

Abstract
The intracellular bacterial pathogen Legionella pneumophila follows a developmental cycle in which replicative forms (RFs) differentiate into infectious stationary-phase forms (SPFs) in vitro and in vivo into highly infectious mature intracellular forms (MIFs). The potential relationships between SPFs and MIFs remain uncharacterized. Previously we determined that L. pneumophila survives, but does not replicate, while it transiently resides (for 1 to 2 h) in food vacuoles of the freshwater ciliate Tetrahymena tropicalis before being expelled as legionellae-laden pellets. We report here that SPFs have the ability to rapidly (<1 h) and directly (in the absence of bacterial replication) differentiate into MIFs while in transit through T. tropicalis, indicating that SPFs and MIFs constitute a differentiation continuum. Mutant RFs lacking the sigma factor gene rpoS, or the response regulator gene letA, were unable to produce normal SPFs in vitro and did not fully differentiate into MIFs in vivo, further supporting the existence of a common mechanism of differentiation shared by SPFs and MIFs. Mutants with a defective Dot/Icm system morphologically differentiated into MIFs while in transit through T. tropicalis. Therefore, T. tropicalis has allowed us to unequivocally conclude that SPFs can directly differentiate into MIFs and that the Dot/Icm system is not required for differentiation, two events that could not be experimentally addressed before. The Tetrahymena model can now be exploited to study the signals that trigger MIF development in vivo and is the only replication-independent model reported to date that allows the differentiation of Dot/Icm mutants into MIFs.
The natural history of Legionella pneumophila indicates that this gram-negative bacterium evolved as an intracellular pathogen of amoebae. In nature, as well as in human-made water systems, reproduction of L. pneumophila takes place primarily in amoebae (16, 31, 37). When L. pneumophila accidentally reaches the alveoli of susceptible humans, it may cause an atypical pneumonia known as Legionnaires' disease, characterized by the intracellular growth of L. pneumophila in alveolar macrophages (reviewed in reference 13). L. pneumophila thus behaves like (although it is not) an obligate intracellular pathogen, emulating its close relatives Coxiella spp. (40, 42) and the many Legionella-like amoebal pathogens (3, 10).
Key to the life cycle of obligate intracellular pathogens is their ability to alternate between a replicative form (RF), which produces intracellular progeny, and a nonreplicative form that is infectious and survives in the environment until a new host is found (36). L. pneumophila is no exception (20), and it alternates between an RF and a mature intracellular form (MIF) that is highly infectious to cells in culture and resistant to environmental stress (12, 21, 22; reviewed in reference 18). At the ultrastructural level, the RF of L. pneumophila shows a typical gram-negative envelope, a moderately dense cytoplasm rich in ribosomes, and cell division. RFs are susceptible to environmental stress and exhibit poor infectivity (11, 30). In contrast, the MIF displays an altered and complex envelope architecture, an electron-dense cytoplasm with large inclusions, and no morphological evidence of cell division (15, 21).
In vitro, L. pneumophila grown in broth or on solid medium differentiates into infectious stationary-phase forms (11, 35) but does not form MIFs (21). Therefore, L. pneumophila RFs produce two differentiated forms, stationary-phase forms (SPFs) in vitro and MIFs in vivo. Although SPFs and MIFs share some traits, such as the ability to infect cells in culture, resistance to heat, and the presence of cytoplasmic inclusions, they show numerous differences: MIFs are 10- to 100-fold more infectious to HeLa cells than SPFs, have a unique envelope ultrastructure, have a very low basal respiration rate, and are resistant to antibiotics, high pH, and lysis by detergents (21). A plausible explanation for these observed similarities and differences between SPFs and MIFs is the possibility that SPFs represent a differentiation intermediate in the RF-to-MIF transition (21), a hypothesis that has remained experimentally unproven. Therefore, we set out to elucidate the developmental relationships between SPFs and MIFs, to better understand the process of L. pneumophila differentiation.
We have previously shown that L. pneumophila is efficiently ingested by the freshwater ciliate Tetrahymena tropicalis and transiently resides in the ciliate's food vacuoles for about 1 to 2 h before being expelled in the form of spherical pellets containing live legionellae (9). We also determined that during its transient residence in T. tropicalis, L. pneumophila does not replicate (9). Here, we report that T. tropicalis organisms feeding on SPFs expel live legionellae with all the morphological characteristics of MIFs, clearly indicating that SPFs have the ability to rapidly (<1 h) and directly (in the absence of bacterial replication) differentiate into MIFs, thus confirming that SPFs constitute differentiation intermediates in the RF-to-MIF transition. In addition, this study focuses on the morphology of the various L. pneumophila forms present in food vacuoles of T. tropicalis feeding on avirulent dot mutants, or regulatory mutants unable to produce normal SPFs in vitro, in an effort to identify some of the factors involved in MIF development.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The L. pneumophila Philadelphia-1 virulent strain Lp1-SVir (streptomycin resistant) has been described previously (29). The L. pneumophila Philadelphia-1 virulent strain Lp02 (a transformable thymidine auxotroph) and its nonvirulent, salt-tolerant derivatives JV303 (dotB mutant) and JV309 (dotA mutant) were obtained from Ralph Isberg, Tufts University Medical School (8, 43). Lp02 derivatives MB445 (rpoS::kan carrying the empty vector pKB5mobAΔ) and MB434 (letA-22-3 carrying the empty plasmid vector pMMBGentmobAΔ) were a gift from Michele Swanson, University of Michigan Medical School, as were the genetically complemented strains MB444 (rpoS::kan carrying the plasmid pRpoS+, which is pKB5mobAΔ harboring a wild-type copy of rpoS) and MB435 (letA-22-3 carrying the plasmid pLetA, which is pMMBGentmobAΔ harboring a wild-type copy of letA) (5, 26, 27). All strains were kept as frozen stocks at −80°C in buffered yeast extract (BYE) broth (see below) with 7% dimethyl sulfoxide. Frozen stocks of virulent strains Lp02, Lp1-SVir, MB444, and MB435 were made from crude lysates of infected HeLa cells after intracellular growth had taken place (23). Frozen stocks of Lp1-SVir were routinely grown for 3 to 5 days at 37°C in a humid incubator, on buffered charcoal-yeast extract agar (BCYE) (39) supplemented with 100 μg/ml of streptomycin. Frozen stocks of Lp02 and its derivatives JV303 and JV309 were grown on BCYE agar containing streptomycin and thymidine (both at 100 μg/ml). Strains MB444 and MB445 were grown on BCYE containing streptomycin (100 μg/ml) and kanamycin (25 μg/ml), but without thymidine. Strains MB434 and MB435 were grown on BCYE containing streptomycin and thymidine (both at 100 μg/ml), kanamycin (25 μg/ml), and gentamicin (50 μg/ml). Bacteria from the 5-day growth on solid media were either harvested and used without subculture to feed T. tropicalis (see below) or subcultured to stationary phase (11) in BYE with the appropriate supplements, when needed. BYE had the same formulation as BCYE, but charcoal and agar were omitted. Before use in feeding experiments, the bacterial cells were washed in modified Tris-buffered Osterhout's solution (TBOS) (9) and resuspended in an appropriate volume of the same solution to achieve an optical density (read at 620 nm) of 1 unit, equivalent to ∼109 legionellae/ml.
Tetrahymena speciation.
We previously identified our pellet-producing ciliate as a species of the genus Tetrahymena within the T. mobilis-T. tropicalis group, based on the DNA sequence of its gene encoding the small subunit rRNA (9). To determine the species, we used the DNA sequence of a fragment of the gene encoding the mitochondrial cytochrome c oxidase (CO1), as reported by Lynn and Strüder-Kypke (33). Total DNA was extracted by a modification of the method of Arroyo et al. (4). Briefly, ∼106 Tetrahymena cells were harvested by centrifugation (700 × g, 15 min) and washed once in 10 mM Tris, 1 mM EDTA, pH 8. The cell pellet was resuspended in 300 μl of lysis buffer (0.4% sodium dodecyl sulfate, 100 mM NaCl, 10 mM EDTA, 10 mM Tris, 0.02% dimethyl sulfoxide, 2.0% sucrose, pH 7) and incubated 1 h at 60°C, followed by 5 min at 93°C. Total DNA was then extracted with phenol-chloroform followed by an additional extraction with chloroform. After precipitation in ethanol at −20°C, the DNA pellet was solubilized in 16 MΩ · cm MilliQ water (Millipore) and used as template in subsequent PCRs. PCR amplification of the CO1 fragment was performed with forward primer 288 and reverse primer FolB as previously described (33), but using a slightly modified PCR amplification protocol, as follows: initial denaturation at 95°C for 5 min; 5 amplification cycles at 95°C for 45 s, 45°C for 1.5 min, and 72°C for 2 min; 30 cycles at 94°C for 45 s, 51°C for 2 min, and 72°C for 2 min; and a final extension at 72°C for 10 min. The amplification product was visualized by ethidium bromide staining after agarose gel electrophoresis and purified using the Marligen/Biosciences Inc. rapid gel extraction kit. DNA sequencing was performed at the Instituto de Investigaciones Biomédicas (Mexico City) using a Genetic Analyzer 310 (PE Applied Biosystems). The sequencing results indicated that our isolate belongs to the T. tropicalis species (33).
Ciliate cultures.
The T. tropicalis isolate used here was originally isolated from a cooling tower biofilm and maintained in axenic culture according to the procedures outlined by Elliot (14), as detailed elsewhere (9). Before use in feeding studies, T. tropicalis cells were gradually transferred from their plate count broth (PCB) (Difco) growth medium (by sequential pelleting at 700 × g for 10 min and resuspension) into increasing concentrations of modified TBOS. Modified TBOS contained (in mg/liter) NaCl (420), KCl (9.2), CaCl2 (4), MgSO4·7H2O (16), MgCl2·6H2O (34), and Tris base (121) and was sterilized by filtration in a bottle-top 0.45-μm filter (Nalgene) after adjusting the pH to 7. The intermediate concentration steps used for the PCB-to-TBOS transition were PCB 50%-TBOS 50%, PCB 25%-TBOS 75%, and PCB 12.5%-TBOS 87.5%.
Feeding experiments.
All feeding experiments were performed in six-well plates (Falcon Plastics) at room temperature (22 to 25°C) with 1.5 × 106 Tetrahymena cells and ∼5 × 108 L. pneumophila cells per well resuspended in 3 ml of modified TBOS. Tris-buffered Osterhout's solution. Samples were taken at different times for electron microscopy by pelleting the ciliate-bacteria mixture at 700 × g for 10 min in 15-ml conical tubes (Falcon Plastics) and removing the supernatant. To produce samples enriched in expelled pellets, the ciliate-bacteria mixture was incubated overnight (16 to 18 h) at room temperature and then centrifuged at 700 × g for 10 min in 15-ml conical tubes. The live ciliates were then allowed to swim back into suspension before removing the supernatant. This operation was repeated three times, after which the recovered sample almost exclusively contained aggregated, ciliate-expelled pellets containing legionellae.
HeLa cells.
HeLa cells were cultured at 37°C, 5% CO2 in 25-cm2 flasks (Falcon Plastics) containing 7 ml of complete minimal essential medium (MEM) as previously described (23). For infection, HeLa cells from flasks were trypsinized, transferred to six-well plates at a ratio of 106 cells per well, and cultured until confluent monolayers were formed. An inoculum of 108 bacteria/well was then added and incubated overnight (∼16 h) to allow bacterial invasion and initiation of intracellular growth. The infected cells were then washed three times with warm PBS and treated with gentamicin (100 μg/ml) for 2 h in MEM. The MEM with gentamicin was removed with two washes of warm PBS and replaced with fresh MEM without antibiotics. Then, HeLa cells were further incubated (for up to 2 to 3 days) and observed in a 1X71 Olympus inverted microscope until legionellae-laden vacuoles were observed. Then, the infected cultures were air dried for Giménez staining and/or harvested to be prepared for electron microscopy.
Light microscopy.
Ciliate cultures and HeLa cells were routinely monitored in a 1X71 Olympus inverted microscope equipped with phase contrast. Giménez staining with carbol fuchsin as primary stain and malachite green as secondary stain was performed as described by McDade (34) except that the malachite green solution was used at 4% instead of 0.8%. The application of Giménez staining to the detection of MIFs has been described elsewhere (21). Giménez-stained preparations were observed in an Olympus BX61 upright microscope equipped with an Evolution QEi monochrome digital camera (Media Cibernetics, San Diego, CA). Micrographs were captured as TIFF files using Image Pro software (Media Cibernetics). Direct counts of HeLa cells in suspension were done in a Neubauer hemocytometer, as was the enumeration of ciliates fixed with Lugol's iodine (9).
Transmission electron microscopy.
Samples of Tetrahymena cells feeding on L. pneumophila, taken at different times, were fixed in 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide, dehydrated with ethanol, and embedded in epoxy resin for thin sectioning, followed by standard staining in uranium and lead salts, as described previously (15). Thin sections were observed in a JEOL JEM-1230 transmission electron microscope equipped with a Hamamatsu ORCA-HR high-resolution (2,000 by 2,000 pixels) digital camera, and images were saved as TIFF files. To enumerate the various morphological forms of L. pneumophila present in our samples, we were careful to include ultrathin sections from at least two separate experiments and record images from at least 50 sectioned cells from each experiment. While quantification was not the foremost aim of this study, micrographs were purposely selected to represent the full scope of our observations, and counting of particular parameters was carried out on the actual micrographs, rather than at the electron microscope.
Immunoblotting to detect MagA.
Approximately 106 ciliates resuspended in 30 ml of modified TBOS were fed with ∼3 × 1010 L. pneumophila Lp1-SVir cells in 25-cm2 cell culture flasks. After a feeding period of 3 h, the ciliates were separated from free bacteria by filtration through SCWP 025 00 Millipore (Bedford, MA) hydrophilic membranes with 8.0-μm pores. The ciliates remaining on the filter were washed once by passing ∼30 ml of modified TBOS through the filter and then resuspended in 30 ml of modified TBOS by gentle pipetting, followed by a static 5-min incubation to allow ciliates to freely swim into suspension. Samples (3 ml total) were taken at various times and split to perform legionellae CFU counts and immunoblotting. Samples for CFU counts (1 ml) were placed in a 1.5-ml microcentrifuge tube and centrifuged at 10,000 × g for 1 min. To lyse ciliates, as well as disperse and break up legionella-laden pellets, the centrifugation pellet was quickly resuspended in 50 μl of 0.5% Triton X-100 in sterile deionized water (dH2O) with vigorous pipetting. Then, 450 μl of dH2O was added, followed by vigorous vortexing for 1 min. The sample was brought to 1 ml by adding 50 μl of 0.5% Triton X-100 in dH2O and 450 μl of dH2O, before performing serial dilutions. Aliquots (100 μl) of the 102 to 106 dilutions were spotted on BCYE plates and incubated at 37°C for 4 to 5 days before counting colonies. Samples for immunoblotting (2 ml) were centrifuged at 10,000 × g in two 1.5-ml microcentrifuge tubes for 1 min, and the centrifugation pellet was kept frozen until results from the bacterial CFU counts were obtained. Loading onto 12% acrylamide gels was adjusted based upon CFU/ml values for each sample, so that the equivalent to 5 × 104 CFU was loaded per lane in 15 μl of sample buffer. Solubilized proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis using a minigel vertical slab apparatus (Bio-Rad), and the separated proteins were transferred onto a nitrocellulose membrane in a Transblot apparatus (Bio-Rad). The nitrocellulose membrane was then immunostained with a hyperimmune rabbit serum raised against purified MagA protein, as described previously (22, 28). The immunostained membrane was scanned to produce a TIFF file that was then analyzed with Gel-Pro Analyzer version 4.5 software (Media Cybernetics, Silver Spring, MD), to obtain integrated optical density (IOD) values (in arbitrary units) for the immunostained MagA bands, using the single-band analysis tool. In a second experiment, we compared the amount of MagA present in Lp02 MIFs purified from Tetrahymena pellets, Lp02 MIFs purified from HeLa cells, and Lp02 SPFs collected by centrifugation from a BYE culture and washed once in dH2O. A suspension enriched in legionella-laden pellets (produced as described above under “Feeding experiments”) was mechanically disrupted by forcefully passing it 20 times through a 27-gauge needle. Released free MIFs and undisrupted small pellets were further fractionated in a continuous Percoll density gradient, produced as described previously (23). Free MIFs were collected from the bottom (most dense) layer. MIFs from HeLa cells were also purified in a continuous Percoll density gradient as previously reported (23). At least ∼2 × 109 bacteria (estimated by optical density) were dissolved in 60 to 100 μl of 2× Laemmli sample buffer, and the amount of total protein was then determined by the detergent-compatible DC protein assay (Bio-Rad). Loading onto 12% acrylamide gels was targeted to 20 μg of total protein per lane, and samples were processed for immunoblotting as described above. Before immunostaining with the MagA-specific antibody, the nitrocellulose membrane was stained with Ponceau-S and a digital image of the stained membrane was obtained as a TIFF file in an Epson ES 1200C scanner. After immunostaining, the IOD of MagA bands was obtained using a Gel-Pro Analyzer. These IOD values were then corrected for loading and efficiency of transfer in relation to the IOD of four arbitrarily selected and apparently constant bands from the Ponceau-S-stained nitrocellulose membrane.
RESULTS
Ultrastructural features of food vacuoles and vacuole maturation.
Our pellet-producing Tetrahymena sp. was speciated as T. tropicalis, based on the mitochondrial cytochrome c oxidase gene sequence (33). As previously reported (9), food vacuoles of diverse morphology and stage of maturation were present concurrently in each ciliate, due to constant food vacuole generation in actively feeding T. tropicalis cells (Fig. 1). Based on fine structural observations, it was clear that normal trafficking (or maturation) of the T. tropicalis food vacuoles was accompanied by a striking remodelling of the vacuoles. It has not escaped our notice that the morphological resemblance of this remodelling process and the early maturation of L. pneumophila-containing vacuoles in mammalian host cells suggests a common underlying mechanism. In the initial stages of feeding, nascent food vacuoles were easily identified at the profusely ciliated cytopharynx. These early vacuoles were characterized by a loose membrane defining a spacious interior (Fig. 1A and B). Based on previously reported pulse-chase experiments with fluorescent legionellae (9), internal vacuoles detached from the cytopharynx began to appear as early as 5 min after addition of the bacterial inoculum, indicating that early vacuoles are short-lived. Mid-stage maturation was depicted by vacuoles bound by a membrane that closely followed the contour of the contained legionellae and the first appearance of membranous material in the vacuole (Fig. 1C and D). Late-stage vacuoles were identified by their roughly spherical shape, a smooth membrane profile, tightly-packed bacteria and membrane remnants, and a characteristic “halo,” or space, between the condensed content and the food vacuole's membrane (Fig. 1E and F). Together, the mid- and late vacuole maturation stages must be completed in 50 to 60 min, because the first pellets expelled by actively feeding T. tropicalis are seen ∼1 h after addition of the bacterial inoculum (9). After 1 h of active feeding, each ultrathin-sectioned ciliate presented more late-stage vacuoles than mid-stage ones, suggesting that mid-stage maturation may be shorter than late-stage vacuole maturation. While our vacuole progression scheme is not strictly based on the time course, it closely agrees with early reports on Tetrahymena pyriformis which described the “dehydration” of the newly formed vacuole and the condensation of the contained bacteria into a tightly packed sphere (see references 14 and 38 and references within).
FIG. 1.

Estimated reconstruction of the morphological progression of Legionella-containing food vacuoles in T. tropicalis, as depicted in the electron microscope. (A and B) Development at the ciliate's cytopharynx of early spacious vacuoles with a smooth membrane and spherical shape. (C) Mid-stage vacuole bound by a circuitous membrane that closely follows the outline of the contained peripheral bacteria (white arrows) and showing the first evidence of bacterial cell degradation resulting in accumulation of membrane material (black arrow). Large bacterial inclusions, characteristic of MIF development, are indicated by the white arrowhead. (D) Mid-stage vacuole in which the lining membrane appears detached from the contained peripheral bacteria and has begun to acquire a smoother contour. The arrowhead points at a prominent inclusion. (E) Late-stage vacuole with a smooth lining membrane and a defined spherical shape. (F) Late-stage spherical-shaped vacuole depicting a “halo” or space between the smooth vacuolar membrane and the tight concentration of bacteria (wrapped by membrane fragments) similar in appearance to expelled pellets.
Morphological features of vacuole-contained legionellae.
Micrographs of early-, mid-, and late-stage legionellae-containing food vacuoles did not show ultrastructural evidence of bacterial cell division, suggesting that the legionellae in each food vacuole represent a captive, isolated, and nonreplicating population. These bacteria, even within early- and mid-stage vacuoles, showed several ultrastructural features typical of MIFs (Fig. 2). The MIF-associated features (previously detailed in references 15 and 21) included prominent cytoplasmic inclusions, thick-layered envelopes, a dark cytoplasm, irregular shapes, and an unapparent periplasm (Fig. 2). In each food vacuole bacteria seemed to be experiencing the same morphological changes at the same time (Fig. 1 and 2), suggesting synchronicity. Furthermore, the legionellae in vacuoles and free expelled pellets projected a bright red color after Giménez staining (not shown), another characteristic of MIFs (21). In addition, lysates obtained from ciliates fed with strain Lp1-SVir confirmed enhanced levels of MagA (Fig. 3), a protein previously reported to be a marker of MIF formation for Philadelphia-1 L. pneumophila strains (28). To avoid any potential skewing of our MagA immunoblotting results by differences in culturability (since the gel shown in Fig. 3 was loaded to equivalent CFU per lane), we ran a second immunoblot assay to compare purified Lp02 MIFs to purified Lp02 SPFs in which the gel's lanes were loaded based on total protein. The IOD values for the immunostained MagA bands in this second experiment were 212 for SPFs and 938 for the MIFs purified from T. tropicalis-expelled pellets. The IOD value of the immunostained MagA band of Lp02 MIFs purified from HeLa cells (used as a reference for enhanced expression of MagA [21]) was 547. From these results we concluded that L. pneumophila rapidly differentiates into MIFs in response to the food vacuole environment of T. tropicalis, in a rather synchronous process that is unlinked to bacterial replication.
FIG. 2.

L. pneumophila cells depicting typical characteristics of MIFs, in both mid-stage food vacuoles of T. tropicalis (A and B) and in expelled pellets (C and D). (A) Lp1-SVir cells depicting smooth (nonwavy) dark outer membranes, inclusions (I), and no apparent periplasm. (B) Lp02 cells showing an electron-dense thick layer in the inner leaflet of the outer membrane (arrow), the apparent lack of an inner membrane and periplasm, and inclusions (I). (C and D) Lp02 cells in pellets fixed 24 h postinoculation displaying thick laminated envelopes formed by the stacking of multiple layers of internal membranes (arrow in panel C), inclusions (I), electron-dense cytoplasm (white arrow in panel D), and irregular shapes (arrows in panel D). Bars, 100 nm (A to C) or 500 nm (D).
FIG. 3.
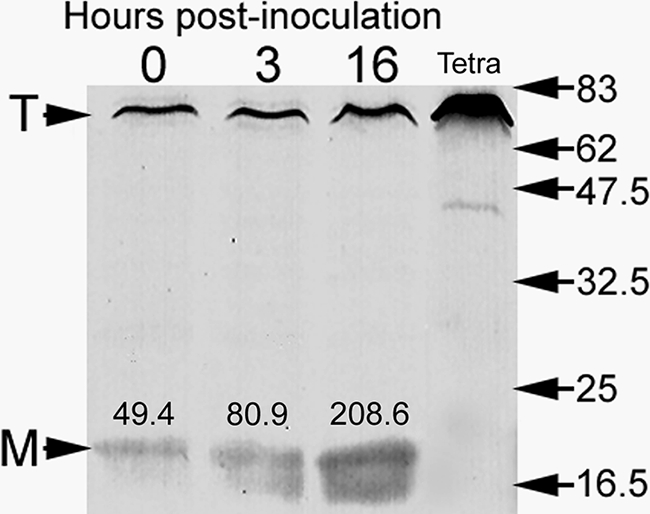
Immunoblot showing that expression of MagA (a protein marker of MIF development in Philadephia-1 strains) is upregulated during the interaction of Lp1-SVir with T. tropicalis. Samples were loaded based on CFU counts (5 × 104/lane). The mean CFU/ml values of the two independent samples pooled at each time point were 6.3 ± 1.52 × 105 (time zero), 8.5 ± 1.95 × 105 (3 h), and 18.3 ± 6.00 × 105 (16 h). The T arrowhead points to a Tetrahymena protein band recognized by the MagA polyclonal antibody. This protein band is well-labeled in samples containing ciliates only (lane labeled Tetra). The M arrowhead points at the position of MagA. Densitometry values in arbitrary IOD units are shown above each MagA band. The positions and molecular weights (×1,000) of broad-range, prestained protein standards are indicated on the right side.
rpoS and letA mutants are extensively digested in T. tropicalis.
The sigma factor RpoS and the response regulator LetA regulate the differentiation of L pneumophila RFs into SPFs, mainly in response to amino acid starvation in vitro (reviewed in references 19 and 35). Therefore, rpoS and letA mutants do not produce normal SPFs (26, 27). To check whether RpoS and LetA are also involved in MIF formation within T. tropicalis, the ciliates were fed the Lp02 derivatives MB445 (rpoS mutant) and MB434 (letA mutant) and their in vacuole ultrastructure was examined.
Unexpectedly, both regulatory mutants were digested to a large extent by T. tropicalis, and abundant membranous material was typically present in both food vacuoles and expelled pellets (Fig. 4). The accumulated membrane has been previously shown to be bacterial in origin and to represent outer membrane remains of digested legionellae (9). However, survivor rpoS mutants seemingly depicted morphological characteristics typical of MIFs, as shown in Fig. 4A, but their very low abundance did not allow us to make reliable observations. The number of letA mutant cells that resisted digestion was negligible. In fact, nearly all expelled pellets of the letA mutant contained no bacterial cells (Fig. 4E). Thus, the characterization of the rpoS and letA mutants' ultrastructures in T. tropicalis was compromised by the very low numbers of morphologically intact cells found.
FIG. 4.

Mutants unable to differentiate in vitro are readily digested by T. tropicalis. In two separate experiments Lp02 derivatives MB445 (rpoS mutant) (A and B) and MB434 (letA mutant) (D and E) did not survive in T. tropicalis vacuoles (A and D) and produced membranous pellets with virtually no surviving cells (B and E). Due to the very low number of morphologically intact bacteria it was not possible to assess bacterial morphology in a quantitative manner, but RpoS mutant survivors produced inclusions (arrow in panel A) and/or developed a smooth (nonwavy) outer membrane and dark cytoplasm (arrowhead in panel A), which are characteristics of MIFs. Genetically complemented rpoS and letA mutants recovered their ability to survive in food vacuoles and differentiate into MIFs. In expelled pellets from two separate samplings, the complemented rpoS and letA mutants (C and F, respectively) showed inclusions, straight dark envelopes, lack of an apparent periplasm, a dark cytoplasm, and irregular shapes. MIF-laden pellets in panels C and F still contained abundant membranous material in the space between bacterial cells (white arrows), suggesting that resistance to digestion did not reach parent strain (Lp02) levels.
To confirm that the inability to survive (and consequently differentiate) in T. tropicalis was indeed attributable to the mutated rpoS and letA genes, the mutants were genetically complemented in trans with plasmid-borne wild-type copies of rpoS and letA, respectively. The complemented mutant strains MB444 (wild-type rpoS) and MB435 (wild-type letA) recovered both the ability to resist in vacuole digestion and differentiate into MIFs and consequently were able to produce MIF-laden pellets in T. tropicalis (Fig. 4C and F). However, it is important to note that the recovery of wild-type MIF morphological traits in the genetically complemented strains was not complete, so that food vacuoles and expelled pellets produced from the genetically complemented mutants still contained abundant outer membrane remains, with 10 to 20% of the complemented cells lacking multilayered envelopes, inclusions, an electron-dense cytoplasm, and/or irregular shapes.
Extended observations of rpoS and letA mutants in HeLa cells.
To assess the ability of rpoS and letA mutants to form MIFs in other cellular models, and perhaps avoid the severe in vacuole degradation observed in T. tropicalis, HeLa cells (in which MIFs were first characterized [15, 23, 21]) were used. The two regulatory mutants were able to infect HeLa cells and proliferate, suggesting that their genetic defect is not growth limiting in mammalian cells. The rpoS mutant produced morphological forms characterized by a wavy outer membrane with sharp ripples and an electron-dense cytoplasm but lacking a multilayered envelope and irregular shapes (Fig. 5A and B). Late in the infection process (∼72 h), rpoS mutants associated with dead HeLa cells depicted a heterogeneous ultrastructure that included morphological characteristics typical of MIFs, i.e., nonwavy outer membranes and inclusions (Fig. 5B, inset). However, in ∼90% of the latter mutants a periplasmic space was still distinguishable, and the inclusions were not as numerous as in Lp02 MIFs. In other words, only ∼10% of the rpoS mutants found associated with dead HeLa cells showed the combined characteristics of MIFs, indicating that in HeLa cells the sigma factor RpoS is required for the full differentiation of L. pneumophila into MIFs. The letA mutant appeared even less able than the rpoS mutant to mature into MIFs or kill the host cell and remained in replicative vacuoles for extended periods (Fig. 5C). More than 95% of the letA mutants clearly showed the two membranes and periplasmic space typical of gram-negative envelopes and prominently depicted a wavy outer membrane with sharp ripples, but inclusion bodies were not found (Fig. 5D). Therefore, in the HeLa cell infection model the letA mutants did not differentiate into MIFs, suggesting that the response regulator LetA is essential for the maturation of RFs into MIFs.
FIG. 5.

Ultrastructure of rpoS and letA mutants in HeLa cells. Legionella-containing vacuoles of Lp02 derivatives MB445 (ΔrpoS) (A and B) and MB434 (letA mutant) (C and D) depict bacterial cells with morphological characteristics similar to previously described intermediate developmental forms of L. pneumophila (15). (A and C) Low-magnification micrographs showing that the mutants largely remained inside replicative vacuoles in heavily infected, but morphologically intact, host cells even after 3 days postinfection, a time at which the parent strain Lp02 had already killed most infected cells and produced abundant MIFs. (B) In morphologically intact HeLa cells, >90% of the rpoS mutant cells had outer membranes with wavy outlines (black arrowhead) and sharp ripples (white arrowhead), but thick layered envelopes were typically absent. In those HeLa cells showing signs of cell death or lysis, the rpoS mutant showed a heterogeneous morphology, which included the above-described morphology (25%), bacteria with inclusions (70%), and those with inclusions (I) and straight outer membranes (35%, inset). (D) Cells of the letA mutant (>95%) presented a single morphology characterized by wavy outer membranes depicted by sharp ripples (arrowhead) and did not develop inclusions. At least 50 infected cells were looked at in two separate samplings (for a total of ∼100 infected cells observed) of ultrathin sections cut from a single block.
We have previously determined that SPFs and MIFs show Giménez-positive (red) staining, with MIFs in particular projecting a bright red color (21). In spite of the incomplete ultrastructural differentiation of the rpoS and letA mutants in HeLa cells, both mutants showed bright Giménez staining (not shown).
dot mutants morphologically differentiate into MIFs.
We have previously shown that L. pneumophila dot mutants are largely digested in T. tropicalis food vacuoles (9). However, it is possible to find some vacuoles and expelled pellets containing undigested dot mutants. Thus, by increasing the sample size of transmission electron microscopy sections observed, we were able to establish that dot mutants displayed morphological features (Fig. 6) similar to those shown in Fig. 2 for the parent strain Lp02 MIFs. This observation indicated that the rapid morphological differentiation of SPFs into MIFs did not require a functional Dot/Icm system.
FIG. 6.

Differentiation of dot mutant SPFs into MIFs as observed by electron microscopy in T. tropicalis food vacuoles (A and B) or expelled pellets (C to E). (A and C) dotA mutant; (B, D, and E) dotB mutant. (A and B) Early development of MIFs was evident in mid-stage food vacuoles for both mutants. While several bacteria appeared degraded (e.g., black arrowhead in panel B), small groups of bacteria could be seen with irregular shapes, dark cytoplasm, and thick envelopes (white arrowheads in panel B), straight outer membranes with no evidence of periplasm, and inclusions (I). (C to E) In pellets, structurally complete bacteria also exhibited features typical of MIFs, e.g., thick, dark, and nonwavy outer membrane (arrow in panel C), multiple internal membranes (arrows in panel D), a laminated envelope with no obvious periplasm (arrows in panel E), dense cytoplasm, and prominent inclusions (I). Bars in panels C to E, 100 nm.
DISCUSSION
The developmental biology of L. pneumophila is beginning to be examined in molecular detail, and the study of SPFs has been crucial for the elucidation of regulatory networks that control the differentiation of L. pneumophila in vitro (reviewed in references 19 and 35). However, in vivo (i.e., intracellular differentiation) progress has been limited by the lack of experimental models where the complete and synchronous differentiation of L. pneumophila into MIFs can be achieved. Here we report that the ciliate T. tropicalis constitutes a useful experimental model to both study MIF development in vivo and explore the developmental relationships that may exist between SPFs and MIFs. By showing that SPFs directly and rapidly differentiate into MIFs while in transit through T. tropicalis, we have now proven that SPFs represent a stable intermediate in the differentiation of RFs to MIFs and have addressed previously unanswered questions in relation to the mechanism of L. pneumophila differentiation in vitro versus in vivo.
MIF development in T. tropicalis was recorded as early as 30 min after the addition of the bacterial inoculum. This rapid and direct in vacuole differentiation of SPFs into MIFs stands in sharp contrast to the differentiation of RFs into MIFs in HeLa cells (15, 23) and amoebae (24), a process preceded by bacterial replication that thus appears to be lengthy. In fact, MIF development may be equally fast in T. tropicalis, HeLa cells, and amoebae, once the right conditions for differentiation are met. In macrophages derived from the U937 human monocytic cell line, L. pneumophila does not fully develop into MIFs (21). We have thus hypothesized that the early apoptotic demise of macrophages results in insufficient time for differentiation to take place (21). In light of the results presented here, this hypothesis may require revision in favor of a possible failure in molecular signaling inside macrophages. Whereas in HeLa cells and amoebae the signals that trigger differentiation are seemingly delivered late in the growth cycle (after bacterial replication), in human macrophages these signals may not be present. The study of L. pneumophila gene expression in response to the vacuolar environment of Tetrahymena, as well as the characterization of such an in vivo environment, may present a rare opportunity to identify the signals that drive the direct differentiation of SPFs into MIFs.
Using the model presented in Fig. 7, we can now speculate about the nature of the signal(s) that triggers MIF development in T. tropicalis. Legionellae-laden food vacuoles always contain varied amounts of outer membrane remains (9) (Fig. 1), suggesting that the contained legionellae are exposed to T. tropicalis digestive factors. The rapid differentiation of SPFs into MIFs, the latter being characterized as the most resilient form of L. pneumophila (1, 6, 7, 21), could be viewed as an attempt by L. pneumophila to enhance survival in the hostile environment of the Tetrahymena food vacuole. If this were the case, the signals that trigger L. pneumophila differentiation might include structural damage to the bacterial cell envelope or the sensing of specific digestive enzymes. It is possible that this type of signal is also delivered in amoeba, particularly if the amoebal Legionella-containing vacuole fuses with lysosomes and becomes acidic late in the growth cycle, as reported to occur in bone marrow-derived mouse macrophages (41). Moreover, because the late Legionella-containing vacuole is not acidified in human macrophages (44), the signal would be incomplete in these cells, potentially explaining why MIFs do not fully develop in U937-derived macrophages. Alternatively, a host factor secreted into the food vacuole by the ciliate, or actively imported into the food vacuole by L. pneumophila, may constitute the signal that, in ciliates, triggers MIF development.
FIG. 7.

Model for the differentiation pathways of L. pneumophila. Steps supported by experimental evidence, previously reported or presented here, are indicated with solid lines and regular text. Speculative steps, not yet supported experimentally (but testable), are indicated by dotted lines and text in italics. White X's indicate closed pathways. We propose a cyclical sequence rather than a linear reversible one, implying that the two main differentiated forms of L. pneumophila, the RF and the MIF, have to follow two different pathways to reach each other. We have thus used the terms maturation and germination to distinguish these two pathways and the differentiation intermediates involved. Maturation refers to the RF-to-MIF differentiation process and germination to the MIF-to-RF differentiation process. We have now shown that SPFs represent a stable maturation intermediate produced in vitro. SPFs have been well-characterized, but their similarity to maturation intermediates produced in vivo remains to be established.
One requirement to be met by the signals that trigger the differentiation of SPFs into MIFs is that they must be different from the signals already present in the spent medium used to grow L. pneumophila in vitro, simply because SPFs do not differentiate into MIFs in vitro (21). It is known that in vitro low concentrations of amino acids trigger (via increased levels of ppGpp) the differentiation of RFs into SPFs (11, 26, 35). Therefore, additional signals (different from a low amino acid concentration) are required in vivo to drive the differentiation of SPFs into MIFs. Similarly, it is reasonable to surmise that additional regulatory molecules different from RpoS and the two-component regulatory system LetA/S are needed for the differentiation of SPFs into MIFs in vivo. Our data suggest that in HeLa cells RpoS and LetA are essential for the differentiation of RFs into MIFs. Consequently, the RF-to-SPF and the RF-to-MIF differentiation pathways share a common mechanism that involves these two factors. That is, RFs of the rpoS and letA mutants do not produce normal SPFs or MIFs. However, the SPF-to-MIF transition seems to be controlled by additional factors, different from RpoS and LetA, simply because the SPF-to-MIF and RF-to-MIF differentiation steps are not reversible. That is, whereas SPFs seem to easily revert to RFs in vitro (by transferring SPFs into fresh nutrient-rich medium), MIFs do not ostensibly revert to SPFs in vitro. In addition, MIFs do not revert to RFs via SPFs in vitro (15). Instead, when MIFs are placed in fresh medium they differentiate into RFs via an intermediate (designated the “germination” intermediate in Fig. 7) that is structurally dissimilar to SPFs (15, 20). Therefore, we propose that differentiation of MIFs into RFs is not achieved by simply tracing back the SPF-to-MIF and RF-to-SPF differentiation steps. To close the L. pneumophila developmental cycle, a differentiation pathway that involves regulatory molecules other than RpoS and LetA seems to be required (Fig. 7), a notion that has also been advanced to explain some aspects of SPF development in vitro (5). Therefore, to distinguish the pathways of L. pneumophila differentiation, in Fig. 7 we propose use of the term “maturation” to describe the differentiation process that results in the formation of MIFs and the term “germination” to describe the MIF-to-RF differentiation process. The use of the term germination does not imply that MIFs behave as spores or that the differentiation process of L. pneumophila conforms with the current models of bacterial germination, but it accurately conveys the notion that MIFs are changing from a resting, nonreplicative form into a metabolically active, vegetative RF.
The extensive degradation experienced by rpoS and letA mutants in T. tropicalis food vacuoles was surprising to us, mainly because we knew that resistance to digestion in T. tropicalis is dependent on a functional Dot/Icm system (9) and we assumed that rpoS and letA mutants would have intact Dot/Icm systems. However, RpoS and LetA turned out to be essential factors for the survival of L. pneumophila in T. tropicalis. In fact, the regulatory mutants had a much worse fate in T. tropicalis than the dot mutants. A possible explanation for this fact is the minor role that RpoS and LetA play in regulating the expression of some dot/icm genes (17, 32, 45). However, it is difficult to envisage that the nearly complete elimination of the rpoS and letA mutants could be caused by a minor alteration in the levels of some Dot/Icm proteins. Instead, the dramatic degradation of letA and rpoS mutants by T. tropicalis suggested a severe defect in the ability of these mutants to resist digestion. Thus, a more compelling argument would be that the function of the Dot/Icm system is developmentally regulated, or that developmentally regulated factors other than the Dot/Icm secretion apparatus play a significant role in resisting digestion. Perhaps the rpoS and letA mutants have structurally intact Dot/Icm systems but lack functional factors or effectors induced only during L. pneumophila differentiation into SPFs and MIFs. Alternatively, the Dot/Icm system may not be fully assembled in RFs and only becomes operational during the drastic morphological rearrangement of the L. pneumophila cell envelope (15) that takes place during differentiation of RFs into SPFs and MIFs. But apart from the underlying mechanisms involved in resistance to digestion, our results pose a very important derivative: that in nature, but not in the human host, L. pneumophila is under a strong selective pressure to differentiate. That is, we determined here that the rpoS and letA mutants were severely digested in T. tropicalis (Fig. 4), and others have shown that these differentiation mutants are unable to grow or survive in amoebae (2, 17, 25, 32), the preferred hosts of L. pneumophila in natural environments. On the other hand we found that the rpoS and letA mutants grew well in HeLa cells (Fig. 5), and others have shown that these differentiation mutants also grow (albeit sometimes showing an early growth defect) in mouse and human macrophages (2, 5, 17, 25). Consequently, the fact that human macrophages fail to coerce L. pneumophila to differentiate into MIFs (the L. pneumophila form that is most infectious to cells in culture) may be a factor to consider in the lack of person-to-person transmission of Legionnaires' disease.
Finally, the T. tropicalis infection model also offered the opportunity to separate (unlink) the processes of L. pneumophila differentiation and intracellular replication. Previously, in all the cellular models of differentiation, a replication phase was required before MIFs could be formed. Therefore, it had been impossible to determine whether dot/icm mutants failed to produce MIFs in vivo (in spite of being capable of differentiating into SPFs in vitro) as a consequence of their inability to replicate intracellularly, or because MIF development requires a functional Dot/Icm system. Using the Tetrahymena differentiation model we have now determined that mutants with a defective Dot/Icm virulence system morphologically differentiate into MIFs. In summary, T. tropicalis has allowed us to unequivocally conclude that SPFs directly differentiate into MIFs, and that the Dot/Icm system is not required for the maturation of L. pneumophila into MIFs, two events that could not be experimentally addressed before. The Tetrahymena model could now be exploited to study the signals that trigger differentiation into MIFs and as the only replication-independent model reported to date that allows the maturation of Dot/Icm mutants and perhaps other intracellular growth-deficient mutants of L. pneumophila.
Acknowledgments
We thank Michele Swanson for her insightful comments. The technical support of Mary Ann Trevors in the preparation of samples for electron microscopy is gratefully acknowledged, as is the help of Kim Jefferies and David Allan in the implementation of immunoblots with MagA-specific antibody.
This work was supported by the Canadian Institutes of Health Research, through operating grant ROP-83334 (R.A.G.) and major equipment maintenance grant PRG-80150 (G.F. and R.A.G.), as well as by the Center for the Management, Utilization and Protection of Water Resources at Tennessee Technological University (S.G.B.).
Footnotes
Published ahead of print on 19 September 2008.
REFERENCES
- 1.Abu Kwaik, Y., L.-Y. Gao, O. S. Harb, and B. J. Stone. 1997. Transcriptional regulation of the macrophage-induced gene (gspA) of Legionella pneumophila and phenotypic characterization of a null mutant. Mol. Microbiol. 24629-642. [DOI] [PubMed] [Google Scholar]
- 2.Abu-Zant, A., R. Asare, J. E. Graham, and Y. Abu Kwaik. 2006. Role for RpoS but not RelA of Legionella pneumophila in modulation of phagosome biogenesis and adaptation to the phagosomal microenvironment. Infect. Immun. 743021-3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adeleke, A., J. Pruckler, R. Benson, T. Rowbotham, M. Halablab, and B. Fields. 1996. Legionella-like amebal pathogens—phylogenetic status and possible role in respiratory disease. Emerg. Infect. Dis. 2225-230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arroyo, R., J. Engbring, J. Nguyen, O. Musatovova, O. Lopez, C. Lauriano, and J. F. Alderete. 1995. Characterization of cDNAs encoding adhesin proteins involved in Trichomonas vaginalis cytoadherence. Arch. Med. Res. 26361-369. [PubMed] [Google Scholar]
- 5.Bachman, M. A., and M. S. Swanson. 2001. RpoS co-operates with other factors to induce Legionella pneumophila virulence in the stationary phase. Mol. Microbiol. 401201-1214. [DOI] [PubMed] [Google Scholar]
- 6.Barker, J., M. R. W. Brown, P. J. Collier, I. Farrell, and P. Gilbert. 1992. Relationship between Legionella pneumophila and Acanthamoeba polyphaga: physiological status and susceptibility to chemical inactivation. Appl. Environ. Microbiol. 582420-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barker, J., H. Scaife, and M. R. Brown. 1995. Intraphagocytic growth induces an antibiotic-resistant phenotype of Legionella pneumophila. Antimicrob. Agents Chemother. 392684-2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berger, K. H., and R. R. Isberg. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 77-19. [DOI] [PubMed] [Google Scholar]
- 9.Berk, S. G., G. Faulkner, E. Garduño, M. C. Joy, M. A. Ortiz-Jiménez, and R. A. Garduño. 2008. Packaging of live Legionella pneumophila into pellets expelled by Tetrahymena sp. does not require bacterial replication and depends on a Dot/Icm-mediated survival mechanism. Appl. Environ. Microbiol. 742187-2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berk, S. G., J. H. Gunderson, A. L. Newsome, A. L. Farone, B. J. Hayes, K. S. Redding, N. Uddin, E. L. Williams, R. A. Johnson, M. Farsian, A. Reid, J. Skimmyhorn, and M. B. Farone. 2006. Occurrence of infected amoebae in cooling towers compared with natural aquatic environments: Implications for emerging pathogens. Environ. Sci. Technol. 407440-7444. [DOI] [PubMed] [Google Scholar]
- 11.Byrne, B., and M. S. Swanson. 1998. Expression of Legionella pneumophila virulence traits in response to growth conditions. Infect. Immun. 663029-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cirillo, J. D., S. Falkow, and L. S. Tompkins. 1994. Growth of Legionella pneumophila in Acanthamoeba castellani enhances invasion. Infect. Immun. 623254-3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dowling, J. N., A. K. Saha, and R. H. Glew. 1992. Virulence factors of the family Legionellaceae. Microbiol. Rev. 5632-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elliot, A. M. (ed.). 1973. Biology of Tetrahymena. Dowden, Hutchinson & Ross, Inc., Stroudsburg, PA.
- 15.Faulkner, G., and R. A. Garduño. 2002. Ultrastructural analysis of differentiation in Legionella pneumophila. J. Bacteriol. 1847025-7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fields, B. S. 1993. Legionella and protozoa: interaction of a pathogen and its natural host, p. 129-136. In J. M. Barbaree, R. F. Breiman, and A. P. Dufour (ed.), Legionella: current status and emerging perspectives. American Society for Microbiology, Washington, DC.
- 17.Gal-Mor, O., and G. Segal. 2003. The Legionella pneumophila GacA homolog (LetA) is involved in the regulation of icm virulence genes and is required for intracellular multiplication in Acanthamoeba castellanii. Microb. Pathogen. 34187-194. [DOI] [PubMed] [Google Scholar]
- 18.Garduño, R. A. 2008. Life cycle, growth cycles and developmental cycle of Legionella pneumophila, p. 65-84. In P. Hoffman, H. Friedman, and M. Bendinelli (ed.), Legionella pneumophila: pathogenesis and immunity. Springer, New York, NY.
- 19.Garduño, R. A., A. Chong, and G. Faulkner. 2008. Developmental cycle—differentiation of Legionella pneumophila, p. 55-73. In K. Heuner and M. Swanson (ed.), Legionella: molecular microbiology. Caister Academic Press, Norfolk, United Kingdom.
- 20.Garduño, R. A., E. Garduño, M. Hiltz, D. Allan, and P. S. Hoffman. 2002. Morphological and physiological evidence for a developmental cycle in Legionella pneumophila, p. 82-85. In R. Marre, Y. AbuKwaik, C. Bartlett, N. Cianciotto, B. S. Fields, M. Frosch, J. Hacker and P. C. Lück (ed.), Legionella. ASM Press, Washington, DC.
- 21.Garduño, R. A., E. Garduño, M. Hiltz, and P. S. Hoffman. 2002. Intracellular growth of Legionella pneumophila gives rise to a differentiated form dissimilar to stationary-phase forms. Infect. Immun. 706273-6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garduño, R. A., E. Garduño, and P. S. Hoffman. 1998. Surface-associated Hsp60 chaperonin of Legionella pneumophila mediates invasion in a HeLa cell model. Infect. Immun. 664602-4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garduño, R. A., F. Quinn, and P. S. Hoffman. 1998. HeLa cells as a model to study the invasiveness and biology of Legionella pneumophila. Can. J. Microbiol. 44430-440. [PubMed] [Google Scholar]
- 24.Greub, G., and D. Raoult. 2003. Morphology of Legionella pneumophila according to their location within Hartmanella vermiformis. Res. Microbiol. 154619-621. [DOI] [PubMed] [Google Scholar]
- 25.Hales, L. M., and H. A. Shuman. 1999. The Legionella pneumophila rpoS gene is required for growth within Acanthamoeba castellanii. J. Bacteriol. 1814879-4889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hammer, B. K., and M. S. Swanson. 1999. Co-ordination of Legionella pneumophila virulence with entry into stationary phase by ppGpp. Mol. Microbiol. 33721-731. [DOI] [PubMed] [Google Scholar]
- 27.Hammer, B. K., E. S. Tateda, and M. S. Swanson. 2002. A two-component regulator induces the transmission phenotype of stationary-phase Legionella pneumophila. Mol. Microbiol. 44107-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hiltz, M. F., G. R. Sisson, A. K. Brassinga, E. Garduño, R. A. Garduño, and P. S. Hoffman. 2004. Expression of magA in Legionella pneumophila Philadelphia-1 is developmentally regulated and a marker of formation of mature intracellular forms. J. Bacteriol. 1863038-3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffman, P. S., C. A. Butler, and F. D. Quinn. 1989. Cloning and temperature-dependent expression in Escherichia coli of a Legionella pneumophila gene coding for a genus-common 60-kilodalton antigen. Infect. Immun. 581731-1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi, A. D., S. Sturgill-Koszycki, and M. S. Swanson. 2001. Evidence that Dot-dependent and -independent factors isolate the Legionella pneumophila phagosome from the endocytic network in mouse macrophages. Cell. Microbiol. 399-114. [DOI] [PubMed] [Google Scholar]
- 31.Kuiper, M. W., B. A. Wullings, A. D. L. Akkermans, R. R. Beumer, and D. van der Kooij. 2004. Intracellular proliferation of Legionella pneumophila in Hartmanella vermiformis in aquatic biofilms grown on plasticized polyvinyl chloride. Appl. Environ. Microbiol. 706826-6833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lynch, D., N. Rieser, K. Gloggler, V. Forsbach-Brik, and R. Marre. 2003. The response regulator LetA regulates the stationary-phase stress response in Legionella pneumophila and is required for efficient infection of Acanthamoeba castellanii. FEMS Microbiol. Lett. 219241-248. [DOI] [PubMed] [Google Scholar]
- 33.Lynn, D. H., and M. C. Strüder-Kypke. 2006. Species of Tetrahymena identical by small subunit rRNA gene sequences are discriminated by mitochondrial cytochrome c oxidase I gene sequences. J. Eukaryot. Microbiol. 53385-387. [DOI] [PubMed] [Google Scholar]
- 34.McDade, J. E. 1979. Primary isolation using guinea pigs and embryonated eggs, p. 70-76. In G. L. Jones and G. A. Hébert (ed.), “Legionnaires'” the disease, the bacterium and methodology. Centers for Disease Control, Bureau of Laboratories, U.S. Public Health Service, U.S. Department of Health, Education, and Welfare, Atlanta, GA.
- 35.Molofsky, A. B., and M. S. Swanson. 2004. Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 5329-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moulder, J. W. 1985. Comparative biology of intracellular parasitism. Microbiol. Rev. 49298-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murga, R., T. S. Forster, E. Brown, J. M. Pruckler, B. S. Fields, and R. M. Donlan. 2001. Role of biofilms in the survival of Legionella pneumophila in a model potable-water system. Microbiology 1473121-3126. [DOI] [PubMed] [Google Scholar]
- 38.Nilsson, J. R. 1987. Structural aspects of digestion of Escherichia coli in Tetrahymena. J. Protozool. 341-6. [Google Scholar]
- 39.Pascule, A. W., J. C. Feeley, R. J. Gibson, L. G. Cordes, R. L. Meyerowitz, C. M. Patton, G. W. Gorman, C. L. Carmack, J. W. Ezzell, and J. N. Dowling. 1980. Pittsburgh pneumonia agent: direct isolation from human lung tissue. J. Infect. Dis. 141727-732. [DOI] [PubMed] [Google Scholar]
- 40.Samuel, J. E., K. Kiss, and S. Varghees. 2003. Molecular pathogenesis of Coxiella burnetii in a genomics era. Ann. N. Y. Acad. Sci. 990653-663. [DOI] [PubMed] [Google Scholar]
- 41.Sturgill-Koszycki, S., and M. S. Swanson. 2000. Legionella pneumophila replication vacuoles mature into acidic, endocytic organelles. J. Exp. Med. 1921261-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vogel, J. P. 2004. Turning a tiger into a house cat: Using Legionella pneumophila to study Coxiella burnetii. Trends Microbiol. 12103-105. [DOI] [PubMed] [Google Scholar]
- 43.Vogel, J., C. Roy, and R. R. Isberg. 1996. Use of salt to isolate Legionella pneumophila mutants unable to replicate in macrophages. Ann. N. Y. Acad. Sci. 797271-272. [DOI] [PubMed] [Google Scholar]
- 44.Wieland, H., F. Goetz, and B. Neumeister. 2004. Phagosomal acidification is not a prerequisite for intracellular multiplication of Legionella pneumophila in human monocytes. J. Infect. Dis. 1891610-1614. [DOI] [PubMed] [Google Scholar]
- 45.Zusman, T., O. Gal-Mor, and G. Segal. 2002. Characterization of a Legionella pneumophila relA insertion mutant and roles of RelA and RpoS in virulence gene expression. J. Bacteriol. 18467-75. [DOI] [PMC free article] [PubMed] [Google Scholar]