplasts.
To assess the distribution of chimeraplasts in the tissue after injection, we injected fluorochrome-coupled MDX1 into the tibialis anterior muscles of mdx mice. The distribution of the fluorescent label was examined in muscle sections at different times after injection and was very characteristic (Fig. 2). Labeled fibers were seen in two contiguous areas—a linear pattern defining the track of the needle and a cluster at the end of the needle track at the actual injection site. This pattern was clearly discernible 4 h after injection and persisted with little apparent change over the next 24 h. We would need to perform more sensitive anatomical analysis to determine whether the pattern of fluorescence truly represented cellular uptake as opposed to a T-tubule distribution. However, the facts that brightly stained fibers exist adjacent to unstained fibers and that the fluorescence was seen homogeneously throughout the cytoplasm in longitudinal and cross sections argue against an extracellular (e.g., T-tubule) distribution. Furthermore, by 48 h after injection, the fluorescence intensity had become localized primarily to nuclei, as has been seen in other studies (7, 9, 10, 24). These observations argue strongly in favor of the interpretation that the pattern of fluorescence seen in Fig. 2 represents cellular uptake. The fluorescent signal was barely detectable 72 h after injection. Presumably, this decline in signal represents the disappearance of the chimeraplast (by both diffusion and degradation) and provides some evidence of the stability of these molecules in the tissue.
Figure 2.
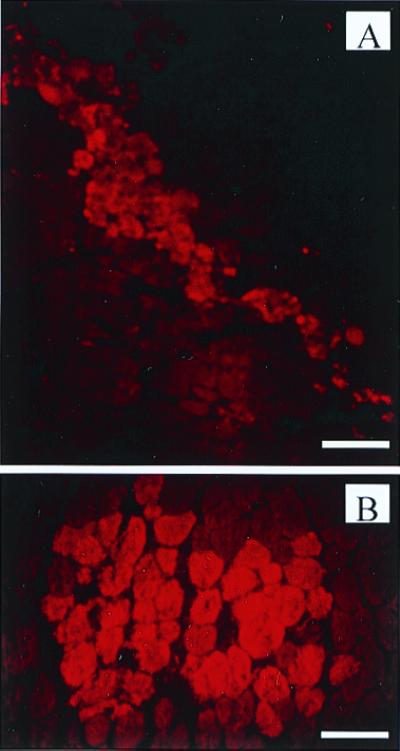
Distribution of injected chimeraplasts. Muscles injected with Cy3-coupled MDX1 were examined for fluorescent label 24 h after injection. (A) The linear pattern of fluorescent fibers representing the track of the needle through the muscle. (Scale bar represents 120 μm.) (B) A cluster of fluorescently labeled fibers at the injection site. (Scale bar represents 60 μm.)
Dystrophin Expression in MDX1-Injected Muscles.
To test the efficacy of MDX1 chimeraplasts to effect gene correction in mdx mouse muscle, we examined muscle sections for dystrophin expression at different times after MDX1 injections. Knowing the distribution of injected chimeraplasts (Fig. 2), we expected to see dystrophin expression only along the needle track and at the injection site, and this was indeed what we found. Fig. 3 shows dystrophin immunostaining around the injection site in two muscles injected with MDX1 2 wk earlier. In each muscle, dystrophin-positive fibers were detected in a pattern similar to the pattern of fluorescent label seen with the fluorochrome-labeled chimeraplast, either along a linear track or in a small cluster. No dystrophin expression was detected within 3 days after injection, but dystrophin-positive fibers were clearly present 1 wk after MDX1 injections. Although we did not do a detailed dose-response study, injections of one-fourth the amount of MDX1 chimeraplast clearly resulted in fewer dystrophin-positive fibers. When control chimeraplasts were injected, no dystrophin-positive fibers were detected [other than the occasional “revertant” fiber (see below)].
Figure 3.

Dystrophin expression in mdx muscles injected with MDX1 chimeraplast. Dystrophin-positive fibers from two MDX1-injected muscles are shown 2 wk after injection. For each muscle, the image on top shows dystrophin immunofluorescence staining (“α-dys”) and the image on bottom is the same region viewed with differential interference contrast (“DIC”) optics to show the surrounding fibers. The arrows in each pair point to the same fiber. (Scale bar represents 37.5 μm.)
To obtain a quantitative measure of the efficacy of this procedure, we set up a series of injections using mice of the same age to be injected with identical amounts of MDX1 and control chimeraplasts, to be injected on the same day, and to be analyzed at the same interval of time after the injections (2 wk). Table 1 shows the results of those analyses. For MDX1 injections, the number of dystrophin-positive fibers in the muscles ranged from a low of nine to a high of 32. These numbers represent a range of about 10–20% of the number of fibers brightly stained by fluorescent chimeraplast 24 h after injection (Fig. 2). Thus, although the absolute number of fibers that expressed dystrophin after single injections was low, the efficiency appeared to be quite high (see Discussion). Injection of control chimeraplasts again did not result in the appearance of any dystrophin-positive fibers (Table 1).
Table 1.
Efficacy of chimeraplast-mediated dystrophin expression
| Chimeraplast | No. of dystrophin-positive fibers |
|---|---|
| MDX1 | 24, 16, 32, 18, 9, 27 |
| Control | 0, 0, 0, 0, 0, 0 |
A series of 12 tibialis anterior muscles of mdx mice were injected with chimeraplasts, 6 with MDX1, and 6 with control. The groups of mice were matched for age and all injection procedures. All mice were injected at 14 days of age and sacrificed 2 wk later. The data presented are from the cross section with the maximum number of dystrophin-positive fibers from each muscle.
As a further test that the dystrophin immunoreactivity found in MDX1-injected muscle represented a correction of the point mutation and thus the expression of full-length dystrophin, we examined the muscles for dystrophin expression by immunoblot analysis. Dystrophin was not detectable by standard Western blot analysis in MDX1-injected muscles, which was not surprising given the low numbers of dystrophin-positive fibers in cryosections. Therefore, we used an antidystrophin antibody to immunoprecipitate any dystrophin that might be present, and we then subjected the immunoprecipitate to immunoblot analysis. Using this approach, we were able to detect a single band at a molecular mass corresponding to full-length dystrophin (427 kDa) in MDX1-injected muscles (Fig. 4). In muscles injected with control chimeraplasts, no such band was detected.
Figure 4.

Full-length dystrophin expression in MDX1-injected muscles. Muscles were analyzed for dystrophin expression 2 wk after injection of MDX1 or control (CON) chimeraplasts. Dystrophin was immunoprecipitated from injected muscles, and subsequent immunoblot analysis revealed a single band corresponding to the molecular mass of full-length dystrophin (427 kDa) in MDX1-injected muscles; no band was seen in muscles injected with control chimeraplasts. A nonspecific band at 115 kDa was seen in both groups of muscles (as well as in uninjected mdx muscle and in C57 muscle, data not shown). Identical results were seen in three different experiments, representing the analysis of six MDX1-injected muscles and six control chimeraplast-injected muscles.
We expected that chimeraplast-induced dystrophin expression would be stable over time because the corrected gene would be under its endogenous regulatory mechanisms. To test this directly, we injected MDX1 and control chimeraplasts into mdx mice and examined the muscles 10 wk later. Of the eight muscles injected (four with MDX1, four with control), clusters of dystrophin-positive fibers were detected only in MDX1-injected muscles. In those muscles, the number of dystrophin-positive fibers (36, 22, 23, and 11) fell into the same range as that seen 2 wk after injection (Table 1), suggesting that dystrophin expression was stable in those fibers.
Test for “Revertant” Fibers.
In mdx mouse muscle as well as in human muscle from patients with DMD, there is an increase in the appearance of rare dystrophin-positive fibers (“revertant” fibers) with age (25). In mdx mice, the molecular basis of this reversion has been postulated to be spontaneous, somatic mutations resulting in either in-frame deletions around and including exon 23 or alternative splicing reactions that would produce transcripts that excluded exon 23. This hypothesis is supported by analysis of revertant fibers with exon-specific antibodies and by nested PCR analysis of dystrophin transcripts (26, 27). The negative results with the control chimeraplasts (Fig. 3 and Table 1) argue against any nonspecific (i.e., sequence-independent) effect of the experimental procedures leading to an increase in the number of revertant fibers. Furthermore, the immunoblot results (Fig. 4) indicate that the dystrophin that is expressed is full length and not a truncated form as would be expected in revertant fibers. Still, to rule out this possibility with greater certainty, we used antibodies directed against the protein products of exons that are rarely expressed in revertant fibers (generally, exons 20–30) (26). An antibody against the protein product of exon 26 showed characteristic dystrophin staining in all C57 muscle fibers (Fig. 5A). This same antibody did not recognize the vast majority of revertant fibers (Fig. 5A), consistent with findings of previous authors that exon 26 is expressed in a small minority of revertant mdx fibers (26, 28). However, in the clusters of dystrophin-positive fibers in MDX1-injected muscles (identified with an antibody against a more distant region of the protein), the exon 26-specific antibody recognized all of the fibers (Fig. 5B). These data provide further evidence that the clusters of dystrophin-positive fibers in MDX1-injected muscles do not represent groups of revertant fibers.
Figure 5.

Dystrophin expression after MDX1 injection is not the result of increased frequency of revertant fiber formation. (Upper) The pattern of dystrophin staining in C57 muscle with the MANDYS-18 antibody (directed against the protein of exon 26) as compared with the MANDYS-8 antibody (directed against a more C-terminal region of the protein). All fibers were stained by both antibodies. (Lower) The same comparison in mdx muscle. Two revertant fibers recognized by the MANDSY-8 antibody were not stained by the MANDYS-18 antibody. (Scale bar represents 37.5 μm.) (B) Serial sections of MDX1-injected muscles were stained with the MANDYS-8 and MANDYS-18 antibody, as labeled. All of the dystrophin-positive fibers as identified by the MANDYS-8 antibody also were recognized by MANDYS-18, providing evidence that these are not revertant fibers. (Scale bar represents 37.5 μm.) To reduce background staining (see Fig. 3 for comparison), immunostaining was done by using a modification of a protocol recently reported to reduce interstitial staining in mdx muscle with mAbs (21).
Molecular Evidence of Correction of the mdx Point Mutation.
The combined immunohistochemical and immunoblot data (Figs. 3 and 4) of dystrophin expression in MDX1-injected muscles are consistent with gene correction by means of nucleotide exchange in a small number of cells. To test for molecular evidence of gene conversion, we used the mdx-ARMS assay (23) to test for what would likely be rare wild-type dystrophin sequences among abundant mdx dystrophin sequences. The PCR primers were designed to distinguish with exquisite specificity, as originally described (23), between the two sequences. As shown in Fig. 6A, the C57 primers amplified a PCR product only in C57 muscle, whereas the mdx primers amplified a product only in mdx muscle. When we used both sets of primers in parallel in MDX1-injected mdx muscle, not only was a major product amplified with the mdx primers as expected, but a minor product also was amplified with the C57 primers (Fig. 6B). In control chimeraplast-injected muscles, a PCR product was detected only with the mdx primers. These results provide molecular evidence of chimeraplast-mediated gene conversion that is specific to the targeting (i.e., MDX1) chimeraplast.
Figure 6.

Evidence of gene conversion by MDX1 chimeraplasts. (A) Total RNA was isolated from C57 or mdx muscle and analyzed for dystrophin transcripts using RT-PCR and the mdx-ARMS assay. Two different 3′ primers, designated “C” and “M,” were designed to amplify only the wild-type (C57) sequence or only the mdx sequence, respectively. The exquisite specificity is shown by the presence of a 191-bp amplification product in C57 muscle only when the “C” primer was used and in mdx muscle only when the “M” primer was used. (B) Total RNA was isolated from mdx muscle injected with either MDX1 or control chimeraplasts. We tested for the presence of transcripts containing the wild-type dystrophin sequence (evidence of nucleotide exchange) using the same procedure as in A. A PCR amplification product using the “C” primer was seen in mdx muscles injected with the MDX1 chimeraplast, indicating the presence of wild-type transcripts, but not in mdx muscles injected with control chimeraplast. (C) Controls for potential PCR artifacts. (Left) RNA was prepared from uninjected mdx muscle, and different amounts of MDX1 were added to each sample, as indicated. Each sample was then subjected to RT-PCR using the “C57” primers to test whether the presence of MDX1 itself could result in artifactual amplification of a PCR product. No amplification product was detected. When the “mdx” primers were used, an amplification product of 191-bp was detected, as in B, reflecting appropriate amplification from the mdx transcripts. (Right) One microgram of MDX1 was either loaded directly onto the gel (left lane, “−PCR”) or was added to PCR solutions with either the “C57” or “mdx” primers (“+PCR” lanes). After the PCR, approximately 20% of each reaction volume (thus approximately 0.2 μg of added chimeraplast) was loaded onto the gel. The RNA/DNA oligonucleotide runs at the position indicated, and no PCR amplification product was detected with either set of primers.
The question has been raised as to whether evidence of nucleotide exchange in other studies could have arisen from PCR artifacts generated from chimeraplasts themselves (29). In some cases, independent methods to confirm the gene correction, such as Southern blot analysis, have been used (6, 11). We designed our experiments to avoid possible chimeraplast-induced PCR artifacts by examining muscles only 2 wk after chimeraplast injection. Previous studies have suggested that chimeraplasts are rapidly degraded within 48–72 h in vivo (11) and in vitro (29), which is consistent with our observations with fluorescently labeled chimeraplasts injected into muscle as described above. Thus, 2 wk after injection of chimeraplasts into muscle, the likelihood of residual chimeraplast accounting for PCR artifacts is remote. However, to test this directly, we tested whether MDX1 could themselves lead to PCR amplification products, and we “spiked” RNA isolated from mdx muscle with different amounts of MDX1 and carried out RT-PCR with the C57 primers. No amplification products were detected in either type of experiment (Fig. 6C).
Discussion
These results demonstrate that chimeraplast-mediated gene conversion can correct the mdx point mutation and lead to the expression of dystrophin in muscle fibers of mdx mice. Our finding of wild-type dystrophin transcripts in MDX1-injected muscle is direct evidence of gene conversion and is consistent with the evidence that the dystrophin that is expressed is full length by immunoblot analysis and includes exons around the position of the mdx point mutation. Chimeraplast-induced single base pair changes in genomic DNA have been demonstrated by direct sequencing of targeted genes in different cell types (6–10, 12, 13). This process is highly sequence-specific and does not occur with oligonucleotides of other configurations, even all DNA oligonucleotides of the same sequence as a corresponding RNA/DNA chimeric oligonucleotide (8, 19). The mechanism of gene correction seems to involve endogenous mismatch repair mechanisms (10, 19). Cole-Strauss et al. (16) recently reported chimeraplast-mediated gene repair in mammalian cell-free extracts. Their data indicate that gene conversion depends not only on accurately designed chimeraplasts, but also on the presence of functional MSH2 protein, a protein that is critical to mismatch repair (30). Variability of endogenous mismatch repair activities among different cell types may explain some of the variability of the efficiency of gene conversion found by investigators using cells from different tissues (6–8) or even using different cell lines of the same lineage (17).
In our studies, the efficacy of chimeraplast-mediated gene conversion is clearly limited in that dystrophin was expressed in a small minority (around 1–2% at best) of the fibers in the tibialis anterior muscle after a single injection of the targeting chimeraplast. The expression of dystrophin in mature fibers within 2 wk of injection of MDX1 suggests that chimeraplast-induced gene correction occurs in postmitotic cells, as has been found by other investigators (7, 9). Although the generation of dystrophin-positive fibers reported here was limited to a small percentage of the fibers in the muscle, the efficacy could easily have been increased simply by performing multiple injections or perhaps by increasing the concentration of chimeraplast. Other investigators have shown a gene conversion efficacy that depends on chimeraplast concentration (7, 9). A major limitation of the ability of injected chimeraplast to effect gene conversion in muscle appears to be the restricted uptake of chimeraplasts into fibers (Fig. 2). We do not know why the distribution is so restricted, but it may be that some minor injury during the injection procedure facilitates chimeraplast uptake in fibers only along the needle track and at the injection site. The cellular mechanisms that determine whether or not a fiber will take up extracellular oligonucleotides require further investigation, as they are obviously critical to the success of this technology. It is likely that these processes relate to mechanisms that have been postulated to underlie the unique ability of skeletal muscle fibers to take up “naked” DNA (31).
Of the limited number of fibers that stained brightly after injection of fluorescently labeled chimeraplast (Fig. 2), a remarkably high percentage (in the range of 10–20%) were found to express dystrophin 2 wk later. This suggests that the gene conversion efficiency is extremely high and is consistent with findings by other investigators using different methods to calculate efficiency (6–9, 24). All of these studies suggest a gene conversion frequency around 10%, with variability over a linear range from somewhat less than 5% to more than 40%. This variability over this linear range is insignificant compared with the many orders of magnitude over which gene conversion using chimeraplast technology is more efficient than homologous recombination (32), another technique of gene conversion involving homologous pairing. Clearly, the efficiency with which chimeraplasts appear to be able to induce gene conversion is very promising for gene therapy.
The application of this technology as a therapeutic approach to human muscular dystrophies requires both theoretical and technical considerations. The exact percentage of patients with DMD with point mutations that would be amenable to chimeraplast-mediated correction is unknown, but is about 20% at most (33). Most patients with DMD have large deletions in the dystrophin gene, and the chimeraplast technology is, at this time, developed for single base alterations only. However, there is a potential application of chimeraplast technology even for large deletions because it is the disruption of the reading frame by deletions that leads to the severe clinical phenotype (34). Deletions that preserve the reading frame result in a milder condition, Becker muscular dystrophy. Theoretically, chimeraplast-mediated base conversion could alter splice sites within the gene to produce truncated messages but with the reading frame restored. Alternatively, although chimeraplasts have been used mostly to induce single base pair changes, they also have been shown either to insert or to delete single bases in genomic sequences (11, 13, 16). Base insertion or base deletion could restore the reading frame in a dystrophin gene rendered nonsensical by a deletion. In either case, the resulting gene would produce a partially functional, truncated protein, thus converting a severe DMD phenotype into a milder Becker phenotype.
Technically, the challenges are more daunting. First, the low efficacy of gene conversion would have to be substantially improved, potentially involving methods to facilitate chimeraplast uptake into muscle cells, enhance transport of chimeraplasts to the nucleus, and enhance the efficiency of chimeraplast-mediated gene “repair.” Second, as with viral-mediated gene therapy, a possible immune response against the “novel” protein would be a consideration, and an efficient and effective method of delivery of the vector to a tissue as massive and distributed as skeletal muscle would have to be developed. Clearly, major advances in systemic delivery will be necessary. In this regard, it is promising that Kren et al. (9) were able to detect high rates of gene conversion in liver genes after i.v. chimeraplast injection. Although those chimeraplasts were delivered complexed to lactosylated polyethylenimine to target their uptake into liver, the fact that the gene conversion was effective after i.v. chimeraplast injection is encouraging.
Despite these limitations, chimeraplast-mediated gene therapy has several advantages over gene therapy using viral vectors. With chimeraplast-mediated therapy, the targeted gene, once corrected, is under its endogenous regulatory mechanisms. Thus, there would be no untoward effects of correcting the targeted gene in cells in which it is not normally expressed. In addition, as supported by our data and those of others (10, 11), expression of the protein product is likely to be stable and not subject to transcriptional silencing that may occur with certain viral vectors. Furthermore, chimeraplast-mediated gene conversion would avoid any immunologic response against viral antigens or the possibility of mutagenesis associated with the insertion of exogenous DNA sequences into the genome. The prospect of the use of chimeraplast technology for the treatment of muscular dystrophies faces many hurdles, but is promising as an approach to therapeutics in these and other genetic disorders of muscle.
Acknowledgments
We thank Dr. Glenn Morris (The North East Wales Institute, Wrexham, U.K.) for providing us with the MANDYS-18 antibody. We also acknowledge Dr. Terry Partridge of the Medical Research Council Clinical Research Centre, Hammersmith Hospital, London, for useful discussions concerning revertant fibers. This work was supported in part by a grant from Kimeragen (Newtown, PA).
Abbreviations
- DMD
Duchenne muscular dystrophy
- ARMS
amplification-resistant mutation system
- RT-PCR
reverse transcriptase–PCR
References
- 1.Emery A E H. In: Neuromuscular Disorders: Clinical and Molecular Genetics. Emery A E H, editor. New York: Wiley; 1998. pp. 1–20. [Google Scholar]
- 2.Hoffman E P, Brown R H, Jr, Kunkel L M. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 3.McArdle A, Edwards R H, Jackson M J. Neuromuscul Disord. 1995;5:445–456. doi: 10.1016/0960-8966(95)00001-4. [DOI] [PubMed] [Google Scholar]
- 4.Tidball J G, Albrecht D E, Lokensgard B E, Spencer M J. J Cell Sci. 1995;108:2197–2204. doi: 10.1242/jcs.108.6.2197. [DOI] [PubMed] [Google Scholar]
- 5.Rando T A, Disatnik M H, Yu Y, Franco A. Neuromuscul Disord. 1998;8:14–21. doi: 10.1016/s0960-8966(97)00124-7. [DOI] [PubMed] [Google Scholar]
- 6.Cole-Strauss A, Yoon K, Xiang Y, Byrne B C, Rice M C, Gryn J, Holloman W K, Kmiec E B. Science. 1996;273:1386–1389. doi: 10.1126/science.273.5280.1386. [DOI] [PubMed] [Google Scholar]
- 7.Kren B T, Cole-Strauss A, Kmiec E B, Steer C J. Hepatology. 1997;25:1462–1468. doi: 10.1002/hep.510250626. [DOI] [PubMed] [Google Scholar]
- 8.Xiang Y, Cole-Strauss A, Yoon K, Gryn J, Kmiec E B. J Mol Med. 1997;75:829–835. doi: 10.1007/s001090050172. [DOI] [PubMed] [Google Scholar]
- 9.Kren B T, Bandyopadhyay P, Steer C J. Nat Med. 1998;4:285–290. doi: 10.1038/nm0398-285. [DOI] [PubMed] [Google Scholar]
- 10.Alexeev V, Yoon K. Nat Biotechnol. 1998;16:1343–1346. doi: 10.1038/4322. [DOI] [PubMed] [Google Scholar]
- 11.Kren B T, Parashar B, Bandyopadhyay P, Chowdhury N R, Chowdhury J R, Steer C J. Proc Natl Acad Sci USA. 1999;96:10349–10354. doi: 10.1073/pnas.96.18.10349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu T, Peterson D J, Tagliani L, St. Clair G, Baszczynski C L, Bowen B. Proc Natl Acad Sci USA. 1999;96:8768–8773. doi: 10.1073/pnas.96.15.8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beetham P R, Kipp P B, Sawycky X L, Arntzen C J, May G D. Proc Natl Acad Sci USA. 1999;96:8774–8778. doi: 10.1073/pnas.96.15.8774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang W H, Modrich P. J Biol Chem. 1993;268:11838–11844. [PubMed] [Google Scholar]
- 15.Modrich P. Science. 1994;266:1959–1960. doi: 10.1126/science.7801122. [DOI] [PubMed] [Google Scholar]
- 16.Cole-Strauss A, Gamper H, Holloman W K, Munoz M, Cheng N, Kmiec E B. Nucleic Acids Res. 1999;27:1323–1330. doi: 10.1093/nar/27.5.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santana E, Peritz A E, Iyer S, Uitto J, Yoon K. J Invest Dermatol. 1998;111:1172–1177. doi: 10.1046/j.1523-1747.1998.00403.x. [DOI] [PubMed] [Google Scholar]
- 18.Sicinski P, Geng Y, Ryder-Cook A S, Barnard E A, Darlison M G, Barnard P J. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- 19.Yoon K, Cole-Strauss A, Kmiec E B. Proc Natl Acad Sci USA. 1996;93:2071–2076. doi: 10.1073/pnas.93.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Disatnik M H, Dhawan J, Yu Y, Beal M F, Whirl M M, Franco A A, Rando T A. J Neurol Sci. 1998;161:77–84. doi: 10.1016/s0022-510x(98)00258-5. [DOI] [PubMed] [Google Scholar]
- 21.Lu Q L, Partridge T A. J Histochem Cytochem. 1998;46:977–984. doi: 10.1177/002215549804600813. [DOI] [PubMed] [Google Scholar]
- 22.Disatnik M H, Rando T A. J Biol Chem. 1999;274:32486–32492. doi: 10.1074/jbc.274.45.32486. [DOI] [PubMed] [Google Scholar]
- 23.Amalfitano A, Chamberlain J S. Muscle Nerve. 1996;19:1549–1553. doi: 10.1002/(SICI)1097-4598(199612)19:12<1549::AID-MUS4>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 24.Bandyopadhyay P, Ma X, Linehan-Stieers C, Kren B T, Steer C J. J Biol Chem. 1999;274:10163–10172. doi: 10.1074/jbc.274.15.10163. [DOI] [PubMed] [Google Scholar]
- 25.Hoffman E P, Morgan J E, Watkins S C, Partridge T A. J Neurol Sci. 1990;99:9–25. doi: 10.1016/0022-510x(90)90195-s. [DOI] [PubMed] [Google Scholar]
- 26.Wilton S D, Dye D E, Laing N G. Muscle Nerve. 1997;20:728–734. doi: 10.1002/(sici)1097-4598(199706)20:6<728::aid-mus10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 27.Thanh L T, Nguyen T M, Helliwell T R, Morris G E. Am J Hum Genet. 1995;56:725–731. [PMC free article] [PubMed] [Google Scholar]
- 28.Lu Q L, Morris G E, Wilton S D, Strong P, Partridge T A. Muscle Nerve. 1998;21:S192. [Google Scholar]
- 29.Zhang Z, Eriksson M, Falk G, Graff C, Presnell S C, Read M S, Nichols T C, Blomback M, Anvret M. Antisense Nucleic Acid Drug Dev. 1998;8:531–536. doi: 10.1089/oli.1.1998.8.531. [DOI] [PubMed] [Google Scholar]
- 30.Umar A, Boyer J C, Thomas D C, Nguyen D C, Risinger J I, Boyd J, Ionov Y, Perucho M, Kunkel T A. J Biol Chem. 1994;269:14367–14370. [PubMed] [Google Scholar]
- 31.Wolff J A, Dowty M E, Jiao S, Repetto G, Berg R K, Ludtke J J, Williams P, Slautterback D B. J Cell Sci. 1992;103:1249–1259. doi: 10.1242/jcs.103.4.1249. [DOI] [PubMed] [Google Scholar]
- 32.Capecchi M R. Science. 1989;244:1288–1292. doi: 10.1126/science.2660260. [DOI] [PubMed] [Google Scholar]
- 33.Bakker E, Van Ommen G. In: Neuromuscular Disorders: Clinical and Molecular Genetics. Emery A E H, editor. New York: Wiley; 1998. pp. 59–85. [Google Scholar]
- 34.Monaco A P, Bertelson C J, Liechti-Gallati S, Moser H, Kunkel L M. Genomics. 1988;2:90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]