

Abstract
Efficient and mild reaction conditions were developed for the catalytic carbonylation of fluorinated epoxides to their corresponding β-lactones. Six new lactones with fluorinated side chains were prepared in high isolated yields. These lactones were polymerized to form a series of new poly(β-hydroxyalkanoate)s with fluorinated side chains, and their properties were examined with respect to their hydrocarbon analogs. Finally, copolymerizations were performed with fluorinated lactones and β-butyrolactone, which resulted in tapered copolymers rather than the expected random copolymers.
1. Introduction
Since the serendipitous discovery of poly(tetrafluoroethylene) by DuPont in 1938, fluorinated polymers have become widespread in specialty and commodity materials.1 The incorporation of fluorinated monomers into polymers can produce materials with distinctive properties relative to their non-fluorinated analogs. In particular, fluorinated polymers often display low coefficients of friction, good chemical resistance, and low surface energies.2 These properties have made fluorinated polymers useful in applications ranging from membranes and coatings1 to medical devices.3
Poly(β-hydroxyalkanoate)s (PHAs) are a class of aliphatic polyesters that are naturally produced by bacteria as a form of energy storage during nutrient-rich periods.4 Due to their biodegradability, and therefore their potential use in biomedical applications, they have received increasing attention in recent years.4,5 The mechanical properties of PHAs are dependent on the monomer composition,6 so polymers with diverse material properties and degradation rates can be produced by incorporating different monomers.7 PHAs containing a wide variety of functional side groups have been prepared by bacterial fermentation, including the incorporation of fluorinated side chains.8 A high content of halogenated side groups has been shown to increase the melting temperature (Tm) and glass transition temperature (Tg) of the PHAs. Unfortunately, the synthesis of PHAs with high degrees of fluorination has been difficult to achieve using fermentation methods,8a,c and attempts to produce highly fluorinated homopolymers by these methods have not been successful.
Recent work in our laboratory has focused on the synthesis of β-lactones through the catalytic carbonylation of epoxides9 and the ring-opening polymerization of these lactones to produce PHAs.10 The availability of epoxides in both racemic and enantiomerically pure11 form has enabled the synthesis of a wide range of β-lactones.9e The ring-opening polymerization of these lactones to produce PHAs creates the opportunity for the synthesis of a number of different polymer microstructures. Herein, we report on the highly efficient carbonylation of epoxides with fluorinated side chains to their respective β-lactones, and the subsequent homo-and co-polymerization of these β-lactones to form fluorinated PHAs.
2. Results and Discussion
2.1 Optimization of epoxide carbonylation
Previous work in our group has identified a number of catalysts that are active for the carbonylation of a functionally diverse array of epoxides.9 However, the carbonylation of fluorinated epoxides has never been reported. We recently discovered the catalyst [(salph)Cr(THF)2]+[Co(CO)4]−) (1, Figure 1, salph = N,N′-bis(3,5-di-tert-butylsalicylidene) 1,2-phenylene-diamine, THF = tetrahydrofuran) that is active for epoxide carbonylation under mild reaction conditions (room temperature and 1 atm CO).9f Additionally, with this catalyst epichlorohydrin, a traditionally difficult substrate, was carbonylated to 4-chloromethylpropiolactone with nearly the same rate and yield as the analogous non-chlorinated 1,2-epoxybutane.9f This led us to explore other halogenated epoxides as carbonylation substrates.
Figure 1.

Structure of epoxide carbonylation catalyst 1.
Subsequent exploration into the substrate scope revealed that epoxides with fluorinated side chains were carbonylated effectively to their respective β-lactones. Though 3,3,3-trifluoro-1,2-epoxypropane suffered from incomplete conversion, epoxides with a methylene spacer between the epoxide and fluorinated side chain were efficiently converted to β-lactones (Figure 2). We were thus intrigued by the possible materials that could be prepared from these fluorinated lactones.
Figure 2.

Efficient carbonylation of epoxides with a methylene group linking the epoxide and fluorinated side chain.
Upon identification of this new substrate class, the reaction conditions were optimized for efficient carbonylation. Our previous work has demonstrated that the choice of reaction solvent is extremely important for epoxide carbonylation efficiency,12 so initial studies focused on identifying the optimal solvent. Table 1 shows the results of the solvent screening. In general, non-coordinating solvents (entries 1 and 2) generally decreased the rate of reaction compared to ether solvents (entries 3–6). However, contrary to other carbonylation catalysts in which the rate of carbonylation was accelerated by THF,12 the strongly donating solvent THF (entry 4) severely inhibited conversion. We believe that solvents such as THF bind too strongly to the Lewis acid and inhibit epoxide coordination. Thus, moderately donating ether solvents such as 1,2-dimethoxyethane (DME, entry 3), 2,5-dimethyltetrahydrofuran (entry 5), and 1,4-dioxane (entry 6) were favorable for the carbonylation of fluorinated epoxides. Because of its high volatility and low cost, DME was selected as the optimal reaction solvent.
Table 1.
Effect of reaction solvent on epoxide carbonylation.
 | ||
|---|---|---|
| Entry | Solvent | Conversion (%)a |
| 1 | Toluene | 36 |
| 2 | 1,2-Difluorobenzene | 18 |
| 3 | 1,2-Dimethoxyethane | 46 |
| 4 | Tetrahydrofuran | 9 |
| 5 | 2,5-DMTHFb | 47 |
| 6 | 1,4-Dioxane | 43 |
Determined by 1H NMR spectroscopy.
2,5-DMTHF = 2,5-dimethyltetrahydrofuran (cis/trans mixture).
The effect of CO pressure on the carbonylation reaction was also examined. Catalyst 1 has been shown to be active and selective for β-lactone formation at CO pressures as low as 1 atm.9f Table 2 shows the effect of lowering the CO pressure on the reaction rate. Although no attenuation of rate was observed at CO pressures as low as 100 psi, dropping the pressure to 1 atm (entry 4) resulted in incomplete conversion to β-lactone. A pressure of 100 psi was chosen to facilitate efficient carbonylation without the requirement of stainless steel, high pressure reactors.
Table 2.
Effect of CO pressure on epoxide carbonylation.
 | ||
|---|---|---|
| Entry | CO Pressure | Conversion (%)a |
| 1 | 800 psi | >99 |
| 2 | 400 psi | >99 |
| 3 | 100 psi | >99 |
| 4 | 1 atm (14.7 psi) | 44 |
Determined by 1H NMR spectroscopy.
2.2 Carbonylation of fluorinated epoxides
A variety of epoxides with fluorinated side chains are either commercially available or accessible through the straightforward epoxidation of the corresponding alkenes. Using the optimized carbonylation conditions, a range of epoxides with different fluorinated groups were carbonylated to their respective β-lactones. Both branched and straight-chain perfluoroalkyl epoxides (Table 3, entries 1, 3–6) were completely converted to β-lactone within 6 h at room temperature. Other functionalities, such as ether (entry 2) and perfluoroaryl (entry 7) groups were also tolerated under the optimized reaction conditions. Analytically pure β-lactone products were readily isolated in high yields (see Experimental Section).
Table 3.
Carbonylation of fluorinated epoxides using 1 at 100 psi CO.
 | |||||
|---|---|---|---|---|---|
| Entry | Epoxide | β-Lactone | Conv. (% Yield)a | ||
| 1 | 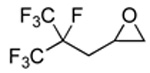 |
2a | 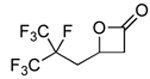 |
3a | 99 (86) |
| 2 | 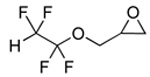 |
2b |  |
3b | 99 (79) |
| 3 |  |
2c |  |
3c | 99 (85) |
| 4 |  |
2d |  |
3d | 99 (88) |
| 5 |  |
2e |  |
3e | 99 (91) |
| 6 | 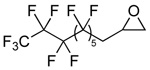 |
2f |  |
3f | 99 (71) |
| 7 | 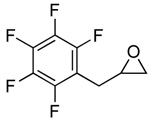 |
2g | 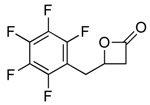 |
3g | 99 (82) |
β-Lactone was the sole product as determined by 1H NMR spectroscopy; % yield of isolated, analytically pure material.
Importantly, optically pure epoxides can be carbonylated to their respective β-lactones with retention of absolute stereochemistry.12a Optically pure epoxide 2d was prepared using the well-established kinetic resolution methodology developed by Jacobsen and coworkers.11c This epoxide was then subjected to the optimized carbonylation conditions to produce 3d in quantitative conversion.
2.3 Polymerization of fluorinated lactones
A number of catalyst systems have been developed for the polymerization of β-lactones to PHAs.10,13 However, the β-diiminate (BDI) Zn alkoxide catalysts developed in our laboratory are among the most active and selective catalysts for the synthesis of high molecular weight PHAs.10 To our satisfaction, our most active catalyst for β-butyrolactone polymerization, [(BDI)ZnOiPr] (4), was also active for the polymerization of fluorinated lactones (Figure 3). During the polymerization at 50 °C, the homogeneous reaction mixture became cloudy with precipitated polymer, and in less than six hours, all monomer had been consumed.
Figure 3.

Polymerization of 3c to produce PHA using a β-diiminate zinc catalyst.
After optimization, most of the polymerizations achieved high conversions within 6 h at 50 °C, though the polymerization of 3c and 3d (entries 3 and 4) only required 2 h for complete conversion of monomer. During the polymerization of every monomer except 3g, polymer precipitated from solution near the end of the reaction. The polymerization of 3f was not practical due to the extremely low solubility of the lactone and polymer in nearly every solvent.14
The atactic polymers (Table 4, entries 1–3, 5) with perfluoroalkyl side chains were amorphous as determined by DSC analysis, though they were stable to >250 °C by TGA. The Tg values of the perfluoroalkyl-substituted PHAs appear to increase with fluorine content and side chain branching. Overall, the fluorinated PHAs display higher Tg values than their hydrocarbon analogs. For example, naturally produced poly(β-hydroxyoctanoate) displays a Tg of −35 °C,6 while 5c, the fluorocarbon analog, has a significantly higher Tg of 4 °C. The only polymer that displayed a melting point was 5d (entry 4). Analysis by 13C NMR spectroscopy showed that 5d is isotactic, an observation consistent with the polymerization of other optically pure lactones by 4.10 The 13C NMR spectra of the atactic polymers contain two peaks in the carbonyl region, assigned to the m and r diads; 5d shows just one sharp peak which corresponds to the m diad. This observation is further supported by the detection of a Tm, as crystallinity has been shown to be a function of tacticity for PHAs.15
Table 4.
Homopolymerization of fluorinated lactones using 4.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Lactone | RF | t (h) | Conv. (%)b | Mn (g/mol)c | Mw/Mnc | Tg (°C)d | Tm (°C)d |
| 1 | 3a |  |
6 | >95 | 17,000 | 1.13 | 14 | nde |
| 2 | 3b |  |
6 | >95 | 20,000 | 1.14 | −12 | nde |
| 3 | 3c |  |
2 | >95 | 18,000 | 1.16 | 4 | nde |
| 4 | 3d |  |
2 | >95 | 16,000 | 1.11 | nde | 82 |
| 5 | 3e |  |
6 | >95 | 8,000 | 1.21 | 24 | nde |
| 6 | 3g |  |
6 | >95 | 23,000f | 1.16 | 53 | nde |
Polymerization conditions: [lactone]/[4] = 100, [lactone] = 1.0 M in toluene, Trxn = 50 °C.
Determined by 1H NMR spectroscopy of crude reaction mixture.
Determined using gel-permeation chromatography in THF; calibrated with polystyrene standards.
Determined by differential scanning calorimetry.
nd = not detected.
Mn = 34,000 determined by end group analysis.
2.4 Copolymerization of fluorinated lactones with BBL
PHA copolymers have been shown to display drastically different properties than their respective homopolymers;6 thus, we were interested in examining the copolymerization of fluorinated lactones with β-butyrolactone. Our initial attempt on an NMR-scale provided a surprising result. Unlike the copolymerization of hydrocarbon-substituted β-lactones where both monomers are consumed at similar rates, the rate of fluorinated lactone polymerization was noticeably faster than BBL incorporation (Figure 4). This difference in polymerization rates produces tapered copolymers rather than random copolymers. Furthermore, the high reactivity of the fluorinated monomers enables production of PHAs with short, fluorinated end blocks, which may have similar surface properties to the fluorinated homopolymers without requiring high ratios of fluorinated monomer.
Figure 4.

Copolymerization of 3a and BBL. Prior to polymerization (a), the two lactone methine peaks were observed in a 1:1 ratio. Within 2 h at 50 °C (b), nearly all of 3a had been polymerized, and after 4 h (c) the polymerization of BBL was proceeding. After 24 h (d), both lactones had been consumed and the polymer methine peaks were observed in a 1:1 ratio.
On a larger scale, the copolymerization of 3g with BBL also produced tapered copolymers (Figure 5).16 The copolymer was determined to be monomodal by GPC analysis, consistent with the incorporation of both monomers into one polymer chain rather than the formation of two homopolymers.
Figure 5.

Copolymerization of 3g and BBL using 4 to produce tapered copolymers.
3. Conclusions
In this work, we efficiently prepared a new class of β-lactones with fluorinated side chains. These lactones were polymerized to form fluorinated PHAs which are inaccessible through biological fermentation routes. Copolymerization of the highly fluorinated β-lactones with BBL formed tapered rather than random copolymers. Further study of the synthesis and properties of these fluorinated copolymers will be the subject of future reports.
4. Experimental
4.1 General Comments
All manipulations of air- and/or water-sensitive compounds were carried out using standard Schlenk line techniques or in an MBraun Unilab drybox under an atmosphere of dry nitrogen, and all solvents were dried according to standard procedures. NMR spectra were recorded using Varian Mercury 300 MHz or Inova 500 MHz spectrometers (1H NMR, 300 MHz; 13C NMR, 125 MHz; 19F NMR, 470 MHz) and referenced against residual solvent shifts for 1H and 13C NMR and hexafluorobenzene for 19F NMR spectra. Mass spectra were acquired using a JEOL GCMate II mass spectrometer operating at 3000 resolving power for high-resolution measurements in positive ion mode and an electron ionization potential of 70 eV. Samples were introduced via a GC inlet using an Agilent HP 6890N GC equipped with a 30 m (0.25 µm i.d.) HP-5ms capillary GC column. The carrier gas was helium with a flow rate of 1 mL/min. The lactones were generally not stable to the GC or MS, and all except 3g underwent decarboxylation to the alkene. Therefore, exact masses of the alkenes were reported, and NMR spectra showing the product lactones can be found in the Supporting Information. Optical rotations were measured on a Perkin-Elmer 241 digital polarimeter and are reported in the following format: [α]TD r (c, solvent), where T = temperature in °C, D refers to the sodium D line (589 nm), r is the measured rotation, and c is the concentration in g/dL. IR spectra of lactones (neat, NaCl plate) were measured on a Mattson RS-10500 Research Series FTIR. Gel permeation chromatography (GCP) analyses were carried out using a Waters instrument, (M515 pump, 717+ Autosampler) equipped with a Waters UV486 and Waters 2410 differential refractive index detectors, and three 5 µm PSS SDV columns (Polymer Standards Service; 50 Å, 500 Å, and Linear M porosities) in series. The GPC columns were eluted with THF at 40 °C at 1 mL/min and were calibrated using 20 monodisperse polystyrene standards. Differential scanning calorimetry was performed on a TA Instruments Q1000 instrument equipped with a LNCS and automated sampler. Typical DSC experiments were made in crimped aluminum pans under nitrogen with a heating rate of 10 °C/min from −100 °C to 230 °C. All epoxides were purchased from Oakwood or Aldrich chemicals, except 2g which was prepared according to an established procedure. Catalysts 19f and 410 were prepared according to literature procedures, though 1 is commercially available from Aldrich (cas # 909553-60-2). All epoxides and liquid lactones were dried for one week over CaH2 and degassed by three freeze-pump-thaw cycles; solid lactones were dried under vacuum following sublimation.
4.2 General procedure for the carbonylation of fluorinated epoxides
This procedure is analogous to the previously published low pressure carbonylation of epoxides.9f In a drybox, 1 (110 mg, 0.20 mmol) was dissolved in DME (3.0 mL). The solution was transferred to an oven-dried Fisher Porter bottle with a magnetic stir bar and the reactor was sealed. Epoxide (6.0 mmol) and DME (3.0 mL) were drawn into a Gastight® syringe, the needle of which was inserted into a septum to avoid air contamination upon removal from the glove box. The reactor and syringe were removed from the glove box and the reactor was immediately pressured with CO (20 psi) and cooled to 0 °C. After 3 minutes, the epoxide solution was added to the stirring catalyst solution under a light flow of CO. The reactor was then pressured with CO (100 psi) and warmed to room temperature. After six hours, the reactor was carefully vented in a fume hood and the lactone was isolated as described below.
4.2.1 4-(2,3,3,3-Tetrafluoro-2-(trifluoromethyl)-propyl)-2-propiolactone (3a)
The general procedure was followed with epoxide 2a (1.4 g, 6.0 mmol). The product was purified by distillation at 60 °C under vacuum and 3a was recovered as a colorless oil (1.3 g, 86%). 1H NMR (δ, CDCl3, 300 MHz) 2.45–2.63 (m, 1H), 2.72–2.90 (m, 1H), 3.29 (dd, 3J = 4.2 Hz, 2J = 16.7 Hz, 1H), 3.73 (dd, 3J = 5.8 Hz, 2J = 16.7 Hz, 1H), 4.83 (m, 1H); 13C NMR (δ, CDCl3, 125 MHz) 33.8 (d, 2JCF = 19.7 Hz), 44.7, 64.3, 90.4 (dsept, 2JCF = 32.4 Hz, 1JCF = 204.4 Hz), 120.5 (qd, 2JCF = 26.8 Hz, 1JCF = 284.7 Hz), 166.2; 19F NMR (δ, CDCl3, 470 MHz) −188.3, − 79.9, −79.5. IR: νCO = 1842 cm−1. HRMS (EI) calculated (C7H5F7O2-CO2) 210.0279; measured 210.0271, fit −3.9 ppm.
4.2.2 4-(1,1,2,2-Tetrafluoroethoxymethyl)-2-propiolactone (3b)
The general procedure was followed with epoxide 2b (1.0 g, 6.0 mmol). The product was purified by distillation at 70 °C under vacuum and 3b was recovered as a colorless oil (0.96 g, 79%). 1H NMR (δ, CDCl3, 300 MHz) 3.42 (dd, 3J = 4.2 Hz, 2J = 16.5 Hz, 1H), 3.61 (dd, 3J = 6.3 Hz, 2J = 16.5 Hz, 1H), 4.23 (dd, 3J = 4.5 Hz, 2J = 11.7 Hz, 1H), 4.37 (dd, 3J = 3.3 Hz, 2J = 11.7 Hz, 1H), 4.76 (dddd, 3J = 4.2 Hz, 3J = 4.5 Hz, 3J = 6.0 Hz, 3J = 7.5 Hz, 1H), 5.78 (tt, 3JHF = 2.6 Hz, 2JHF = 54.0 Hz, 1H); 13C NMR (δ, CDCl3, 125 MHz) 39.8, 63.5, 67.3, 107.6 (tt, 2JCF = 41.1 Hz, 1JCF = 249.8 Hz), 117.3 (tt, 2JCF = 28.4 Hz, 1JCF = 268.4 Hz), 166.7; 19F NMR (δ, CDCl3, 470 MHz) −139.9, −139.7, −94.7. IR: νCO = 1839 cm−1. HRMS (EI) calculated (C6H6F4O3-CO2) 158.0355; measured 158.0350, fit −3.0 ppm.
4.2.3 4-(2,2,3,3,4,4,5,5,5-Nonafluoropentyl)-2-propiolactone (3c)
The general procedure was followed with epoxide 2c (1.7 g, 6.0 mmol). The product was purified by concentration of the crude reaction mixture followed by sublimation (35 °C; cold finger cooled to 0 °C) and 3c was recovered as a white solid (1.6 g, 85%). 1H NMR (δ, CDCl3, 300 MHz) 2.44–2.67 (m, 1H), 2.71–2.94 (m, 1H), 3.34 (dd, 3J = 4.3 Hz, 2J = 16.7 Hz, 1H), 3.78 (dd, 3J = 5.8 Hz, 2J = 16.7 Hz, 1H), 4.88 (dddd, 3J = 3.9 Hz, 3J = 4.1 Hz, 3J = 5.6 Hz, 3J = 6.1 Hz, 1H); 13C NMR (δ, CDCl3, 125 MHz) 36.1 (t, 2JCF = 21.6 Hz), 44.6, 63.8, 166.3 (The fluorine-substituted carbons could not be definitively identified.); 19F NMR (δ, CDCl3, 470 MHz) −129.1, −127.5, −116.5, −84.1. IR: νCO = 1844 cm−1. mp = 41–42 °C. HRMS (EI) calculated (C8H5F9O2-CO2) 260.0247; measured 260.0250, fit 1.1 ppm.
4.2.4 (R)-4-(2,2,3,3,4,4,5,5,5-Nonafluoropentyl)-2-propiolactone (3d)
The general procedure was followed with epoxide 2d (1.7 g, 6.0 mmol). The product was purified as in 3c and lactone 3d was recovered as a white solid (1.6 g, 88%). 1H NMR (δ, CDCl3, 300 MHz) 2.44–2.67 (m, 1H), 2.70–2.93 (m, 1H), 3.34 (dd, 3J = 4.3 Hz, 2J = 16.7 Hz, 1H), 3.77 (dd, 3J = 5.8 Hz, 2J = 16.7 Hz, 1H), 4.88 (dddd, 3J = 3.9 Hz, 3J = 4.1 Hz, 3J = 5.6 Hz, 3J = 6.1 Hz, 1H); 13C NMR (δ, CDCl3, 125 MHz) 36.1 (t, 2JCF = 21.6 Hz), 44.6, 63.8, 166.2 (The fluorine-substituted carbons could not be definitively identified.); 19F NMR (δ, CDCl3, 470 MHz) −128.9, −127.3, −116.3, −83.9. IR: νCO = 1831 cm−1. mp = 33–34 °C. [α]25D −10.8 (c = 1.0, CH2Cl2). (HRMS (EI) calculated (C8H5F9O2-CO2) 260.0247; measured 260.0237, fit −3.9 ppm.
4.2.5 4-(2,2,3,3,4,5,5,5-Octafluoro-4-(trifluoromethyl)pentyl)-2-propiolactone (3e)
The general procedure was followed with epoxide 2e (2.0 g, 6.0 mmol). The product was purified by distillation at 60 °C under vacuum and 3e was recovered as a slightly pink oil (1.9 g, 91%). 1H NMR (δ, CDCl3, 300 MHz) 2.45–2.67 (m, 1H), 2.71–2.94 (m, 1H), 3.34 (dd, 3J = 4.3 Hz, 2J = 16.7 Hz, 1H), 3.78 (dd, 3J = 5.9 Hz, 2J = 16.7 Hz, 1H), 4.88 (dddd, 3J = 4.0 Hz, 3J = 4.2 Hz, 3J = 5.6 Hz, 3J = 6.1 Hz, 1H); 13C NMR (δ, CDCl3, 125 MHz) 36.1 (t, 2JCF = 21.9 Hz), 44.5, 64.0, 90.1 (dsept, 2JCF = 34.4 Hz, 1JCF = 223.5 Hz), 112.3 (tdt, 2JCF = 26.6 Hz, 2JCF = 36.0 Hz, 1JCF = 266.4 Hz), 117.3 (tt, 2JCF = 34.5 Hz, 1JCF = 258.1 Hz), 119.0 (qd, 2JCF = 27.1 Hz, 1JCF = 289.9 Hz), 166.5; 19F NMR (δ, CDCl3, 470 MHz) −189.2, −119.5, −116.3, −75.3. IR: νCO = 1842 cm−1. HRMS (EI) calculated (C9H5F11O2-CO2) 310.0215; measured 310.0203, fit −4.0 ppm.
4.2.6 4-(2,2,3,3,4,4,5,5,6,6,7,7,8,8,9,9,9-Heptadecafluorononyl)-2-propiolactone (3f)
The general procedure was followed with epoxide 2f (0.95 g, 2.0 mmol). The product was purified by column chromatography (silica gel eluted with CH2Cl2) and 3f was recovered as a white solid (0.72 g, 71%). 1H NMR (δ, CDCl3, 300 MHz) 2.45–2.67 (m, 1H), 2.72–2.94 (m, 1H), 3.35 (dd, 3J = 4.3 Hz, 2J = 16.7 Hz, 1H), 3.78 (dd, 3J= 5.9 Hz, 2J = 16.7 Hz, 1H), 4.88 (dddd, 3J = 4.0 Hz, 3J = 4.2 Hz, 3J = 5.6 Hz, 3J = 6.0 Hz, 1H); 13C NMR (δ, CDCl3, 125 MHz) 36.3 (t, 2JCF = 21.5 Hz), 44.7, 63.8, 166.1 (The fluorine-substituted carbons could not be definitively identified.); 19F NMR (δ, CDCl3, 470 MHz) −129.0, −126.2, −125.6, −124.8, −124.8, −124.5, −116.0, −83.6. IR: νCO = 1815 cm−1. mp = 84–85 °C. HRMS (EI) calculated (C12H5F17O2-CO2) 460.0120; measured 460.0119, fit −0.3 ppm.
4.2.7 4-(Pentafluorophenylmethyl)-2-propiolactone (3g)
The general procedure was followed with epoxide 2g (1.3 g, 6.0 mmol). The product was purified by concentration of the crude reaction mixture followed by sublimation (60 °C; cold finger cooled to −78 °C) and 3g was recovered as a white solid (1.2 g, 82%). (δ, CDCl3, 300 MHz) 3.16–3.32 (m, 3H), 3.60 (dd, 3J = 5.8 Hz, 2J = 16.6 Hz, 1H), 4.71 (dddd, 3J = 4.1 Hz, 3J = 4.3 Hz, 3J = 5.8 Hz, 3J = 6.0 Hz, 1H); 13C NMR (δ, CDCl3, 125 MHz) 27.7, 43.2, 68.4, 108.7 (t, 2JCF = 18.6 Hz), 137.7, 140.7, 145.5, 166.6; 19F NMR (δ, CDCl3, 470 MHz) −164.3, −157.4, −145.3. IR: νCO = 1819 cm−1. mp = 72–74 °C. HRMS (EI) calculated (C10H5F5O2) 252.0210; measured 252.0219, fit 3.7 ppm.
4.3 General procedure for the polymerization of fluorinated lactones
In a drybox, catalyst 4 (4.9 mg, 0.01 mmol) and lactone (1.0 mmol) were weighed into separate dry, 4 ml vials. A stir bar was added to the vial with 4. Dry toluene (0.5 mL) was added to each of the vials, and the catalyst solution was transferred to the vial with lactone. The vial was then capped with a Teflon-lined cap, removed from the drybox, and immediately submerged into an oil bath preheated to 50 °C. After the reaction mixture was stirred for 6 h, the vial was cooled to room temperature and the polymerization was quenched with the addition of 5 drops MeOH. The polymers were isolated as described below.
4.3.1 Poly( β-hydroxy-5,6,6,6-tetrafluoro-5-(trifluoromethyl)hexanoate) (5a)
The general procedure was followed with lactone 3a (250 mg, 1.0 mmol). The polymer precipitated from the reaction mixture and was filtered, washed twice with toluene, and dried under vacuum. 1H NMR (δ, (CD3)2CO, 300 MHz) 2.61–2.95 (m, 4H), 5.51–5.67 (m, 1H); 13C NMR (δ, (CD3)2CO, 125 MHz) 32.6 (d, 2JCF = 19.0 Hz), 39.3, 65.7, 92.1 (dsept, 2JCF = 32.9 Hz, 1JCF = 205.0 Hz), 121.8 (qd, 2JCF = 28.0 Hz, 1JCF = 286.2 Hz), 169.1, 169. 2.
4.3.2 Poly( β-hydroxy-4-(1,1,2,2-tetrafluoroethoxy)butyrate) (5b)
The general procedure was followed with lactone 3b (200 mg, 1.0 mmol). The polymer precipitated from the reaction mixture and was filtered, washed twice with toluene, and dried under vacuum. 1H NMR (δ, (CD3)2CO, 300 MHz) 2.77–2.86 (m, 2H), 4.19–4.32 (m, 2H), 5.42–5.54 (m, 1H), 6.22 (tt, 3JHF = 2.6 Hz, 2JHF = 52.5 Hz, 1H); 13C NMR (δ, (CD3)2CO, 125 MHz) 35.4, 65.4, 69.0, 108.9 (tt, 2JCF = 41.8 Hz, 1JCF = 249.1 Hz), 118.3 (tt, 2JCF = 29.3 Hz, 1JCF = 267.7Hz), 169.5, 169.6.
4.3.3 Poly( β-hydroxy-5,5,6,6,7,7,8,8,8-nona-fluorooctanoate) (5c)
The general procedure was followed with lactone 3c (300 mg, 1.0 mmol). The polymer precipitated from the reaction mixture and was filtered, washed twice with toluene, and dried under vacuum. 1H NMR (δ, (CD3)2CO, 300 MHz) 2.61–2.97 (m, 4H), 5.66–5.79 (m, 1H); 13C NMR (δ, (CD3)2CO, 125 MHz) 34.5 (t, 2JCF = 29.84 Hz), 39.2, 64.7, 169.1, 169.1.
4.3.4 Poly((R)-β-hydroxy-5,5,6,6,7,7,8,8,8-nona-fluorooctanoate) (5d)
The general procedure was followed with lactone 3d (300 mg, 1.0 mmol). The polymer precipitated from the reaction mixture and was filtered, washed twice with toluene, and dried under vacuum. 1H NMR (δ, (CD3)2CO, 300 MHz) 2.64–2.94 (m, 4H), 5.66–5.78 (m, 1H); 13C NMR (δ, (CD3)2CO, 125 MHz) 34.4 (t, 2JCF = 20.9 Hz), 39.1, 64.7, 169.1.
4.3.5 Poly( β-hydroxy-5,5,6,6,7,8,8,8-octa-fluoro-7-(trifluoromethyl)octanoate) (5e)
The general procedure was followed with lactone 3e (350 mg, 1.0 mmol). The polymer precipitated from the reaction mixture and was filtered, washed twice with toluene, and dried under vacuum. 1H NMR (δ, (CD3)2CO, 300 MHz) 2.63–2.93 (m, 4H), 5.64–5.80 (m, 1H); 13C NMR (δ, (CD3)2CO, 125 MHz) 34.6, 39.1, 64.8, 91.0, 119.8, 169.0.
4.3.6 Poly( β-hydroxy-4-pentafluorophenyl-butyrate) (5g)
The general procedure was followed with lactone 3g (250 mg, 1.0 mmol). The polymer, which did not precipitate from toluene solution, was precipitated by addition of the solution to hexanes (4 mL). The precipitated polymer was filtered and washed twice with hexanes and dried under vacuum. 1H NMR (δ, (CD3)2CO, 300 MHz) 2.57–2.76 (m, 2H), 3.01–3.22 (m, 2H), 5.31–5.48 (m, 1H); 13C NMR (δ, (CD3)2CO, 125 MHz) 27.1, 38.3, 69.7, 111.6, 138.3, 141.0, 146.5, 169.6, 169.6, 169.7.
4.4 General procedure for the polymerization of fluorinated lactones
The general procedure was analogous to that of the homopolymerization. A solution of both lactones was prepared in toluene and added to a a solution of 4. The copolymerization proceeded for 24 h before being quenched with MeOH.
Supplementary Material
Acknowledgements
This work was supported by the National Science Foundation (CHE-0243605) and the Department of Energy (DE-FG02-05ER15687), and by the National Institutes of Health through a Chemical/Biology Interface (CBI) Training Grant (to J.W.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Grainger DW, Stewart CW. Fluorinated Surfaces, Coatings, and Films. In: Castner DG, Grainger DW, editors. ACS Symposium Series 787. Washington, DC: American Chemical Society; 2001. pp. 1–14. [Google Scholar]
- 2.Imae T. Curr. Opin. Colloid In. 2003;8:307–314. [Google Scholar]
- 3.Reddy VS, Bruma M, Fitch J, Cassidy P. Fluoropolymers 1: Synthesis. In: Houghton G, Cassidy PE, Johns K, Davidson T, editors. Topics in Applied Chemistry. New York: Kluwer Academic/Plenum; 1999. pp. 3–10. [Google Scholar]
- 4.Müller H-M, Seebach D. Angew. Chem. Int. Ed., Engl. 1993;32:477–502. [Google Scholar]
- 5.Lenz RW, Marchessault RH. Biomacromolecules. 2005;6:1–8. doi: 10.1021/bm049700c. [DOI] [PubMed] [Google Scholar]
- 6.Williams SF, Martin DP. In: Biopolymers. Steinbuchel A, editor. Vol. 4. Wiley VCH; 2004. pp. 91–120. [Google Scholar]
- 7.For the properties of PHA copolymers, see:Kunioka M, Tamaki A, Doi Y. Macromolecules. 1989;22:694–697.Noda I, Green PR, Satkowski MM, Schectman LA. Biomacromolecules. 2005;6:580–586. doi: 10.1021/bm049472m.For the degradation of copolymers, see:Doi Y, Kanesawa Y, Kunioka M, Saito T. Macromolecules. 1990;23:26–31.
- 8.Kim O, Gross RA, Hammar WJ, Newmark RA. Macromolecules. 1996;29:4572–4581.Mesiano AJ, Beckman EJ, Russell A. J. Biotechnol. Prog. 2000;16:64–68. doi: 10.1021/bp990138p.Takagi Y, Yasuda R, Maehara A, Yamane T. Eur. Polym. J. 2004;40:1551–1557.For examples of other fluorinated β-lactones, see:McBee ET, Kim YS, Braendlin HP. J. Am. Chem. Soc. 1962;84:3154–3157.Anello LG, Price AK, Sweeney RF. J. Org. Chem. 1968;33:2692–2696.Lavallée C, Leborgne A, Spassky N, Prud’homme RE. J. Polym. Sci., Part A. 1986;25:1315–1328.
- 9.For a review of epoxide carbonylation, see:Church TL, Getzler YDYL, Byrne CM, Coates GW. Chem. Commun. 2007:657–674. doi: 10.1039/b613476a.For recent examples, see:Getzler YDYL, Mahadevan V, Lobkovsky EB, Coates GW. J. Am. Chem. Soc. 2002;124:1174–1175. doi: 10.1021/ja017434u.Mahadevan V, Getzler YDYL, Coates GW. Angew. Chem. Int. Ed. 2002;41:2781–2784. doi: 10.1002/1521-3773(20020802)41:15<2781::AID-ANIE2781>3.0.CO;2-S.Schmidt JAR, Mahadevan V, Getzler YDYL, Coates GW. Org. Lett. 2004;6:373–376. doi: 10.1021/ol036244g.Schmidt JAR, Lobkovsky EB, Coates GW. J. Am. Chem. Soc. 2005;127:11426–11435. doi: 10.1021/ja051874u.Kramer JW, Lobkovsky EB, Coates GW. Org. Lett. 2006;8:3709–3712. doi: 10.1021/ol061292x.
- 10.Rieth LR, Moore DR, Lobkovsky EB, Coates GW. J. Am. Chem. Soc. 2002;124:15239–15248. doi: 10.1021/ja020978r. [DOI] [PubMed] [Google Scholar]
- 11.For a recent, comprehensive review of asymmetric epoxidation, see:Xia Q-H, Ge H-Q, Ye C-P, Liu Z-M, Su K-X. Chem. Rev. 2005;105:1603–1662. doi: 10.1021/cr0406458.For kinetic resolution of epoxides, see:Tokunaga M, Larrow JF, Kakiuchi F, Jacobsen EN. Science. 1997;227:936–938. doi: 10.1126/science.277.5328.936.Schaus SE, Brandes BD, Larrow JF, Tokunaga M, Hansen KB, Gould AE, Furrow ME, Jacobsen EN. J. Am. Chem. Soc. 2002;124:1307–1315. doi: 10.1021/ja016737l.
- 12.For solvent effects in epoxide carbonylation to β-lactones, see:Church TL, Getzler YDYL, Coates GW. J. Am. Chem. Soc. 2006;128:10125–10133. doi: 10.1021/ja061503t.For solvent effects in epoxide carbonylation to anhydrides, see:Rowley JM, Lobkovsky EB, Coates GW. J. Am. Chem. Soc. 2007;129:4948–4960. doi: 10.1021/ja066901a.
- 13.(a) Tanahashi N, Doi Y. Macromolecules. 1991;24:5732–5733. [Google Scholar]; (b) Hori Y, Suzuki M, Yamaguchi A, Nishishita T. Macromolecules. 1993;26:5533–5534. [Google Scholar]; (c) Schechtman LA, Kemper JJ. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 1999;40(1):508–509. [Google Scholar]
- 14.The polymerization of 3f in CHCl3 produced some polymer which was slightly soluble in hexafluoroisopropanol. However, characterization was difficult due to the polymer’s insolubility.
- 15.Tanahashi N, Doi Y. Macromolecules. 1991;24:5732–5733. [Google Scholar]
- 16.The copolymerization of 3g and BBL was analogous to 3a and BBL as monitored by 1H NMR spectroscopy.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.