

Abstract
A gene encoding a putative AraC-type transcriptional regulator was identified on the 153-kb pathogenicity island (PAI) found among virulent Enterococcus faecalis strains. In an effort to understand the function of this regulator, designated PerA (for pathogenicity island-encoded regulator), we first examined the expression of the perA gene in the original PAI strain MMH594 and in an unrelated clinical isolate E99 by reverse transcription-PCR. Interestingly, expression analysis revealed no detectable perA transcript in MMH594, whereas a transcript was observed in strain E99. Nucleotide sequence analysis revealed that this altered expression between the two strains was attributable to the differential location of an IS1191 element within the putative promoter region upstream of the perA gene. In order to determine the role of this putative regulator in E. faecalis pathogenesis, a perA-deficient mutant was created in strain E99, and the wild-type and mutant pair were compared for phenotypic differences. In in vitro biofilm assays, the mutant strain showed a significantly higher level of growth medium-specific biofilm formation compared to the wild type. However, in a murine intraperitoneal infection model, the mutant strain was significantly less pathogenic. The mutant was also attenuated for survival within macrophages in vitro. These findings highlight the importance of PerA as a regulator of biofilm formation and survival within macrophages and is likely a regulator controlling determinants important to pathogenesis.
Enterococcus faecalis is a member of the commensal flora of the gastrointestinal tract of a multitude of organisms ranging from insects to humans. However, E. faecalis has also emerged as a leading cause of nosocomial infections that are refractory to antibiotic therapy in many instances (13). A pathogenicity island (PAI) encoding a number of factors previously shown to contribute to enterococcal pathogenesis has been identified in a hospital ward outbreak strain of E. faecalis, MMH594 (29). This 153-kb PAI consists of 129 open reading frames (ORFs), including several with previously ascribed roles in virulence (13, 29). These include genes coding for the broad-spectrum toxin cytolysin, the enterococcal surface protein (Esp), and aggregation substance (29). In addition, are genes specifying factors that have been hypothesized to be advantageous in gastrointestinal tract colonization, including a bile acid hydrolase, new carbohydrate utilization pathways, and a Gls24-like starvation-inducible protein. Located approximately 1.5 kb 3′ to the esp gene on the MMH594 PAI is an ORF potentially encoding a member of the AraC/XylS family of transcriptional regulatory proteins. This araC-type gene was designated perA (for pathogenicity island-encoded regulator).
Members of the AraC/XylS transcriptional regulator family have been characterized from a wide variety of prokaryotes including both gram-negative and gram-positive bacteria. The characteristic feature of this family is a conserved stretch of approximately 100 amino acids at the C-terminal end that serves as the DNA-binding domain (10). Within this domain, two helix-turn-helix motifs are responsible for binding to adjacent major grooves that constitute the DNA-binding site (10). In addition, most members contain a domain responsible for dimerization and interaction with a ligand effector molecule. The main regulatory roles ascribed to this family are carbon metabolism, stress response, and virulence and/or pathogenesis. A number of family members have been demonstrated to be involved in pathogenesis, regulating the elaboration of virulence factors of microorganisms that infect plants and mammals (10). A well-characterized AraC family member that is involved in pathogenesis is ToxT, which has been shown to activate expression of the toxin coregulated pilus and cholera toxin in Vibrio cholerae (6). ToxT has further been shown to perform a dual regulatory task: inhibition of repression and direct activation (reviewed in reference 8). The predicted product of the perA gene from the MMH594 PAI is similar to the MsmR protein from Streptococcus pyogenes. MsmR has been shown to transactivate the expression of the major fibronectin-binding adhesion protein, as well as positively regulate the cytolysin-mediated translocation system genes (24).
The close proximity of the perA gene to a number of well-characterized virulence determinants on the E. faecalis pathogenicity island, as well as the fact that AraC/XylS members are known to regulate virulence factors in other pathogens, suggests a possible role in the regulation of these virulence determinants. as well as other uncharacterized PAI genes. Moreover, it has recently been demonstrated that an AraC-type ORF encoded on the putative E. faecium PAI regulates Esp and is involved in biofilm formation (R. Willems et al., unpublished data). In E. faecalis, the fsr regulatory locus has been implicated in regulation of biofilm formation through the control of gelatinase (15), and three ArgR family regulators were also identified as involved in biofilm formation (18). Further, the EbpR regulator, which shows similarities to a AtxA/MgaA family member, has been implicated in biofilm formation through the activation of genes encoding a piluslike structure (1). However, the involvement of AraC/XylS family members in biofilm formation has not been evaluated in E. faecalis.
In the present study, expression of the perA gene was evaluated in E. faecalis strain MMH594, the strain possessing the prototype PAI, as well as in the urinary tract isolate E99. In order to examine the potential role of the perA gene in biofilm formation and pathogenesis, the perA gene was insertionally inactivated in strain E99, and the resultant perA mutant strain was compared to the wild type in in vitro biofilm and macrophage survival assays and in a mouse peritonitis model.
MATERIALS AND METHODS
Bacterial strains, media, and reagents.
All strains and plasmids used in the present study are listed in Table 1. E. faecalis strain E99 is a clinical isolate from the urine of a patient at the VA medical center in Little Rock, AK (39). E. faecalis strain MMH594 is a bloodstream isolate that caused multiple infections in a large medical center and is the prototype for the enterococcal PAI (16, 29). Escherichia coli strain XL1-Blue (Stratagene, La Jolla, CA) was used as a host for cloning and plasmid purification and cultivated in Luria-Bertani broth. Enterococcal strains were cultivated in brain heart infusion (BHI), Todd-Hewitt broth (THB) containing 1% glucose, or Trypticase soy broth (TSB) plus 0.75% glucose. Antibiotics used for selection were obtained from Sigma Chemical (St. Louis, MO) and included kanamycin (25 μg/ml), erythromycin (50 μg/ml), spectinomycin (500 μg/ml), and gentamicin (500 μg/ml) for E. faecalis strains. The antibiotics used for the selection of E. coli strains included erythromycin (400 μg/ml) and spectinomycin (150 μg/ml).
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| MMH594 | Clinical blood isolate | 16 |
| E99 | Clinical urine isolate | 39 |
| DBS01 | perA insertion mutant | This study |
| DBS01(pGT101) | Contains plasmid pGT101 (perA insertion mutant complemented with plasmid-encoded perA) | This study |
| Plasmids | ||
| p3ERM | Suicide vector (erythromycin resistance marker) | 2, 14 |
| p3ERMperA | Suicide vector used to generate a SCO mutant deficient in perA expression (DBS01) | This study |
| pAT28 | Shuttle vector (spectinomycin resistance marker) | 40 |
| pGT101 | pAT28 containing a full-length copy of the perA gene to complement the mutant strain DBS01 | This study |
RNA isolation and RT-PCR.
In order to assess perA expression, strains E99 and MMH594 were cultivated at 37°C overnight in BHI supplemented with appropriate antibiotics. The bacteria were diluted 1:1,000 into fresh, prewarmed medium and harvested at predetermined optical density values (600 nm; 0.05 for logarithmic and 1.0 for stationary phase). Total RNA extraction was performed as previously described (33), with the following modifications. After the initial RNA precipitation, the RNA pellet was resuspended in 50 μl of diethyl pyrocarbonate-treated water. The RNA concentration was determined by measuring the A260 with an ND-1000 spectrophotometer (NanoDrop, Wilmington, DE). Removal of contaminating genomic DNA was carried out by processing 25 μg of total RNA with a DNA-free kit (Ambion Austin, TX) according to the manufacturer's instructions, in a total volume of 20 μl. Generation of cDNA was carried out by using a Superscript first-strand synthesis system for reverse transcription-PCR (RT-PCR; Invitrogen, Carlsbad, CA) according to the manufacturer's instructions and 5 μg of RNA as a template. Parallel reactions were carried out with or without SuperScript II (SSII; reverse transcriptase) to ensure the absence of contaminating genomic DNA. The cDNA generated was used as a template and subjected to 30 cycles of PCR using the perA specific primers perA-FF and perA-RR (Table 2). In addition, all cDNA samples were subjected to 30 cycles of PCR using the 16S rRNA specific primers 16S RT-F and 16S RT-R (Table 2) to ensure the integrity of the extracted RNA.
TABLE 2.
Primers used in this study
| Primer | Sequence (5′-3′)a |
|---|---|
| perA-13F | GAGACTGCAGCATTACATGTATGCTTAGGGATACC |
| perA-14R | GAGAGAATTCCCACAGGAACAACACCATTACTTAG |
| perA-compF | GAGAGGATCCGAATACGGATAAAGAAGTTTTATT |
| perA-compR | GAGAGAATTCTTAATGATATGAAGTTTTAAGTG |
| perA-FF | GAGAGAATTCACGGGAAAGATTGAAGACTT |
| perA-RR | GAGACTGCAGTAGGAGTCGTCCCTGTTATC |
| 16S RT-F | AGCCGGAATCGCTAGTAATCG |
| 16S RT-R | TCGGGTGTTACAAACTCTCGTG |
Restriction sites are underlined.
DNA manipulations.
All PCRs generating a product for use in cloning or sequencing were performed using LA Taq polymerase (TaKaRa Biomedicals, Shiga, Japan) and purified from a 0.8% low-melting-point agarose gel utilizing a Wizard Preps DNA purification system (Promega, Madison, WI). Restriction endonucleases for use in cloning were obtained from New England Biolabs (Beverly, MA).
Insertional inactivation of the perA gene in strain E99.
In order to generate a insertional mutant in the perA gene, a 550-bp internal fragment of the perA gene was amplified using the primer pair perA-13F and perA-14R (Table 2) and E99 genomic DNA as a template. The resultant product was treated with PstI and EcoRI and ligated to the similarly restricted suicide vector p3ERM (2, 14). The resulting plasmid construct, p3ERMperA, was then introduced into electrocompetent E99 by electroporation as previously described (32). Erythromycin-resistant clones were selected, and insertion of the vector into the perA gene was verified by Southern hybridization analysis and PCR. Southern analysis demonstrated a single insertion in the perA locus, ruling out the possibility that the observed phenotypes were due to insertion in other locations. The perA insertion mutant strain was designated DBS01. To rule out a polar effect of insertion of the p3ERMperA plasmid into the perA gene, expression analysis of the gene located immediately 3′ of perA on the PAI revealed no differences in expression between E99 and the perA mutant strain DBS01 using RNA derived from the logarithmic phase of growth (data not shown). The mutant DBS01 was complemented in trans using pGT101, a derivative of the shuttle vector pAT28 (40) possessing a full-length copy of the perA gene, including the putative E99 native promoter region. The plasmid pGT101 was constructed by PCR amplification of the perA gene using the primer pair perA-compF and perA-compR (Table 2), restriction of the amplicon with BamHI and EcoRI, and ligation of the product to similarly restricted pAT28.
Biofilm assay.
In order to determine whether the PerA regulator influences biofilm formation, a crystal violet staining assay was used essentially as described by Tendolkar et al. (38). E. faecalis strains E99, DBS01, and DBS01(pGT101) were grown at 37°C for 16 h in TSB plus 0.75% glucose or in THB plus 1% glucose and the appropriate selective antibiotics. Following this growth period, bacterial cells were centrifuged at 6,000 × g for 10 min, and the cell pellet resuspended in 5 ml of fresh medium. The optical densities of the bacterial suspensions were measured by using a UV-1201 UV-VIS spectrophotometer (Shimadzu, Kyoto, Japan) and normalized to an optical density at 600 nm of 1.0. Bacterial cultures were then diluted 1:40 in fresh TSB plus 0.75% glucose or in THB plus 1% glucose and the appropriate antibiotics. From the diluted culture, 200 μl was dispensed into 12 wells in a single row of a sterile 96-well flat-bottom polystyrene microtiter plate (Corning, Inc., Corning, NY). After incubation at 37°C for 24 h, the medium was aspirated, and the wells were washed three times with sterile phosphate-buffered saline (PBS). The plates were inverted and allowed to dry for 1 h at room temperature. For biofilm quantification, wells were stained with 200 μl of 0.2% aqueous crystal violet solution for 15 min and washed thrice with sterile PBS to remove the excess crystal violet. Crystal violet bound to the biofilm was extracted with 200 μl of an 80:20 (vol/vol) mixture of ethyl alcohol and acetone, and the absorbance of the extracted crystal violet was measured at 595 nm with an ELX-800 (Bio-Tek Instruments, Inc., Winooski, VT) automatic microplate reader. All biofilm assays were performed in triplicate, with 12 replicates for each strain per assay.
Intraperitoneal mouse infection model.
Bacteria were cultivated overnight at 37°C in THB containing 1% glucose supplemented with appropriate antibiotics. The bacteria were collected by centrifugation at 6,000 × g for 10 min, washed twice in PBS (pH 7.4), and resuspended in 5% hog gastric mucin (pH 7.0; Pfaltz and Bauer, Waterbury, CT) at a concentration of 1.0 × 109 CFU/ml. Six-week-old female BALB/c mice (Harlan Sprague-Dawley, Indianapolis, IN) were administered 200 μl of the suspension (2.0 × 108 CFU) via intraperitoneal injection. Control mice were also injected with 200 μl of 5% hog gastric mucin without bacteria to ensure both the sterility of the mucin and the absence of antibiotic-resistant bacteria in the tissues to be sampled. Control animals were treated in a manner identical to the experimental groups in all respects. Groups of 10 mice per strain were sacrificed at 24, 48, and 72 h by CO2 asphyxiation. From each animal, samples of blood were taken by cardiac puncture, and samples of the liver and spleen were taken by dissection. Liver and spleen samples from each time point were placed into preweighed 2-ml screwcap microcentrifuge tubes containing 0.5 ml of 1.0-mm zirconia/silica beads (BioSpec Products, Bartlesville, OK). Tubes were weighed to determine the size of the tissue sample, and 1 ml of PBS (pH 7.4) was then added. Tubes were shaken horizontally at 5,000 rpm for 1 min in a Mini-Beadbeater (BioSpec Products). Bacteria were enumerated via serial dilution and plating of the blood and tissue homogenates on BHI agar containing appropriate antibiotics. Bacterial concentrations were expressed as CFU/ml for blood and as CFU/g for the liver and spleen. The nonparametric Wilcoxon/Kruskal-Wallis test was used to determine significance levels.
Transmission electron microscopy.
RAW 264.7 macrophages (American Type Culture Collection [ATCC], Manassas, VA) were cultivated in Dulbecco modified Eagle medium (DMEM; ATCC 30-2002) plus 10% fetal bovine serum (FBS; ATCC 30-2020) to confluence in T-25 flasks. Cells were harvested, and viability determined by trypan blue dye exclusion staining. Approximately 106 cells were seeded into each well of six-well plates, followed by incubation at 37°C under 5% CO2 for 24 h prior to infection. E. faecalis strains E99, DBS01, and DBS01(pGT101) were grown in THB containing 1% glucose and supplemented with the appropriate antibiotics for 16 h, centrifuged, and resuspended in PBS (pH 7.4). RAW 264.7 cells were infected at a multiplicity of infection (MOI) of 100 for each bacterial strain for 1 h at 37°C under 5% CO2, washed twice with PBS, and further incubated with DMEM plus 10% FBS containing 16 μg of vancomycin per ml and 150 μg of gentamicin per ml. At 0, 2, 24, 48, and 72 h postinfection, the cells were harvested; centrifuged at 1,000 × g for 5 min; and resuspended in 500 μl of a fixative solution consisting of 4% paraformaldehyde, 2% glutaraldehyde (Electron Microscopy Services, Hatfield, PA), and 0.1 M sodium cacodylate (Sigma, St. Louis, MO). Fixed cells were dehydrated and embedded in Epon/Araldite resin, cured, and sectioned. Sections (100 nm thick) were placed on glow-discharged 300 Cu Mesh grids and lead/uranyl acetate stained using Sato's lead. Cells were then imaged on a Hitachi H-7600 transmission electron microscope.
Macrophage survival assay.
Survival of E. faecalis strains in macrophages was assessed essentially as described earlier (37). RAW 264.7 macrophages were seeded into six-well plates at approximately 106 cells per well and incubated at 37°C under 5% CO2 for 24 h prior to infection. E. faecalis strains E99, DBS01, and DBS01(pGT101) and E. coli XL1-Blue were grown in THB containing 1% glucose and supplemented with the appropriate antibiotics for 16 h, centrifuged, and resuspended in PBS (pH 7.4). Triplicate wells of RAW 264.7 cells were infected at an MOI of 100 for each bacterial strain for 1 h at 37°C under 5% CO2, washed twice with PBS, and further incubated with DMEM plus 10% FBS containing 16 μg of vancomycin per ml and 150 μg of gentamicin per ml for 2, 24, 48, and 72 h. Importantly, E. faecalis strains E99, DBS01, and DBS01(pGT101) demonstrate equivalent susceptibilities to the bactericidal activity of these two antibiotics. At each time point, the macrophages were washed twice with PBS (pH 7.4) and harvested in 1 ml of PBS (pH 7.4), the viability was assessed by trypan blue staining, and the macrophages were counted with a hemacytometer. Macrophages were then lysed by adding 100 μl of a saponin cell lysis solution (saponin [40 mg/ml], polypropylene glycol [P-2000; 8 ml/liter], sodium polyanetholsulfonate [9.6 mg/ml]) to release intracellular bacteria. Bacteria were quantified by serial dilution and plating. The number of viable bacteria at each time point was expressed as CFU per 105 macrophages. Experiments were performed three times, and the means and standard errors were determined for each time point. The statistical significance of the results was determined by performing pairwise comparisons at each time point using the Wilcoxon/Kruskal-Wallis test.
RESULTS
Characterization of the perA gene.
BLAST analysis of the E. faecalis strain MMH594 PAI ORFs revealed an ORF encoding a product with 44% identity and 67% similarity to a putative AraC/XylS-type transcriptional regulator from S. pyogenes strain MGAS8232 (accession no. NP_608022). The predicted product of this ORF also displayed 28% identity and 52% similarity to the MsmR protein from S. pyogenes (accession no. ABM66482). This putative araC/XylS-type ORF is located ∼1.5 kb 3′ to the esp (for enterococcal surface protein) gene on the MMH594 PAI and was designated perA (for pathogenicity island-encoded regulator). The deduced PerA consists of 331 amino acids with a putative helix-turn-helix DNA-binding domain located within the C-terminal 100 amino acids. In order to determine perA gene expression in MMH594, RT-PCR was conducted using RNA isolated from bacteria grown to logarithmic and stationary phases in BHI broth. Interestingly, RT-PCR did not yield a detectable product from RNA derived from strain MMH594 grown in BHI to either stationary (Fig. 1) or logarithmic (data not shown) phase. Tendolkar et al. (39) have recently characterized a high-biofilm-forming clinical isolate of E. faecalis, designated E99 and identified a novel locus encoding putative surface proteins responsible for the high-biofilm-forming phenotype. This strain also possesses portions of the PAI, including the perA gene. It was therefore of interest to examine the expression of the perA gene in this high-biofilm-forming infection-derived isolate. RT-PCR analysis revealed expression of the perA gene in the stationary phase of growth in BHI (Fig. 1). The predicted PerA protein in E99 is identical to that of MMH594 with the exception of 71 additional N-terminal amino acids in the E99 polypeptide. Alignment of the C-terminal 100 amino acids of the E99 PerA with the consensus AraC helix-turn-helix motif (10, 20) revealed 33% identity and 38% similarity.
FIG. 1.
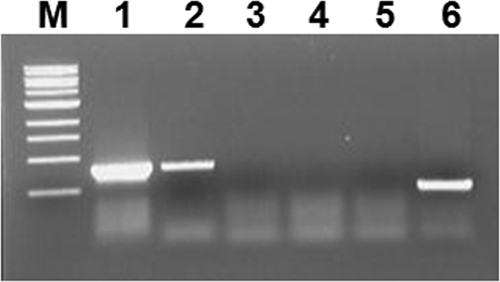
Expression of perA in MMH594 and E99 by RT-PCR. Primers perA-FF and perA-RR (Table 2), specific to perA, were used in all reactions. RT(−) controls indicate samples are free of contaminating genomic DNA. Lanes: 1, E99 DNA; 2, E99 RT(+); 3, MMH594 RT(+); 4, E99 RT(−); 5, MMH594 RT(−); 6, MMH594 DNA. Lane M is a 1-kb size marker from New England Biolabs.
Sequencing of the regions 5′ to the perA gene in both MMH594 and E99 revealed that the observed difference in expression of perA between the two strains was likely due to an IS1191 element disrupting the promoter region in MMH594 but not in E99 (Fig. 2). In MMH594 the 3′ end of IS1191 is located 203 bp from the putative start codon of perA. In E99 the IS1191 element is flipped and located 369 bp from the putative start codon assigned for perA in MMH594. This change in location exposes potential consensus promoter sequences in proximity to perA. The sequence from the 3′ end of the esp gene to the 5′ end of perA is similar between the two strains with the exception of an additional nucleotide “T” in the E99 sequence immediately 5′ to the predicted start codon in MMH594. This results in a frameshift mutation extending the predicted perA ORF in E99 by 213 bp and resulting in an additional 71 N-terminal amino acids in the putative polypeptide (Fig. 2).
FIG. 2.
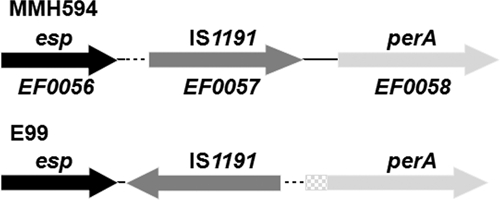
Position of esp, IS1191, and perA in MMH594 (29) and E99. The sequence containing a potential consensus promoter is denoted by the dashed line, and the predicted extended perA sequence as part of the ORF is denoted by the checkered box.
PerA influences the capacity to form biofilms in vitro in a growth medium-dependent manner.
As previously shown, E99 is a high-biofilm-forming strain that possesses a novel locus primarily responsible for this phenotype (39). Moreover, it has been demonstrated that an AraC-type ORF encoded on the putative E. faecium PAI regulates Esp and is involved in biofilm formation (Willems et al., unpublished). We therefore questioned whether PerA is involved in the regulation of biofilm formation in strain E99. To address this hypothesis, a single-crossover mutant deficient in perA transcription was generated in E99. The resultant mutant strain was designated DBS01, and no perA transcript was detectable by RT-PCR (data not shown). Further, the mutant strain was complemented in trans with a plasmid-encoded copy of the perA gene, and the resultant strain was designated DBS01(pGT101). The degree of biofilm formation by the wild-type strain E99 was then compared to the mutant strain DBS01, as well as to DBS01(pGT101) in TSB plus 0.75% glucose. This medium was chosen because under these conditions it has been shown that Esp enhances the ability to form biofilms by ∼5-fold (38). However, we detected no difference in the biofilm density between strain E99 and DBS01 (P = 0.34) (data not shown), suggesting that the perA gene does not play a role under these conditions. We therefore evaluated additional laboratory media for possible medium-dependent effects. After growth of the three strains in THB plus 1% glucose, we observed a statistically significant increase in biofilm formation by the DBS01 strain relative to both E99 (P < 0.0001) and the mutant complemented strain DBS01(pGT101) (P < 0.0001) (Fig. 3). No significant difference was observed between E99 and DBS01(pGT101) (P = 0.23) (Fig. 3). No differences in in vitro growth rates were observed between the three strains in BHI, TSB plus 0.75% glucose, or THB plus 1% glucose (data not shown). These results show that inactivation of the perA gene in strain E99 results in approximately a 30% increase in biofilm formation after growth in THB plus 1% glucose, suggesting that the PerA regulator likely represses factors that contribute to this process in a medium-dependent fashion.
FIG. 3.
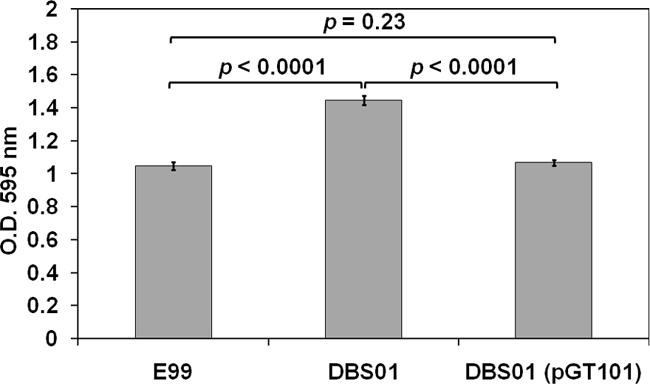
Biofilm density after growth in THB plus 1% glucose. Biofilm formation by E. faecalis strains E99, DBS01, and DBS01(pGT101) on polystyrene microtiter plates was assessed by crystal violet staining. Biofilm assays were performed in triplicate, with 12 replicates for each strain per assay. The error bars represent the means ± the standard errors. A Student t test was used to determine significance levels.
PerA contributes to pathogenesis in a mouse peritonitis model.
Given that PerA plays a role in repressing biofilm formation in a medium-dependent manner and given the location of the perA gene on the E. faecalis PAI in proximity to factors that have previously been ascribed a role in virulence, we next sought to determine whether PerA is involved in E. faecalis pathogenesis. The wild-type E99 and the DBS01 strains were then compared in an in vivo mouse peritonitis model of systemic E. faecalis infection. Preliminary experiments revealed that administration of 2.0 × 108 CFU of either wild type or DBS01 per mouse did not result in significant mortality. However, symptoms such as decreased locomotor activity and fur ruffling were observed among mice infected with wild-type E99 but were considerably less apparent in mice administered strain DBS01. These differences in symptoms were consistent with the bacterial load recovered from hepatic and splenic tissues at 24 and 48 h postinfection in large-scale experiments. Comparisons between E99 and DBS01 revealed striking differences in bacterial concentration in the liver and spleen at 24 and 48 h, with differences normalizing by 72 h (Fig. 4). At 24 h the hepatic bacterial load was nearly 104 CFU/g higher in the mice infected with wild-type E99, while the splenic bacterial load was nearly 103CFU/g higher (Fig. 4A). At 48 h the hepatic bacterial load was 102 CFU/g higher and the splenic load was 103 CFU/g higher in mice infected with wild-type E99 (Fig. 4B). By 72 h, mice infected with E99 still exhibited a 1.5 log CFU/g higher bacterial load in the spleen (Fig. 4C). At all time points, mice administered strain DBS01 exhibited a slightly higher but not significantly different bacterial load in the blood. Given the sample size (n = 10), and the difference at all three time points, the effect appears to be reproducible; however, the difference in the bacteremia exhibited by the two groups was not statistically significant.
FIG. 4.
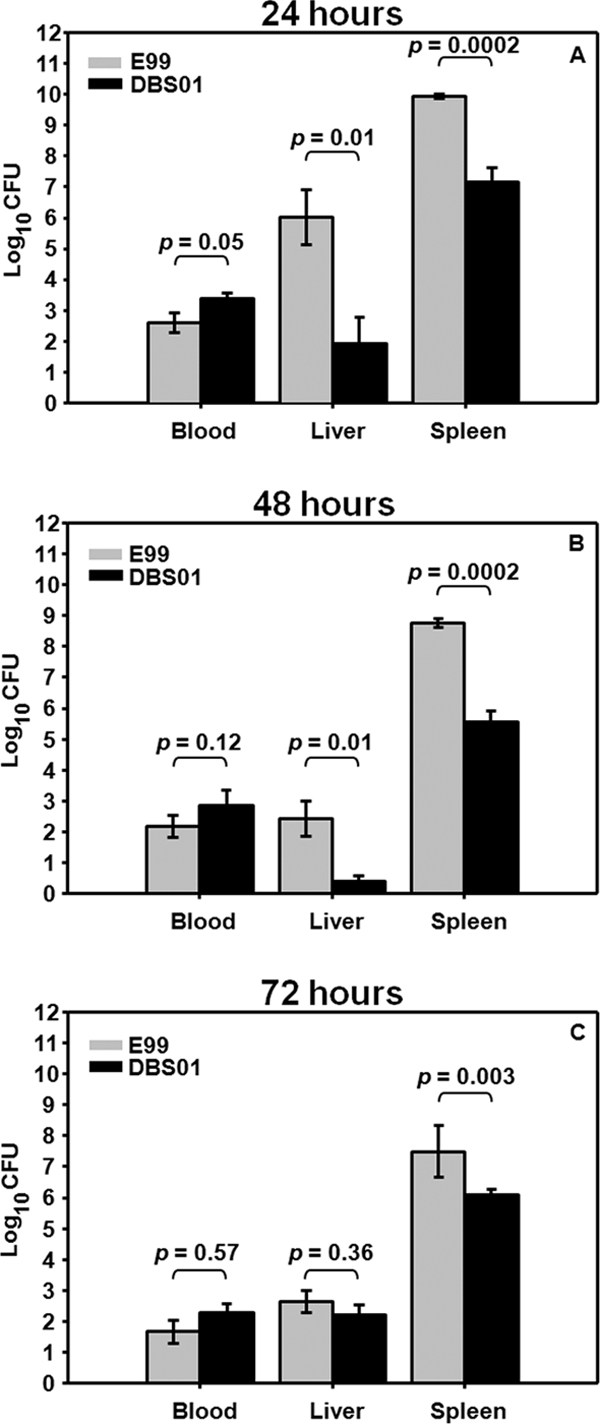
Bacterial load at 24 (A), 48 (B), and 72 (C) h in the liver, spleen, and blood for mice given E. faecalis strains via intraperitoneal injection. Error bars represent means ± the standard errors. A nonparametric Wilcoxon/Kruskal-Wallis test was used to determine significance levels.
PerA contributes to survival of E. faecalis in mouse RAW 264.7 macrophages.
In order to test the hypothesis that the increased bacterial load of wild-type E99 in hepatic and splenic tissues might be due to an increased capacity to survive in phagocytes relative to the perA mutant, we evaluated the relative ability of the wild-type E99 and DBS01 strains, as well as DBS01 complemented with a plasmid-encoded copy of the perA gene, to persist in mouse RAW 264.7 cells over a 72-h period. Macrophages were infected at an MOI of 100. Examination of infected macrophages by transmission electron microscopy immediately after the 1-h infection period (t = 0) revealed similar numbers of bacteria in sections of macrophages infected with either E99, DBS01, or DBS01(pGT101), indicating similar levels of phagocytosis (Fig. 5A, B, and C). Moreover, no significant differences in the number of viable bacteria were observed among the three strains at 2 h postinfection [P = 0.86 for E99 versus DBS01; P = 0.60 for E99 versus DBS01(pGT101); P = 0.66 for DBS01 versus DBS01(pGT101)] (Fig. 6). All three strains were subject to macrophage killing over the 72-h time period; however, the mutant strain was recovered from macrophages at significantly lower levels than strain E99 at 24, 48, and 72 h (P < 0.05 for all time points) (Fig. 6). By 72 h, there was approximately an order of magnitude fewer viable DBS01 bacteria recovered from macrophages than the wild-type E99 or DBS01(pGT101) strains (Fig. 6). No statistically significant differences were observed between strain E99 and the complemented strain at any time point (P = 0.86 for 24 h; P = 0.09 for 48 h; P = 0.22 for 72 h). As a control, macrophages were also infected with E. coli strain XL1-Blue. As expected, this strain was rapidly cleared by the RAW cells over the course of the experiments, and after 72 h only approximately 30 CFU per 105 macrophages were recovered (data not shown).
FIG. 5.

Transmission electron microscopy of RAW 264.7 macrophages at 0 h (A, B, and C), 2 h (D, E, and F), 24 h (G, H, and I), 48 h (J, K, and L), and 72 h (M, N, and O) after infection with E. faecalis strains E99 (A, D, G, J, and M), DBS01 (B, E, H, K, and N), and DBS01(pGT101) (C, F, I, L, and O). Arrows indicate bacterial cells inside macrophages.
FIG. 6.
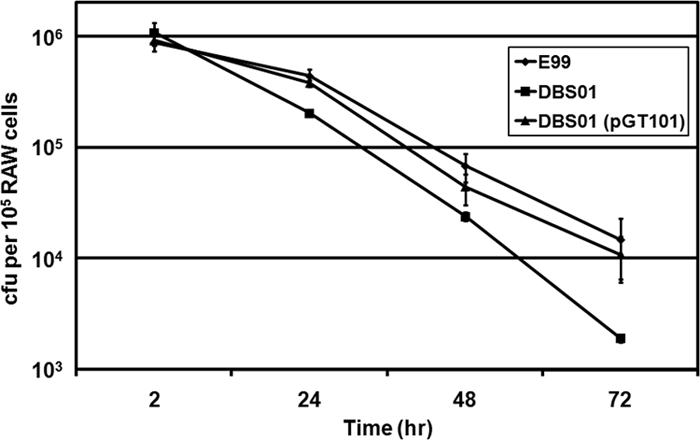
Intracellular survival of E. faecalis strains E99, DBS01, and DBS01(pGT101) in RAW 264.7 macrophages at 2, 24, 48, and 72 h postinfection. The number of viable bacteria at each time point is expressed as CFU per 105 macrophages. Experiments were performed three times, and the means and standard errors are reported for each time point. A nonparametric Wilcoxon/Kruskal-Wallis test was used to perform pairwise analyses at each time point.
Imaging of infected macrophages by TEM over the 72-h period confirmed the results of the macrophage survival assays. Strains E99, DBS01, and DBS01(pGT101) are visible in phagocytic vacuoles at similar numbers at 2 h postinfection (Fig. 5D, E, and F). However, for the 24-h (Fig. 5G, H, and I), 48-h (Fig. 5J, K, and L), and 72-h (Fig. 5M, N, and O) time points, fewer macrophages showing intact DBS01 bacteria were observed (Fig. 5H, K, and N) relative to E99 (Fig. 5G, J, and M) or the complemented strain (Fig. 5I, L, and O). Moreover, macrophages infected with DBS01 showed fewer bacteria per macrophage than macrophages infected with either E99 or DBS01(pGT101). These results show that PerA plays a significant role in the ability of strain E99 to survive in macrophages and likely regulates factors important to the ability of E. faecalis to resist killing by macrophages. Further, these results correlate with the decreased pathogenicity of the DBS01 strain in the mouse peritonitis model and identify a potential mechanism for the increased clearance of the DBS01 strain in the liver and spleen.
DISCUSSION
A number of enterococcal factors that contribute to the severity of infection in several animal models have been characterized, including the bifunctional cytolysin (reviewed in references 5 and 13), the Esp (30, 31), aggregation substance (4, 21, 28), gelatinase (9, 34, 36), and an endocarditis and biofilm-associated pilus (Ebp) (25, 35). Moreover, virulent isolates of enterococci possess a PAI that encodes many of these factors, including a number of factors putatively related to virulence and/or colonization (29). The prototype PAI, initially described in strain MMH594 (16, 29), also possesses an ORF (EF0058) potentially specifying an AraC/XylS-type transcriptional regulator (PerA). In the present work, we sought to characterize this ORF and elucidate its role in the biology and pathogenesis of E. faecalis.
In strain MMH594, perA is located ∼1.5 kb 3′ to the esp gene, with an IS1191 element situated between the two genes. Examination of the intergenic region 3′ to the IS1191 element and 5′ to the putative perA coding sequence did not reveal consensus bacterial promoter elements. RT-PCR confirmed that MMH594 did not express the perA regulator under the conditions examined. However, screening of a collection of 53 geographically diverse clinical and non-infection-derived isolates containing a broad array of insertions and deletions in the PAI revealed the presence of the putative perA gene in 51 of these isolates (S. McBride et al., unpublished data). These results suggest that insertion of the IS1911 has disrupted the promoter region of the perA gene in strain MMH594; however, the overall impact of the apparent silencing of perA expression in this strain is currently unknown. Expression of the perA gene was detected in strain E99, and the product was predicted to possess an extended amino acid sequence at the N terminus relative to that from MMH594, although the impact this has on regulatory function remains unclear. There are no identifiable conserved sequences located within the N-terminal region of PerA in MMH594 or E99, and there are no similarities in this region with other members of the AraC/XylS family of transcriptional regulators.
The ability to form biofilms is an important aspect of the E. faecalis infection process (3, 22, 25). Mohamed et al. (22) suggested that endocarditis isolates were more likely to form biofilms than were nonendocarditis isolates, and disruption of the ebp locus in strain OG1RF resulted in a decrease in the ability to form biofilms and concomitant attenuation in an endocarditis model (25). A number of E. faecalis regulators have been demonstrated to be involved in the formation of biofilms, including the fsr regulatory locus that controls the expression of gelatinase (GelE), a factor required for this process (15), as well as three different members of the ArgR family of transcriptional regulators (18). Given the proximity of the perA gene to esp, which has been shown to contribute to biofilm formation in a glucose-dependent manner, we postulated that PerA might be involved in regulating biofilm formation. Crystal violet staining of biofilms revealed that the PerA regulator influences biofilm formation in a growth medium-dependent manner, i.e., a role for PerA in biofilm formation was observed after growth in THB plus 1% glucose but not TSB plus 0.75% glucose. Given that Esp enhances biofilm formation in both types of media (data not shown), a likely explanation for these observations is that in THB plus 1% glucose factors other than Esp are being regulated by PerA and playing a role in biofilm formation. This idea is supported by preliminary observations which suggest that esp expression may actually be downregulated ∼2-fold in the perA mutant (data not shown). Although it remains unclear what factors in the growth medium are influencing PerA regulation of biofilm formation, it has previously been demonstrated that Rbf, a member of the AraC/XylS family of transcriptional regulators, regulates biofilm formation in S. aureus in a glucose- and NaCl-dependent manner and impacts biofilm formation at the multicellular aggregation stage (20). Experiments are currently under way to identify the additional factors that may be subject to PerA regulation.
Given the location of the perA gene on the E. faecalis PAI, it was of particular interest to explore the role of PerA in pathogenesis. In order to determine the effect of PerA, a murine model of intraperitoneal and systemic infection was used (7, 17). The results clearly demonstrate that PerA contributes significantly to the pathogenicity of E99 in this particular model of systemic E. faecalis infection and identify PerA as a new regulator and the only AraC/XylS-type transcriptional regulator in E. faecalis currently ascribed a role in pathogenesis. Based on these results, we postulated that the increased bacterial load of wild-type E99 in hepatic and splenic tissues might be due to the ability to survive the oxidative burst within phagocytes. AraC-type family regulators are known to play a role in the stress response (10). An AraC-type regulator in Salmonella enterica, designated RamA, confers resistance to oxidative killing in vitro but not in vivo (41). E. faecalis is well recognized for its inherent ability to survive intracellular killing by phagocytes, and this trait may provide a mechanism for E. faecalis to cause systemic disease. Gentry-Weeks et al. (11) reported that E. faecalis persists in murine peritoneal macrophages for 72 h. Gilmore et al. (13) have proposed that nosocomial enterococcal infection arises from colonization of new gastrointestinal tract niches by hospital isolates of enterococci after treatment with antibiotics with little or no anti-enterococcal activity. These strains expand and translocate across the gastrointestinal tract and are then trafficked to a regional lymph node by macrophages (46, 47). Resistance to killing would therefore provide a mechanism to persist at extraintestinal sites, and thus understanding the mediators of this phenotype is important to the development of new therapies.
Studies of the regulation of the response to oxidative stress in E. faecalis have revealed a number of different regulators, including HypR, a member of the LysR family (45); a PerR-like polypeptide (44); and a PrfA-like protein designated Ers (12, 27). Verneuil et al. (45) demonstrated that a hypR mutant strain was significantly more sensitive to a hydrogen peroxide challenge and phagocytic killing than the wild-type strain and was attenuated in a mouse peritonitis model (43). Recently, a thiol peroxidase designated Tpx was identified as a target for HypR regulation, and a tpx deletion mutant was found to be impaired in macrophage survival (19). Verneuil et al. (44) also characterized a regulator with similarities to the peroxide regulator PerR in Bacillus subtilis. Interestingly, although a perR mutant strain displayed increased resistance to hydrogen peroxide relative to a wild-type strain, it did not exhibit any difference from the wild type in the ability to survive in mouse peritoneal macrophages (44). The authors of that study also demonstrated that the PerR is important for virulence, apparently through a mechanism different from the response to oxidative stress (44). However, the PerR regulon in E. faecalis remains undefined.
Giard et al. (12) recently identified a transcriptional regulator of E. faecalis, designated Ers (for enterococcal regulator of survival). This regulator was classified as a member of the Crp/Fnr family and showed a high degree of similarity to a PrfA-like regulator implicated in virulence of Streptococcus pyogenes (12, 26). These authors further demonstrated a role of this regulator in macrophage survival and resistance to oxidative stress and subsequently identified several genes that represent regulatory targets, including genes encoding members of the arginine deiminase system, a gene encoding citrate lyase, and a gene, EF_1459, encoding a hypothetical protein (27). However, the function of these gene products in macrophage survival is unclear, since targeted mutations in these genes did not result in any difference in macrophage survival ability (27).
Muller et al. evaluated the involvement of 8 of the two-component regulatory systems in E. faecalis in mediating resistance to killing by macrophages (23). Mutations in the cognate regulator component of the two-component system Err04-Ehk04 resulted in significant attenuation in the ability to survive in macrophages. Mutations in the Err05 and the Err06 genes seemed to influence survival as well. However, these results are difficult to interpret given that these mutants seemed to be poorly phagocytosed relative to the wild-type JH2-2 strain, i.e., these mutants were detected at 1 to 2 orders of magnitude lower than the wild type at the beginning of the assay (23). In a similar study, Verneuil et al. (42) showed that a mutant strain lacking the manganese-containing superoxide dismutase (sodA) was more susceptible to killing by murine peritoneal macrophages. This indicates a mechanism by which E. faecalis can resist the respiratory burst within macrophages. Whether the ability of PerA to influence survival within macrophages is due to its effect on resistance to oxidative killing remains to be investigated.
The identities of the regulatory targets of PerA in E. faecalis strain E99 are currently unknown. Given the location of the perA gene on the E. faecalis PAI, we have hypothesized that PAI genes, such as those encoding the enterococcal cytolysin, Esp, and aggregation substance, might represent PerA regulatory targets. Gentry-Weeks et al. (11) were not able to demonstrate a role for the cytolysin in macrophage survival; however, it was unclear whether this toxin was actually expressed intracellularly. Although the role of the esp gene has not been directly assessed in macrophage survival, aggregation substance, a surface-associated protein that promotes conjugative plasmid transfer by mediating cell to cell contact, has been shown to play a role in resisting macrophage killing (37). The authors of that study showed that strains expressing aggregation substance were more adherent and internalized to a greater degree than aggregation substance negative strains but were more resistant to phagocytic killing apparently through inhibition of the respiratory burst (37).
In summary, we have identified a gene on the E. faecalis PAI that encodes a putative transcriptional regulator involved in regulation of biofilm formation, pathogenesis, and intracellular macrophage survival. Experiments are currently under way to further elucidate the regulatory role of PerA in these processes in E. faecalis.
Acknowledgments
This study was supported, in part, by NIH grant R01 AI059673 to N.S. and an American Heart Association Postdoctoral Fellowship Award to P.S.C.
We thank Nikki Craig, Miriam Kebaya, Toby Allen, Xingmin Wang, Tamara Hunt, and Roman Wolf for technical support; Joe Wilkerson at the Oklahoma Medical Research Foundation for performing the transmission electron microscopy; and Mark Huycke for helpful discussions.
Editor: J. N. Weiser
Footnotes
Published ahead of print on 29 September 2008.
REFERENCES
- 1.Bourgogne, A., K. V. Singh, K. A. Fox, K. J. Pflughoeft, B. E. Murray, and D. A. Garsin. 2007. EbpR is important for biofilm formation by activating expression of the endocarditis and biofilm-associated pilus operon (ebpABC) of Enterococcus faecalis OG1RF. J. Bacteriol. 1896490-6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Callegan, M. C., B. D. Jett, L. E. Hancock, and M. S. Gilmore. 1999. Role of hemolysin BL in the pathogenesis of extraintestinal Bacillus cereus infection assessed in an endophthalmitis model. Infect. Immun. 673357-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carniol, K., and M. S. Gilmore. 2004. Signal transduction, quorum-sensing, and extracellular protease activity in Enterococcus faecalis biofilm formation. J. Bacteriol. 1868161-8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow, J. W., L. A. Thal, M. B. Perri, J. A. Vazquez, S. M. Donabedian, D. B. Clewell, and M. J. Zervos. 1993. Plasmid-associated hemolysin and aggregation substance production contribute to virulence in experimental enterococcal endocarditis. Antimicrob. Agents Chemother. 372474-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coburn, P. S., and M. S. Gilmore. 2003. The Enterococcus faecalis cytolysin: a novel toxin active against eukaryotic and prokaryotic cells. Cell Microbiol. 5661-669. [DOI] [PubMed] [Google Scholar]
- 6.Cotter, P. A., and V. J. DiRita. 2000. Bacterial virulence gene regulation: an evolutionary perspective. Annu. Rev. Microbiol. 54519-565. [DOI] [PubMed] [Google Scholar]
- 7.Dupont, H., P. Montravers, J. Mohler, and C. Carbon. 1998. Disparate findings on the role of virulence factors of Enterococcus faecalis in mouse and rat models of peritonitis. Infect. Immun. 662570-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Egan, S. M. 2002. Growing repertoire of AraC/XylS activators. J. Bacteriol. 1845529-5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelbert, M., E. Mylonakis, F. M. Ausubel, S. B. Calderwood, and M. S. Gilmore. 2004. Contribution of gelatinase, serine protease, and fsr to the pathogenesis of Enterococcus faecalis endophthalmitis. Infect. Immun. 723628-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gallegos, M. T., R. Schleif, A. Bairoch, K. Hofmann, and J. L. Ramos. 1997. Arac/XylS family of transcriptional regulators. Microbiol. Mol. Biol. Rev. 61393-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gentry-Weeks, C. R., R. Karkhoff-Schweizer, A. Pikis, M. Estay, and J. M. Keith. 1999. Survival of Enterococcus faecalis in mouse peritoneal macrophages. Infect. Immun. 672160-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giard, J. C., E. Riboulet, N. Verneuil, M. Sanguinetti, Y. Auffray, and A. Hartke. 2006. Characterization of Ers, a PrfA-like regulator of Enterococcus faecalis. FEMS Immunol. Med. Microbiol. 46410-418. [DOI] [PubMed] [Google Scholar]
- 13.Gilmore, M. S., P. S. Coburn, S. R. Nallapareddy, and B. E. Murray. 2002. Enterococcal virulence, p. 301-354. In M. S. Gilmore, D. B. Clewell, P. Courvalin, G. M. Dunny, B. E. Murray, and L. B. Rice (ed.), The enterococci: pathogenesis, molecular biology, and antibiotic resistance. ASM Press, Washington, DC.
- 14.Hancock, L. E., and M. S. Gilmore. 2002. The capsular polysaccharide of Enterococcus faecalis and its relationship to other polysaccharides in the cell wall. Proc. Natl. Acad. Sci. USA 991574-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hancock, L. E., and M. Perego. 2004. The Enterococcus faecalis fsr two-component system controls biofilm development through production of gelatinase. J. Bacteriol. 1865629-5639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huycke, M. M., C. A. Spiegel, and M. S. Gilmore. 1991. Bacteremia caused by hemolytic, high-level gentamicin-resistant Enterococcus faecalis. Antimicrob. Agents Chemother. 351626-1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ike, Y., H. Hashimoto, and D. B. Clewell. 1984. Hemolysin of Streptococcus faecalis subspecies zymogenes contributes to virulence in mice. Infect. Immun. 45528-530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kristich, C. J., V. T. Nguyen, T. Le, A. M. Barnes, S. Grindle, and G. M. Dunny. 2008. Development and use of an efficient system for random mariner transposon mutagenesis to identify novel genetic determinants of biofilm formation in the core Enterococcus faecalis genome. Appl. Environ. Microbiol. 743377-3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.La Carbona, S., N. Sauvageot, J. C. Giard, A. Benachour, B. Posteraro, Y. Auffray, M. Sanguinetti, and A. Hartke. 2007. Comparative study of the physiological roles of three peroxidases (NADH peroxidase, alkyl hydroperoxide reductase, and thiol peroxidase) in oxidative stress response, survival inside macrophages and virulence of Enterococcus faecalis. Mol. Microbiol. 661148-1163. [DOI] [PubMed] [Google Scholar]
- 20.Lim, Y., M. Jana, T. T. Luong, and C. Y. Lee. 2004. Control of glucose- and NaCl-induced biofilm formation by rbf in Staphylococcus aureus. J. Bacteriol. 186722-729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCormick, J. K., H. Hirt, C. M. Waters, T. J. Tripp, G. M. Dunny, and P. M. Schlievert. 2001. Antibodies to a surface-exposed, N-terminal domain of aggregation substance are not protective in the rabbit model of Enterococcus faecalis infective endocarditis. Infect. Immun. 693305-3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohamed, J. A., W. Huang, S. R. Nallapareddy, F. Teng, and B. E. Murray. 2004. Influence of origin of isolates, especially endocarditis isolates, and various genes on biofilm formation by Enterococcus faecalis. Infect. Immun. 723658-3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muller, C., M. Sanguinetti, E. Riboulet, L. Hebert, B. Posteraro, G. Fadda, Y. Auffray, and A. Rince. 2008. Characterization of two signal transduction systems involved in intracellular macrophage survival and environmental stress response in Enterococcus faecalis. J. Mol. Microbiol. Biotechnol. 1459-66. [DOI] [PubMed] [Google Scholar]
- 24.Nakata, M., A. Podbielski, and B. Kreikemeyer. 2005. MsmR, a specific positive regulator of the Streptococcus pyogenes FCT pathogenicity region and cytolysin-mediated translocation system genes. Mol. Microbiol. 57786-803. [DOI] [PubMed] [Google Scholar]
- 25.Nallapareddy, S. R., K. V. Singh, J. Sillanpaa, D. A. Garsin, M. Hook, S. L. Erlandsen, and B. E. Murray. 2006. Endocarditis and biofilm-associated pili of Enterococcus faecalis. J. Clin. Investig. 1162799-2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reid, S. D., A. G. Montgomery, and J. M. Musser. 2004. Identification of srv, a PrfA-like regulator of group A streptococcus that influences virulence. Infect. Immun. 721799-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Riboulet-Bisson, E., M. Sanguinetti, A. Budin-Verneuil, Y. Auffray, A. Hartke, and J. C. Giard. 2008. Characterization of the Ers regulon of Enterococcus faecalis. Infect. Immun. 763064-3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schlievert, P. M., P. J. Gahr, A. P. Assimacopoulos, M. M. Dinges, J. A. Stoehr, J. W. Harmala, H. Hirt, and G. M. Dunny. 1998. Aggregation and binding substances enhance pathogenicity in rabbit models of Enterococcus faecalis endocarditis. Infect. Immun. 66218-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shankar, N., A. S. Baghdayan, and M. S. Gilmore. 2002. Modulation of virulence within a pathogenicity island in vancomycin-resistant Enterococcus faecalis. Nature 417746-750. [DOI] [PubMed] [Google Scholar]
- 30.Shankar, N., C. V. Lockatell, A. S. Baghdayan, C. Drachenberg, M. S. Gilmore, and D. E. Johnson. 2001. Role of Enterococcus faecalis surface protein Esp in the pathogenesis of ascending urinary tract infection. Infect. Immun. 694366-4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shankar, V., A. S. Baghdayan, M. M. Huycke, G. Lindahl, and M. S. Gilmore. 1999. Infection-derived Enterococcus faecalis strains are enriched in esp, a gene encoding a novel surface protein. Infect. Immun. 67193-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shepard, B. D., and M. S. Gilmore. 1995. Electroporation and efficient transformation of Enterococcus faecalis grown in high concentrations of glycine. Methods Mol. Biol. 47217-226. [DOI] [PubMed] [Google Scholar]
- 33.Shepard, B. D., and M. S. Gilmore. 1999. Identification of aerobically and anaerobically induced genes in Enterococcus faecalis by random arbitrarily primed PCR. Appl. Environ. Microbiol. 651470-1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sifri, C. D., E. Mylonakis, K. V. Singh, X. Qin, D. A. Garsin, B. E. Murray, F. M. Ausubel, and S. B. Calderwood. 2002. Virulence effect of Enterococcus faecalis protease genes and the quorum-sensing locus fsr in Caenorhabditis elegans and mice. Infect. Immun. 705647-5650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh, K. V., S. R. Nallapareddy, and B. E. Murray. 2007. Importance of the ebp (endocarditis- and biofilm-associated pilus) locus in the pathogenesis of Enterococcus faecalis ascending urinary tract infection. J. Infect. Dis. 1951671-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh, K. V., X. Qin, G. M. Weinstock, and B. E. Murray. 1998. Generation and testing of mutants of Enterococcus faecalis in a mouse peritonitis model. J. Infect. Dis. 1781416-1420. [DOI] [PubMed] [Google Scholar]
- 37.Sussmuth, S. D., A. Muscholl-Silberhorn, R. Wirth, M. Susa, R. Marre, and E. Rozdzinski. 2000. Aggregation substance promotes adherence, phagocytosis, and intracellular survival of Enterococcus faecalis within human macrophages and suppresses respiratory burst. Infect. Immun. 684900-4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tendolkar, P. M., A. S. Baghdayan, M. S. Gilmore, and N. Shankar. 2004. Enterococcal surface protein, Esp, enhances biofilm formation by Enterococcus faecalis. Infect. Immun. 726032-6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tendolkar, P. M., A. S. Baghdayan, and N. Shankar. 2006. Putative surface proteins encoded within a novel transferable locus confer a high-biofilm phenotype to Enterococcus faecalis. J. Bacteriol. 1882063-2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trieu-Cuot, P., C. Carlier, C. Poyart-Salmeron, and P. Courvalin. 1990. A pair of mobilizable shuttle vectors conferring resistance to spectinomycin for molecular cloning in Escherichia coli and in gram-positive bacteria. Nucleic Acids Res. 184296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Straaten, T., L. Zulianello, D. A. van, D. L. Granger, R. Janssen, and J. T. van Dissel. 2004. Salmonella enterica serovar Typhimurium RamA, intracellular oxidative stress response, and bacterial virulence. Infect. Immun. 72996-1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verneuil, N., A. Maze, M. Sanguinetti, J. M. Laplace, A. Benachour, Y. Auffray, J. C. Giard, and A. Hartke. 2006. Implication of (Mn)superoxide dismutase of Enterococcus faecalis in oxidative stress responses and survival inside macrophages. Microbiology 1522579-2589. [DOI] [PubMed] [Google Scholar]
- 43.Verneuil, N., A. Rince, M. Sanguinetti, Y. Auffray, A. Hartke, and J. C. Giard. 2005. Implication of hypR in the virulence and oxidative stress response of Enterococcus faecalis. FEMS Microbiol. Lett. 252137-141. [DOI] [PubMed] [Google Scholar]
- 44.Verneuil, N., A. Rince, M. Sanguinetti, B. Posteraro, G. Fadda, Y. Auffray, A. Hartke, and J. C. Giard. 2005. Contribution of a PerR-like regulator to the oxidative-stress response and virulence of Enterococcus faecalis. Microbiology 1513997-4004. [DOI] [PubMed] [Google Scholar]
- 45.Verneuil, N., M. Sanguinetti, B. Y. Le, B. Posteraro, G. Fadda, Y. Auffray, A. Hartke, and J. C. Giard. 2004. Effects of the Enterococcus faecalis hypR gene encoding a new transcriptional regulator on oxidative stress response and intracellular survival within macrophages. Infect. Immun. 724424-4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wells, C. L., R. P. Jechorek, and S. L. Erlandsen. 1990. Evidence for the translocation of Enterococcus faecalis across the mouse intestinal tract. J. Infect. Dis. 16282-90. [DOI] [PubMed] [Google Scholar]
- 47.Wells, C. L., R. P. Jechorek, M. A. Maddaus, and R. L. Simmons. 1988. Effects of clindamycin and metronidazole on the intestinal colonization and translocation of enterococci in mice. Antimicrob. Agents Chemother. 321769-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]