

Abstract
The locus of enterocyte effacement (LEE) pathogenicity island of enterohemorrhagic and enteropathogenic Escherichia coli (EHEC and EPEC, respectively) comprises a cluster of operons encoding a type III secretion system and related proteins, all of which are essential for bacterial colonization of the host intestines. The LEE1 operon encodes Ler, which positively regulates many EPEC and EHEC virulence genes located in the LEE region and elsewhere in the chromosome. In addition, Ler is a specific autorepressor of LEE1 transcription. To better understand the function of Ler, we screened for Ler mutants defective in autorepression. We isolated 18 different point mutations in Ler, rendering it defective in autorepression and in DNA binding. Among these mutants were those defective in positive regulation as well as in autorepression, dominant-negative mutants, and a mutant deficient in oligomerization. Importantly, a group of Ler autorepression mutants complemented an EPEC ler deletion mutant for transcription activation in a dosage-dependent manner, suggesting that Ler and possibly other autorepressors have an intrinsic compensatory mechanism that enables them to sustain mutations. In addition, the phenotypes of the different mutants identified by the screen define a novel domain in Ler that is required for oligomerization.
Enterohemorrhagic Escherichia coli and enteropathogenic E. coli (EPEC) represent a major global health problem, causing hemorrhagic colitis and hemolytic-uremic syndrome (enterohemorrhagic E. coli) and watery diarrhea of infants (EPEC). Their virulence is mediated by the concerted activity of an array of virulence factors. A major player among these factors is a type III protein secretion system (TTSS), through which these pathogens inject (translocate) toxic proteins (effectors) into host cells, thereby causing “attaching and effacing” intestinal lesions (22). The attaching-and-effacing histopathology is characterized by a localized destruction of the brush border microvilli and an assembly of highly organized pedestal-like actin structures in the epithelial cells beneath the intimately attached bacteria (11). The genes coding for the TTSS components, as well as for several of the effectors translocated by the TTSS, are clustered in a 35-kb chromosomal region, termed the locus of enterocyte effacement (LEE) (18, 19). In the LEE, genes are organized into five major operons (designated LEE1 to LEE5) and several smaller transcriptional units (reviewed in reference 16). Ler, encoded by the first gene in the LEE1 operon, positively regulates the expression of most of the LEE genes. Transcription activation by Ler is achieved by alleviating abundant DNA binding protein H-NS-mediated repression of the LEE promoters (6, 31). Ler activates and represses additional virulence genes located outside the LEE (1, 9, 17).
In addition to its role in activating the transcription of the LEE genes, Ler also functions as an autorepressor, by repressing the transcription of the LEE1 promoter (PLEE1) (3). Autorepression is a common motif in bacterial regulatory networks, shared by over 40% of the transcription factors in E. coli K12 (29). In addition to autorepression, Ler expression (PLEE1 activity) is tightly controlled by a plethora of regulators, including IHF, Fis, PerC, BipA, GrlA, GrlR, GadX and quorum sensing (7, 12-15, 21, 24, 25, 27), attesting to its central role in virulence gene regulation.
In this work, we subjected ler to random mutagenesis and screened for mutants defective in autorepression. We isolated and characterized 18 different point mutations that interfere with autorepression. All of the mutations caused a significant decrease in DNA binding. Most of the mutants were defective in transcription activation of the LEE genes, and some exhibited a dominant-negative effect over wild-type Ler. One of the mutants was defective in oligomerization, harboring a mutation in a region never predicted to be involved in oligomerization. Four mutants were able to successfully complement a ler-deleted strain, activating LEE transcription in a dosage-dependent manner. The latter results suggest that Ler harbors an intrinsic compensatory mechanism that enables it to sustain mutations, and this mechanism may be general to autorepressed genetic systems.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth media.
The bacterial strains and plasmids used in this study are listed in Table 1. Strains were grown in Luria-Bertani (LB) broth at 30°C and diluted 1:50 into Dulbecco's modified Eagle's medium (DMEM) or modified Casamino-DMEM (3). When needed, LB was supplemented with ampicillin (Amp) at 100 μg/ml, kanamycin (Kan) at 40 μg/ml, or 20 mM (NH4)2SO4 (to repress LEE1 expression).
TABLE 1.
List of strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| E. coli strains | ||
| E2348/69 | EPEC wild type | J. Kaper |
| TU1403 | E2348/69 carrying a chromosomal ler-His6 | 3 |
| DF2 | E2348/69 ler::kan | 12 |
| A70 | CC102 (ara Δ(lac-proB)XIIIthi F′-lacI378 lacZ461 proA+B+) mutS215::Tn10 | S. Altuvia |
| W3110 | F− λ−thyA36 deoC2 IN(rrnD-rrnE)1 | A. Oppenheim |
| Plasmids | ||
| pIR1 | pKK177-3 derivative containing gfpmut3 | 12 |
| pGY1 | pIR1 containing a transcriptional fusion of PLEE1, ler, and gfpmut3 (PLEE1-ler-gfpmut3) | This study |
| pGY2180 | pGY1 carrying PLEE1-ler(L16P)-gfpmut3 | This study |
| pGY2206 | pGY1 carrying PLEE1-ler(L23R)-gfpmut3 | This study |
| pGY2189 | pGY1 carrying PLEE1-ler(L56P)-gfpmut3 | This study |
| pGY2190 | pGY1 carrying PLEE1-ler(Y77H)-gfpmut3 | This study |
| pGY2191 | pGY1 carrying PLEE1-ler(Y77C)-gfpmut3 | This study |
| pGY2192 | pGY1 carrying PLEE1-ler(G82V)-gfpmut3 | This study |
| pGY2207 | pGY1 carrying PLEE1-ler(G82E)-gfpmut3 | This study |
| pGY2208 | pGY1 carrying PLEE1-ler(W85R)-gfpmut3 | This study |
| pGY2209 | pGY1 carrying PLEE1-ler(G87S)-gfpmut3 | This study |
| pGY2210 | pGY1 carrying PLEE1-lerG87D)-gfpmut3 | This study |
| pGY2211 | pGY1 carrying PLEE1-ler(G89D)-gfpmut3 | This study |
| pGY2193 | pGY1 carrying PLEE1-ler(P92S)-gfpmut3 | This study |
| pGY2194 | pGY1 carrying PLEE1-ler(W94R)-gfpmut3 | This study |
| pGY2195 | pGY1 carrying PLEE1-ler(L95P)-gfpmut3 | This study |
| pGY2212 | pGY1 carrying PLEE1-ler(A98V)-gfpmut3 | This study |
| pGY2196 | pGY1 carrying PLEE1-ler(G102R)-gfpmut3 | This study |
| pGY2213 | pGY1 carrying PLEE1-ler(G102E)-gfpmut3 | This study |
| pGY2214 | pGY1 carrying PLEE1-ler(L109P)-gfpmut3 | This study |
| pGY2501 | pGY1 carrying PLEE1-ler(E4Stop)-gfpmut3 | This study |
| pGY3682 | pGY1 carrying PLEE1-ler(N50Stop)-gfpmut3 | This study |
| pGY3683 | pGY1 carrying PLEE1-ler(G65Stop)-gfpmut3 | This study |
| pSA10 | pKK177-3 derivative containing lacIq | 23 |
| pGY2746 | pSA10 carrying ler | This study |
| pGY2742 | pSA10 carrying ler(L23R) | This study |
| pGY2743 | pSA10 carrying ler(G82E) | This study |
| pGY2744 | pSA10 carrying ler(A98V) | This study |
| pGY2745 | pSA10 carrying ler(G102R) | This study |
| pGY3576 | pSA10 carrying ler(G89D) | This study |
| pQE-70 | Expression of C-terminally His6-tagged proteins | Qiagen |
| pDF5 | pACYC184 containing lacIq and ler expressed from the tac promoter | 12 |
| pDF11 | pQE-70-based, expressing Ler-His6 from the PLEE1promoter | This study |
Plasmid construction.
The primers used for plasmid construction are listed in Table S1 in the supplemental material. To construct pGY1, a DNA fragment containing PLEE1 and ler (starting from position −159 compared to the transcriptional start site determined in reference 21) was amplified using primers specified in Table S1 in the supplemental material; this amplified fragment was digested by XbaI and BamHI and cloned into pIR1 digested by the same enzymes. pGY1 derivatives carrying ler mutants and the plasmids carrying the truncated ler variants N50Stop and G65Stop (pGY3682 and pGY3683, respectively) were generated using the QuikChange site-directed mutagenesis kit (Stratagene) with the primers specified in Table S1 in the supplemental material. For pGY3682 and pGY3683, an additional step of PCR and self-ligation to delete the ler C-terminal coding region after the newly inserted stop codon followed. To construct a plasmid expressing Ler under the transcriptional regulation of Ptac (pGY2746), a DNA fragment containing ler was amplified from pGY1, digested by EcoRI and SalI, and cloned into pSA10, which was digested by the same enzymes. In order to generate plasmids expressing LerL23R, LerG82E, LerA98V, LerG102R, and LerG89D under the transcriptional regulation of Ptac (pGY2742, pGY2743, pGY2744, pGY2745, and pGY3576, respectively), similar fragments were amplified from the relevant pGY1 derivatives (pGY2206, pGY2207, pGY2212, pGY2196, and pGY2211, respectively) and cloned in a similar manner. To construct pDF11, which contains ler fused in-frame to a His6 tag at the C terminus (expressed from its native promoter and ribosome binding site), a DNA fragment containing PLEE1 and ler was amplified using the primers specified in Table S1 in the supplemental material; this amplified fragment was digested by XhoI and BamHI and cloned into pQE-70, which was digested by the same enzymes.
Fluorescence microscopy.
To test for the formation of actin pedestals and gfp expression from PLEE1, HeLa cells were seeded and grown overnight on glass coverslips in 24-well plates. The cells, in 1 ml of DMEM, were infected with 10 μl of overnight LB standing bacterial cultures. Infection was stopped after 3.5 h by fixing the infected cells in phosphate-buffered saline (PBS) containing 2% paraformaldehyde for 30 min. The fixed cells were washed with PBS, permeabilized for 5 min with 0.1% Triton X-100 in PBS, and washed as before. The actin filaments were stained by overlaying the coverslips with 20 μl (0.1 μg/ml in Tris-buffered saline) of phalloidin-rhodamine (Sigma). After 1 h of incubation, the samples were washed, mounted on glass slides, and analyzed.
Measurement of gene expression.
Strains containing the gfp-expressing plasmids pIR1 and pGY1 and the pGY1 derivatives carrying ler mutants were grown overnight at 30°C in LB supplemented with 20 mM (NH4)2SO4. The cultures were washed and diluted 1:50 in Casamino-DMEM and then grown in 96-well plates in a microplate reader at 37°C (SPECTRAFluor Plus; Tecan). The fluorescence intensity (filter set at a 485-nm excitation wavelength and a 535-nm emission wavelength) and optical density at 600 nm (OD600) were read and collected using Magellan version 5.0 software (Tecan). To determine the protein levels, strains were grown in DMEM at 37°C up to an OD600 of 0.3 to 0.4. When indicated, different concentrations of IPTG (isopropyl-β-d-thiogalactopyranoside) were added. The cultures were centrifuged and the bacteria lysed by boiling in sodium dodecyl sulfate (SDS) loading buffer. Protein concentrations in the samples were adjusted, and then the samples were subjected to immunoblot analysis. Blots were developed with polyclonal anti-Tir, anti-EspB, or anti-Ler antibodies.
Genetic screen to identify mutants in autorepression.
The pGY1 plasmid was introduced into an E. coli mutS strain (A70). Plasmids were prepared from an overnight culture of the mutS strain, introduced into EPEC ler::kan, and plated to give ∼500 individual colonies per plate, and colonies that exhibited high green fluorescence were picked (108 colonies). Those that did not express Ler or expressed truncated Ler proteins were identified by immunoblot analysis with an anti-Ler antibody and omitted, leaving 45 mutant strains. The plasmids from these strains were prepared, and the LEE1 regulatory region and the ler coding region were sequenced. Each of these plasmids was found to contain a single mutation, and several of them carried the same mutation. The screen rendered 17 distinct mutations; each was regenerated on a plasmid that had not been previously introduced into the mutS strain. For this, a QuikChange site-directed mutagenesis kit (Stratagene) was used with the primers specified in Table S1 in the supplemental material. The L23R mutation added to this group was previously termed LerL29R (3, 27), and we renamed it LerL23R, given the identification of the second methionine of Ler as its translation start site.
Analysis of the Ler translation start site.
The pDF11 plasmid containing the ler native promoter and regulatory region and ler fused to a His6 tag at the region encoding the C terminus was introduced into wild-type EPEC. Bacteria were grown in DMEM at 37°C to an OD600 of 0.4, and the Ler-His6 protein was purified by metal affinity chromatography using Talon beads (Clontech), followed by SDS-polyacrylamide gel electrophoresis (PAGE) gel and blotting onto a polyvinylidene difluoride membrane, and then subjected to N-terminal amino acid sequencing.
Protein-DNA binding assay.
A modified enzyme-linked DNA/protein interaction assay (ELDIA) (2) was employed. Briefly, three types of 5′-biotinylated PCR fragments were used: the LEE1 regulatory region (prepared using the primers BioLEE1reg2-F and 5R), the LEE2 regulatory region (prepared using the primers BioLEE2reg2-F and 31R; see Table S1 in the supplemental material) and the etk (yccC) coding region (a negative control; prepared using the primers BioYccC-F and YccC-R [see Table S1 in the supplemental material]). A total of 100 μl of 500 pmol/ml of each of the fragments in PT buffer (PBS supplemented with 0.05% Tween 20 and 1 mM EDTA) were bound separately to streptavidin-coated 96-well plates (Sigma) by incubating for 1 h. Unbound DNA was removed by washing with PT. Whole-cell extracts were prepared by sonicating (in 50 mM Tris-HCl [pH 7.4], 70 mM KCl, 5 mM EDTA, 1 mM dithiothreitol) EPEC ler::kan bacteria harboring the plasmids pIR1 and pGY1 or pGY1 derivatives, and glycerol and Tween 20 were added to final concentrations of 6% and 0.05%, respectively. The extracts were cleared by centrifugation. A total of 100 μl of the cell extracts were added to the fragment-bound wells for 1 h and then unbound proteins were removed by washes with PT buffer. Polyclonal anti-Ler antibodies (100 μl) were added for 1 h, the wells were washed with PT, and then 100 μl of alkaline phosphatase-conjugated secondary anti-rabbit immunoglobulin G antibodies (Sigma) were added for 1 h. The wells were washed with PT and later with a 1 M Tris (pH 9.5) and 0.5 mM MgCl2 buffer. Finally, 100 μl of this buffer containing 10 mg/ml p-nitrophenyl phosphate were added to each well, and the plate was immediately inserted into a microplate reader (SPECTRAFluor Plus; Tecan). The OD405 was read every minute, and data were collected by Magellan version 5.0 software (Tecan). The slopes of the OD405 values at the first 25 min (before the reaction was saturated) were chosen to represent the level of binding.
Ler oligomerization assay.
The oligomerization of wild-type Ler was tested using the pDF5 plasmid, from which two forms of Ler are expressed: the native Ler and Ler fused at its N terminus to a 16-amino-acid peptide containing His6 (6His-10AA-Ler; the exact sequence of the tag is MRGSHHHHHHPRRLFI-Ler). E. coli W3110 containing pDF5 was grown under expression conditions, proteins were extracted, and 6His-10AA-Ler was purified by metal affinity chromatography. The purified protein was analyzed by SDS-PAGE and Coomassie staining or by transfer into nitrocellulose, which was stained with Coomassie or used for immunoblot analysis using anti-His antibodies.
The oligomerization of the Ler mutants was assessed by introducing the pGY1-derived plasmids into TU1403, a strain containing a chromosomal fusion of ler to His6 at the region encoding the C terminus. Bacteria were grown in DMEM at 37°C to an OD600 of 0.4, sonicated, and subjected to metal affinity chromatography using Talon beads (Clontech). Ler-His6 expressed from the chromosome was expected to bind the beads, along with the Ler mutants that were capable of oligomerization. The beads were treated with 8 M urea solution. Under these conditions Ler-His6 remained bound to the beads, but protein-protein interactions were disrupted, eluting the proteins bound to Ler-His6. The residual Ler-His6 in the eluted samples were reabsorbed and completely removed from the preparations with fresh Talon beads in 8 M urea solution. The eluted material was subjected to immunoblot analysis with anti-Ler antibodies.
RESULTS
Determination of the Ler N terminus.
Before utilizing the mutagenesis approach for analyzing Ler activity, we first wished to determine its native N terminus, as it was ambiguous as documented in the databases and never determined experimentally. The putative N-terminal sequence of EPEC Ler according to GenBank (AF022236) is MRRLFIMNMETNSHTTSPYIQ, containing methionine residues at positions 1, 7, and 9 (which are in boldface type). To experimentally determine the native N terminus, we constructed a plasmid (pDF11) expressing ler fused to a His6 tag at its C terminus, under the regulation of its native LEE1 promoter (PLEE1-ler-His6). The Ler-His6 protein was purified by metal affinity chromatography. N-terminal amino acid sequencing analysis of the purified tagged Ler revealed only one protein, whose N-terminal sequence was MNMETNSHTTSPYIQ. This redefined the second methionine (M7) as M1, the translation start site of Ler (see Fig. S1 in the supplemental material). The designations of all the mutations in this paper are according to this newly defined translation start site.
Isolation of Ler mutants defective in autorepression.
To establish a method that will allow rapid screening of a large number of Ler mutants defective in transcription regulation, we constructed the medium-copy-number plasmid pGY1. This plasmid contains a bicistronic operon of ler and gfp expressed from the native LEE1 promoter (PLEE1-ler-gfp) (Fig. 1A). Mutagenesis was accomplished by introducing pGY1 into a mismatch-repair-deficient E. coli mutant (mutS, A70) and growing the bacteria overnight to allow the accumulation of plasmids with random spontaneous point mutations. The plasmids were then recovered from the mutS strain and introduced into EPEC ler::kan, and the transformants were plated to give ∼500 individual colonies per plate. Colonies containing plasmids deficient in autorepression were expected to overexpress gfp, exhibiting increased fluorescence, as PLEE1 was no longer subjected to Ler autorepression (Fig. 1A). About 100 colonies exhibiting strong fluorescence were isolated, representing 0.1 to ∼1.0% of the population. The corresponding cultures were analyzed for Ler production by immunoblot analysis with an anti-Ler antibody, and their corresponding plasmids were recovered and sequenced. In all the selected colonies, mutations were mapped to the ler coding region and none were found in the LEE1 regulatory region. Plasmids expressing a truncated Ler or Ler with more than one mutation and redundant mutations were eliminated from further analysis. The remaining 17 mutations were located throughout the ler coding region (Fig. 1B). The 17 ler mutants, each containing a single mutation, were further analyzed and were shown to be strongly defective in PLEE1 autorepression by immunoblot analysis of their Ler production (data not shown). To reconfirm these results, and to make sure that the effect was not due to other mutations in regions that were not sequenced, we regenerated each of the 17 mutations in vitro by site-directed mutagenesis on the pGY1 plasmid. We also added to this collection the previously described LerL23R mutant (3). These plasmids, designated the pGY1 derivatives (Table 1), were introduced into EPEC ler::kan and showed the derepression of PLEE1 activity similarly to that seen in the original isolates (Fig. 1C and D and data not shown; see also Fig. S2 in the supplemental material). In some cases, the derepression of gfp expression appears stronger than that of ler (Fig. 1C and D), which might reflect reduced reactivity of the mutated Ler with the anti-Ler antibody or reduced stability of the mutated Ler. To conclude, we obtained 18 distinct point mutations in ler, each causing a reduction in autorepression.
FIG. 1.

Identification and initial characterization of 18 ler mutants in autorepression. (A) The pGY1 plasmid, harboring a ler transcriptional fusion to gfp under the control of the native ler promoter (PLEE1), was introduced into a mutS mismatch-repair-deficient strain. The mutated plasmids were recovered and introduced into an EPEC ler::kan strain, and mutants in autorepression were identified as fluorescent colonies. Eighteen mutants were further analyzed. These mutations (B, marked in red) are scattered throughout the Ler amino acid sequence. The predicted coiled-coil and DNA binding motifs are in yellow and light blue, respectively. The core DNA binding domain is underlined. (C) The 18 mutants were reconstructed in pGY1 and the expression of green fluorescent protein, representing the transcription from PLEE1, was measured at late exponential-growth phase. (D) Protein levels of the Ler mutants were analyzed using immunoblot analysis with an anti-Ler antibody.
All the Ler autorepression-deficient mutants show low affinity to the LEE1 and LEE2 regulatory regions.
We next tested the capacity of the different Ler mutants to bind DNA, using a modified ELDIA (2). Whole bacterial extracts, which were generated from EPEC ler::kan harboring pGY1-derived plasmids expressing the different Ler mutants, were added to wells previously coated with different DNA sequences (the LEE1 and LEE2 regulatory regions and the etk coding region, which served as a negative control). The unbound proteins were washed, and the amount of bound Ler was determined using anti-Ler antibodies and secondary alkaline-phosphatase-conjugated antibodies and normalized according to the expression level from PLEE1 by the different mutants (Table 2). Whole bacterial extracts are used in this assay for better reflection of the Ler binding capacity in vivo. All the Ler mutants exhibited a reduced affinity to the DNA binding sites in the LEE1 and the LEE2 regulatory regions (Fig. 2). Since several of the mutations were located outside the predicted DNA binding domain (Fig. 1B), these results suggest that in addition to this domain, other Ler domains are also required for DNA binding, possibly by promoting Ler oligomerization or by affecting the stability of the protein.
TABLE 2.
Summary of the characterization of Ler mutants deficient in autorepression
| Mutant | No. of occurrencesa | Transcription levelb | Secretion of EspB | Pedestal formation | Dominant negativec | Groupd |
|---|---|---|---|---|---|---|
| LerL16P | 4 | 11 | − | − | ? | ? |
| LerL23R | 9 | + | + | − | CID | |
| LerL56P | 2 | 8 | − | − | − | N |
| LerY77H | 3 | 9 | − | − | − | N |
| LerY77C | 2 | 10 | − | − | − | N |
| LerG82V | 1 | 10 | − | − | + | DN |
| LerG82E | 2 | 12 | + | + | − | CID |
| LerW85R | 1 | 6 | − | − | + | DN |
| LerG87S | 1 | 9 | − | − | + | DN |
| LerG87D | 1 | 7 | − | − | + | DN |
| LerG89D | 2 | 8 | − | − | + | DN |
| LerP92S | 1 | 8 | − | − | + | DN |
| LerW94R | 2 | 9 | − | − | + | DN |
| LerL95P | 1 | 9 | − | − | + | DN |
| LerA98V | 5 | 11 | + | + | − | CID |
| LerG102R | 1 | 7 | + | + | − | CID |
| LerG102E | 1 | 10 | − | − | − | N |
| LerL109P | 1 | 11 | − | − | + | DN |
| Wild type | 1 | + | + | − |
The number of identical mutants picked in the screen. The LerL23R mutant was not identified by the screen; it was described in reference 3.
Fluorescence/OD600 compared to that of the wild type at exponential phase.
Expression of EspB and Tir (from the LEE4 and LEE5 promoters, respectively) when the plasmid harboring the Ler mutant was introduced into a wild-type EPEC strain.
N, null; DN, dominant negative; CID, compensation by increased dosage (see Discussion).
FIG. 2.

DNA binding by the Ler autorepression-defective mutants. The binding of Ler mutants to the LEE1 and LEE2 regulatory regions and to the yccC (etk) coding region (used as a negative control) was tested using an ELDIA. Whole bacterial extracts, generated from EPEC ler::kan containing plasmids encoding the different Ler mutations (pGY1 and pGY1-derived plasmids) were used. As a positive control, we used pGY1 expressing native Ler (Ler WT), and as a negative control, we used a strain containing the vector, which is not expressing Ler (Vector). The mutations of the different Ler proteins are indicated below the columns. DNA binding is expressed as a percentage of the maximum binding. Protein levels in the extracts used for the experiments were analyzed by immunoblot analysis using the anti-Ler antibody, shown below the graph. Similar results were obtained in three independent experiments.
Ler forms oligomers.
Ler oligomerization was predicted to be involved in DNA binding (27), but this prediction was never tested. To examine this assumption, we tested the binding of native Ler to His6-Ler, both expressed from the pDF5 plasmid. Proteins were purified from E. coli W3110 containing pDF5 by metal affinity chromatography. The purified proteins were analyzed by SDS-PAGE and Coomassie staining to give two highly purified bands (Fig. 3A; see also Fig. S3 in the supplemental material). A comparison of the immunoblot analyses, using anti-His antibodies with Coomassie-stained nitrocellulose membrane, revealed that the larger protein was His6-Ler (Fig. 3B). The smaller copurified protein was subjected to N-terminal amino acid sequencing, and the obtained sequence was MNMETNSHTT, identifying it as native Ler (see Fig. S1 in the supplemental material). These results suggest that Ler forms homooligomers. The Ler oligomers contained only His6-Ler and Ler, as no other proteins were copurified with the complex (Fig. 3).
FIG. 3.
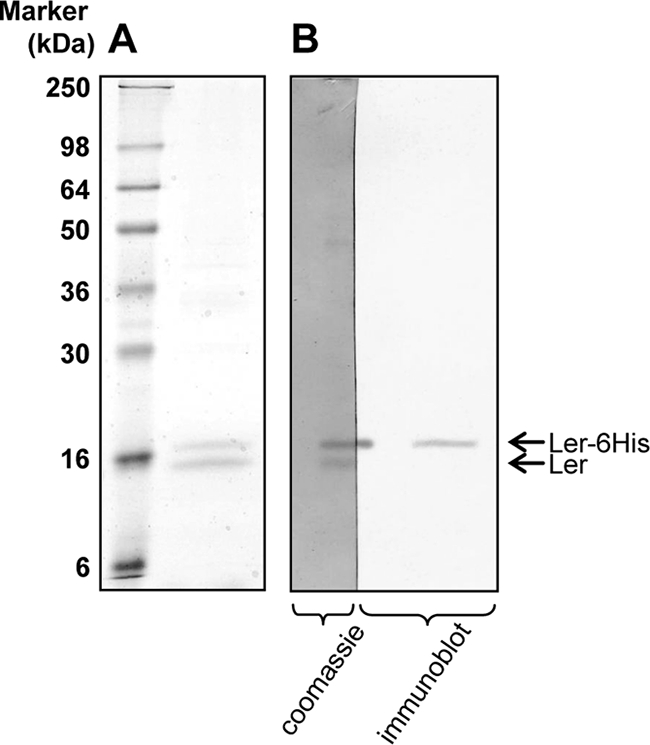
Ler forms homooligomers. (A) The pDF5 plasmid, from which two forms of Ler are expressed (the native Ler and Ler fused at its N terminus to a 16-amino-acid peptide containing His6 [MRGSHHHHHHPRRLFI]-Ler) was introduced into E. coli W3110. The expressed His6-Ler was purified by metal affinity chromatography, and proteins retained on the column were eluted and analyzed by SDS-PAGE and Coomassie staining (right lane). The left lane contains size markers, and the different sizes are indicated. Two highly purified protein bands are located in the right lane near the 16-kDa marker. (B) Proteins from two lanes of a similar gel were transferred onto a nitrocellulose membrane, and the membrane was cut along one of the lanes. The left half of the membrane was developed by Coomassie staining and thus appears darker. The right half of the membrane was used for immunoblot analysis and developed with anti-His6 antibody. Only the upper band reacted with the anti-His6 antibody.
Identification of a mutant deficient in oligomerization.
We next subjected all the Ler mutants to an oligomerization assay, in which we introduced the different pGY1-derived plasmids into an EPEC strain containing the ler gene fused to His6 at the region encoding the C terminus (Ler-His6). The coprecipitation of the different Ler mutants with Ler-His6 was tested by metal affinity chromatography and immunoblot analysis with anti-Ler antibodies as described in Materials and Methods. Interestingly, the mutants in the Ler coiled-coil domain (L16P and L23R) still bound to Ler-His6 (Fig. 4A), implying that this domain does not function as the sole oligomerization domain of Ler. A possible explanation for that can be that the N-terminal coiled-coil domain might be involved in other protein-protein interactions, possibly in higher-order Ler oligomerization or interactions of Ler with other proteins. An alternative possibility is that the coiled-coil domain retained some oligomerization capacity despite these mutations. Only one mutant (L56P) did not coprecipitate with Ler-His6, identifying a domain around residue L56 as essential for oligomerization or as a residue essential for N-terminal oligomerization.
FIG. 4.

Oligomerization of the Ler autorepression-defective mutants and truncation analysis. (A) Oligomerization of the Ler autorepression-defective mutants was assessed by introducing the plasmids encoding them into an EPEC strain chromosomally encoding Ler-His6. Ler-His6 and associated proteins were pulled down from cleared extracts with Talon beads, and proteins associated with Ler-His6 were eluted with 8 M urea. Under these conditions, Ler-His6 remained attached to the beads. The eluted samples were further treated with fresh Talon beads to remove any residual Ler-His6 and were then analyzed by SDS-PAGE, followed by an immunoblot analysis with anti-Ler antibodies. pGY1 expressing native Ler (wild-type) was used as a positive control, and the strain expressing Ler-His6 was used as a negative control. The different mutations are indicated below the corresponding lanes. (B) Schematic representation of the N50Stop and G65Stop truncated Ler variants. (C) EPEC wild-type (top panel) or EPEC ler::kan (bottom panel) containing the plasmids pIR1 (vector), pGY1 expressing wild-type ler (wild-type), and pGY1-derived plasmids expressing a truncated Ler containing only the first 50 amino acids [ler(N50stop)] or only the first 65 amino acids [ler(G65stop)]. To test the ability of the different plasmids to restore or block EspB secretion (the latter is via dominant negativity), the supernatant of bacterial cultures of these strains grown in LEE-inducing conditions was subjected to immunoblot analysis with anti-EspB antibody.
A domain around L56 mediates a dominant-negative effect on wild-type Ler.
To further examine the role of the domain around L56 in oligomerization, we generated two truncated variants of Ler, in which N50 or G65 was changed into a stop codon (LerN50Stop and LerG65Stop, respectively). The LerN50Stop variant contained only the coiled-coil domain, and the LerG65Stop variant contained the coiled-coil domain and the central domain, including L56. Both LerN50Stop and LerG65Stop lack the C-terminal DNA binding domain (Fig. 4B), and in the case of the latter, we also experimentally confirmed its DNA binding deficiency (see Fig. S4 in the supplemental material). We hypothesized that if these truncated variants were able to oligomerize, they would have exerted a dominant-negative effect on wild-type Ler. To test this prediction, plasmids harboring each of the two truncated variants were introduced into wild-type EPEC and EPEC ler::kan, and the expression of the LEE-encoded genes (data not shown) and the secretion of EspB (Fig. 4C) were then examined. The latter served as a readout for TTSS gene expression and the assembly of a functional TTSS. As expected, neither of the truncated variants complemented the EPEC ler::kan strain to restore the expression of the TTSS genes and EspB secretion (Fig. 4C and data not shown). However, the LerG65Stop variant, but not the LerN50Stop variant, showed a dominant-negative effect on wild-type Ler (Fig. 4C). These results show that amino acids 50 to 65 have a role in the ability of LerG65Stop to interfere with wild-type Ler function, probably through the formation of oligomers deficient in DNA binding. These results further support the notion that the central domain of Ler is required for oligomerization in vivo.
Complementation of EPEC ler::kan by four of the autorepression-deficient Ler mutants.
The EPEC ler::kan mutant strain is defective in producing most of the LEE-encoded proteins and consequently cannot assemble a functional TTSS. Complementation of this mutant with a plasmid expressing wild-type ler restores LEE gene expression and the assembly of a functional TTSS (12, 21). We therefore tested the capacity of the different ler mutants (expressed from the pGY1-derived plasmids) to complement EPEC ler::kan. To assess the complementation and the assembly of a functional TTSS, we applied three assays: (i) we examined the production of the LEE-encoded proteins EspB and Tir (encoded by the LEE4 and LEE5 operons, respectively), (ii) we tested for TTSS-dependent EspB secretion to the growth medium, and (iii) we examined the bacterial capacity to induce the formation of actin pedestals in infected HeLa cells. Two categories of mutants were found in these experiments. The first included 14 mutants (the L16P, L54P, Y77H, Y77C, G82V, W85R, G87S, G87D, G89D, P92S, W94R, L95P, G102E, and L109P mutants), which were unable to complement EPEC ler::kan in the secretion of EspB and the induction of actin pedestal formation (Fig. 5A to C). Some of these mutants showed a residual ability to activate transcription, but this was not sufficient to restore EspB secretion and the induction of pedestal formation, either because the expression level of the TTSS proteins was not sufficient or because not all of the TTSS proteins were produced (e.g., Y77C and G102E; Fig. 5A). The second category included four mutants, LerL23R, -G82E, -A98V, and -G102R, which successfully complemented the EPEC ler::kan strain according to all of the aforementioned assays (Fig. 5A to C). These four mutations are located in different regions of the Ler protein (Fig. 1B).
FIG. 5.

Complementation experiments with the Ler autorepression defective mutants. Plasmids expressing each of the Ler mutants (pGY1 or a pGY1 derivative) were introduced into EPEC ler::kan. The capacity of the mutated Ler to restore the expression of Tir and EspB (expressed from the promoters PLEE5 and PLEE4, respectively) (A), EspB secretion (B), and the formation of actin pedestals (C) was determined. (A) Expression of Tir and EspB was tested using immunoblot analysis with antibodies raised against these proteins. (B) To test for EspB secretion, the different strains were used to infect HeLa cells for 3 h. The growth medium was recovered, cleared, and analyzed by immunoblot analysis using anti-EspB antibodies. The ler mutations in the complementing plasmids are indicated below the lanes. The names of the LEE-encoded proteins are indicated on the left-hand side of the blots, and the specific operon carrying each gene is in parentheses. The EspB protein in the bacterial lysate (in panel A) is the upper band out of the seen doublet. The origin of the lower band that cross-reacted with the anti-EspB antibody is not known, and it was seen also in strains deleted of the espB gene (data not shown). (C) To test for the formation of actin pedestals, EPEC ler::kan containing different complementing plasmids was used to infect HeLa cells for 3 h. The infected cells were fixed and actin filaments were stained with rhodamine-phalloidin. A few representative examples are shown, including an uncomplemented ler::kan mutant (no plasmid), a ler::kan mutant complemented with pGY1 (wild-type), and mutants complemented with pGY1-ler(L23R) (L23R) or with pGY1-ler(G89D) (G89D). Phase-contrast images of the infected cells are shown in the first panel, GFP produced by the bacteria in the second panel (green), and actin in the third panel (red). The fourth panel shows overlays of the red and green images. Arrows indicate some actin pedestals.
Identification of dominant-negative Ler mutants.
To further characterize the ler mutants, we tested the mutant alleles for a dominant-negative effect on wild-type Ler. The pGY1-derived plasmids were introduced into wild-type EPEC, and the production of the LEE proteins was tested as an indication for wild-type Ler activity (Fig. 6). The results show that some ler mutants had no effect on wild-type Ler, while others were clearly dominant negative. These include the G82V, W85R, G87S, G87D, G89D, P92S, W94R, L95P, and L109P mutants, all residing within the putative Ler DNA binding region. Since all of the dominant-negative mutants were able to form oligomers with wild-type Ler (Fig. 4A) and did not bind DNA (Fig. 2), we suggest that they cause the dominant-negative effect by forming heterooligomers with wild-type Ler that cannot bind DNA. Interestingly, some of the mutants (LerL16P, -Y77H, -Y77C, and -G102E) were able to oligomerize and did not bind DNA but were not dominant negative. It is possible that in these cases, heterooligomers, but not homooligomers, retained the DNA binding capacity.
FIG. 6.

Identification of dominant-negative Ler autorepression-defective mutants. pGY1 or a pGY1-derivative plasmid expressing each of the Ler mutants was introduced into wild-type EPEC. The expression of Tir and EspB (natively expressed from the Ler-activated promoters PLEE5 and PLEE4, respectively) was determined using immunoblot analysis with antibodies raised against these proteins. The ler mutations are indicated below the lanes. The names of the LEE-encoded proteins are indicated on the left-hand side of the blots and the specific operon carrying each gene is written in parentheses.
The increased dosages of the L23R, G82E, A98V, and G102R Ler mutants are responsible for the recovery of their biological activity.
As was mentioned earlier, four mutants, LerL23R, -G82E, -A98V, and -G102R, were found to be defective in autorepression but otherwise restored the wild-type phenotype upon complementation of the EPEC ler::kan strain (Fig. 5). This intriguing phenotype led us to further investigation of these four mutants. A possible explanation for this phenotype is that these Ler mutants possess residual DNA binding activity and that upon their overexpression (due to reduced autorepression), they reach a concentration that compensates for their reduced affinity to DNA (Fig. 7A). This compensatory mechanism hypothesis predicts that the ability of these four mutants to complement EPEC ler::kan is dosage dependent. To test this prediction, the four mutations (L23R, G82E, A98V, and G102R) were cloned into the pSA10 expression vector, placing them under a tightly regulated tac promoter (Ptac) that can be induced for expression by IPTG. These pSA10-derived plasmids were then introduced into the EPEC ler::kan strain, and the amount of expressed Ler was adjusted by using different IPTG concentrations. The capacity of the expressed Ler to restore the expression of the LEE genes and to enable the assembly of a functional TTSS was tested under the different expression levels. The TTSS-dependent EspB secretion to the growth medium was used as an indicator of TTSS activity. Wild-type Ler was used as a positive control and LerG89D, a mutant that did not complement EPEC ler::kan (Fig. 5 and Table 2), was used as a negative control. We found that a very low concentration of wild-type Ler (its basal level in the absence of IPTG) was sufficient to restore EspB secretion, and maximal secretion was evident upon induction with 5 μM IPTG (Fig. 7B). In the case of the L23R, G82E, A98V, and G102R Ler mutants, an increased amount of IPTG (20 to 50 μM), resulting in an ∼5- to >10-fold increase in the Ler concentration, was required to restore EspB secretion. The increase in the Ler concentration varied between the mutants, indicating that some mutations also affected Ler translation or its stability (Fig. 7C). Our model predicts that at high concentrations, these Ler mutants restore some of their DNA binding functionality, enabling them to activate transcription of the LEE promoters. To test this prediction, we determined the DNA binding affinity of wild-type Ler and LerL23R, revealing that LerL23R still specifically binds to the LEE2 regulatory region but at a lower affinity and thus that higher concentrations of the mutant were needed in order to bind the regulatory LEE2 DNA (see Fig. S5 in the supplemental material). In the case of the G89D mutant, which served as a negative control, even strong overexpression could not restore EspB secretion (Fig. 7B). These results support the hypothesis that in the case of the L23R, G82E, A98V, and G102R Ler mutants, a compensatory mechanism involving an increased Ler dosage is operating. These mutants will therefore be referred to as the CID mutants, for compensation by increased dosage.
FIG. 7.

Increased dosage of the CID mutants is required for their function. (A) A schematic representation of the compensatory mechanism: an autorepressor represses its own transcription (from the Pautorep promoter) and activates transcription from other promoters (Pact). A mutant autorepressor can either retain a partial function like the CID mutants or no function. A CID mutation (middle panel) alleviates autorepression, causing an increase in the concentration of the mutated regulator, which in turn compensates for its reduced DNA binding affinity, enabling it to activate the transcription from Pact. This situation demonstrates the compensatory mechanism embedded in autorepressed regulatory systems. When a mutation completely abolishes the regulator's function (see the “Null mutant” bottom panel), even a high concentration cannot compensate for the mutation. (B) The L23R, G82E, A98V, and G102R CID mutants were cloned under the regulation of the Ptac promoter, from which their expression level could be controlled (plasmids pGY2742, pGY2743, pGY2744, and pGY2745, respectively). Wild-type Ler and LerG89D (a DN mutant) were also subjected to the same analysis (plasmids pGY2746 and pGY3576, respectively). The plasmids were electroporated into EPEC ler::kan, and the strains were grown with increasing concentrations of IPTG (0, 1, 5, 10, 20, and 50 μM). The secretion of EspB, representing the expression and assembly of a functional TTSS, was tested by subjecting the supernatant of the cultures to an immunoblot analysis using anti-EspB antibody. (C) The expression level of Ler was determined by subjecting the pellet of the cultures to an immunoblot analysis using anti-Ler antibody (only two or three IPTG concentrations for each Ler mutant are shown).
DISCUSSION
A simple and rapid method for identifying important residues in regulators.
We have described here a simple method that allows rapid, high-throughput screens for regulators defective in autorepression. Since autorepression is a very common network motif in bacteria, this approach can be easily adopted for the functional analysis of many other regulators. Eight of the mutations picked up by our screen occurred more than once (and up to five times for A98V), indicating that our screen was close to saturation. The 18 Ler mutations causing the deficiency in autorepression described in this paper (Table 2) were not located in a distinct domain in Ler, revealing that there is no specific Ler domain that functions as an “autorepression domain” (Fig. 8). This implies that Ler binds to different DNA regions by the same mechanism, and it is the binding context that defines the outcome: gene induction or repression. This is probably the case with most other regulators which are also autorepressors. Therefore, the described screening approach should reveal residues important to the fundamental aspects of most regulators: oligomerization and DNA binding.
FIG. 8.

A schematic of Ler domains. Based on the comparison to H-NS together with our results, we suggest the following model for Ler domains: an N terminus that functions either as a high-order oligomerization domain or as a domain that interacts with other proteins, a central domain that functions as a homooligomerization domain, and a C-terminal DNA binding domain. The three domains are essential for DNA binding and transcription regulation.
Mutants' classification.
We found that all the isolated mutants are deficient in autorepression and in binding DNA of the regulatory regions of both repressed and activated promoters (PLEE1 and PLEE2, respectively). However, these mutants differ in other aspects, and these differences were used for their classification (Table 2). Most of the mutations in the Ler putative DNA binding domain resulted in a dominant-negative form of Ler. Other mutants could not support the activation of the LEE promoters, but they were not dominant negative. Interestingly, the third class (the CID mutants) restored a wild-type phenotype upon complementation of the EPEC ler::kan mutant with a plasmid encoding these alleles under their native promoter.
The intrinsic compensatory mechanism associated with Ler CID mutants.
The analysis of the CID mutants elucidated an interesting auto-compensatory mechanism, which possibly functions in the case of other autorepressors. The binding of Ler, as well as other DNA binding proteins, is affected by two factors: Kd (dissociation constant) and concentration. Importantly, autoregulation is expected to be the link between the two: strong DNA binding (low Kd) should result in strong autorepression and a low concentration of the regulator. In contrast, weaker DNA binding (increased Kd) due to a point mutation should result in decreased autorepression and an increase in the regulator concentration. The latter increase in concentration might compensate for the reduced binding affinity to allow efficient binding of the regulator to other promoters (Fig. 7A). In the case of Ler, the latter promoters include those of operons LEE2 and LEE5. Consequently, this compensatory mechanism might rescue the biological function of the mutated regulator. Our data support the notion that CID mutants restored the wild-type phenotype (Fig. 5) due to an increase in the steady-state level of the mutated Ler. This compensatory mechanism possibly protects regulators such as Ler from deleterious mutations. This mechanism is predicted to be particularly efficient in cases of cooperativity in DNA binding, as shown for Ler (our unpublished data), where a small increase in the regulator concentration may result in strong DNA binding. This proposed mechanism is not expected to function in cases of a severe reduction in DNA binding as seen in our results (Fig. 7A, bottom panel).
Different putative Ler domains.
Ler is a paralog of H-NS, which induces DNA condensation (26). H-NS and related proteins consist of an N-terminal oligomerization domain and a C-terminal DNA binding domain separated by a flexible linker (28). The N-terminal oligomerization domain of H-NS, which has low sequence similarity with Ler, contains three α-helical segments: the first two stabilize the structure, and the third and longest α-helix forms the core of a coiled-coil configuration (5, 10, 28). This coiled-coil region is essential for H-NS oligomerization (30, 32). Using PSIPRED (20), we found that the first two helices of H-NS have no counterparts in Ler, whereas a counterpart to the coiled-coil region was found in the Ler N terminus, as previously predicted (27). Two of the isolated Ler mutations (L16P and L23R) were located within this N-terminal coiled-coil domain. Unexpectedly, neither of these mutations prevented oligomerization, suggesting that this motif is not the sole determinant of Ler oligomerization. It is possible that in Ler, this domain is required for higher-order oligomerization or for the interaction of Ler with other proteins, possibly regulators. Importantly, these mutations caused a strong deficiency of DNA binding of the homooligomers, but they did not impose a dominant-negative effect upon wild-type Ler. How this domain contributes to DNA binding is not yet clear.
The H-NS C-terminal DNA binding domain is conserved and highly similar to that of Ler (27), containing the consensus DNA binding motif: Y-X(6)-[GS]-[ED]-X(0,2)-T-W-[TS]-G-[QR]-G-[RK]-X-P-X(4,5)-A-X(3,4)-G (4, 8). It was consequently suggested that this domain might be the Ler DNA binding domain (27). In agreement with this prediction, most of the mutants isolated in our screen were clustered in this region. These mutations were either dominant-negative mutations, null mutations, or CID mutations (Fig. 8). Interestingly, in two cases, the same residue could be mutated to give different phenotypes. The G82V mutation caused a dominant-negative effect, whereas the G82E mutation was a CID mutation. Another case is G102R, which was a CID mutation, while G102E had a null phenotype.
The central flexible linker of H-NS is not essential for oligomerization but has been suggested to be involved in higher-order oligomerization (5). The central regions of H-NS and Ler are very different; still, we identified that the central region of Ler, around L56, is essential for oligomerization. L56P is the only mutation that completely prevented oligomerization. Moreover, truncation analysis suggests that this region is essential for the dominant-negative effect, suggesting that it is involved in oligomerization in vivo. These differences in the oligomerization mechanism and the dissimilarity between the N-terminal and central regions of H-NS and Ler may reflect some functional differences. Ler alleviates the H-NS silencing of only a few specific promoters that are flanked by one or two Ler DNA binding regions. Therefore, Ler probably does not function by forming Ler-H-NS heterodimers. In contrast, other proteins, closely related to H-NS, like StpA, can function by forming StpA-H-NS heterooligomers (32).
In conclusion, the results described in this report, together with the comparison of Ler to H-NS, suggest that Ler has three domains (Fig. 8): a C-terminal domain that functions as a DNA binding domain, a central domain that functions as a homo-oligomerization domain, and an N-terminal domain that may have a role in interacting with other proteins or in higher-order oligomerization. All three domains are essential for the DNA binding activity of Ler and for its role as a regulator.
Supplementary Material
Acknowledgments
We thank G. Frankel for providing antibodies, S. Altuvia for providing the mutS strain, D. Friedberg for plasmid construction, and A. Gaton for solving the N-terminal sequence of Ler.
This work was supported by grants from the Israel-United States Binational Science Foundation, the Center of Study of Emerging Disease, the EraNet-PathoGenomic program (to I.R.), and the Abisch-Frenkel Foundation (to C.N. and I.R.). T.B. was supported by a Boehringer Ingelheim Fonds scholarship and G.Y. by the Einstein scholarship, sponsored by the Isaac Kaye Foundation.
Footnotes
Published ahead of print on 3 October 2008.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Abe, H., A. Miyahara, T. Oshima, K. Tashiro, Y. Ogura, S. Kuhara, N. Ogasawara, T. Hayashi, and T. Tobe. 2008. Global regulation by horizontally transferred regulators establishes the pathogenicity of Escherichia coli. DNA Res. 1525-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Benotmane, A. M., M. F. Hoylaerts, D. Collen, and A. Belayew. 1997. Nonisotopic quantitative analysis of protein-DNA interactions at equilibrium. Anal. Biochem. 250181-185. [DOI] [PubMed] [Google Scholar]
- 3.Berdichevsky, T., D. Friedberg, C. Nadler, A. Rokney, A. Oppenheim, and I. Rosenshine. 2005. Ler is a negative autoregulator of the LEE1 operon in enteropathogenic Escherichia coli. J. Bacteriol. 187349-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bertin, P., F. Hommais, E. Krin, O. Soutourina, C. Tendeng, S. Derzelle, and A. Danchin. 2001. H-NS and H-NS-like proteins in Gram-negative bacteria and their multiple roles in the regulation of bacterial metabolism. Biochimie 83235-241. [DOI] [PubMed] [Google Scholar]
- 5.Bloch, V., Y. Yang, E. Margeat, A. Chavanieu, M. T. Auge, B. Robert, S. Arold, S. Rimsky, and M. Kochoyan. 2003. The H-NS dimerization domain defines a new fold contributing to DNA recognition. Nat. Struct. Biol. 10212-218. [DOI] [PubMed] [Google Scholar]
- 6.Bustamante, V. H., F. J. Santana, E. Calva, and J. L. Puente. 2001. Transcriptional regulation of type III secretion genes in enteropathogenic Escherichia coli: Ler antagonizes H-NS-dependent repression. Mol. Microbiol. 39664-678. [DOI] [PubMed] [Google Scholar]
- 7.Deng, W., J. L. Puente, S. Gruenheid, Y. Li, B. A. Vallance, A. Vazquez, J. Barba, J. A. Ibarra, P. O'Donnell, P. Metalnikov, K. Ashman, S. Lee, D. Goode, T. Pawson, and B. B. Finlay. 2004. Dissecting virulence: systematic and functional analyses of a pathogenicity island. Proc. Natl. Acad. Sci. USA 1013597-3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dorman, C. J., J. C. Hinton, and A. Free. 1999. Domain organization and oligomerization among H-NS-like nucleoid-associated proteins in bacteria. Trends Microbiol. 7124-128. [DOI] [PubMed] [Google Scholar]
- 9.Elliott, S. J., V. Sperandio, J. A. Giron, S. Shin, J. L. Mellies, L. Wainwright, S. W. Hutcheson, T. K. McDaniel, and J. B. Kaper. 2000. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 686115-6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Esposito, D., A. Petrovic, R. Harris, S. Ono, J. F. Eccleston, A. Mbabaali, I. Haq, C. F. Higgins, J. C. Hinton, P. C. Driscoll, and J. E. Ladbury. 2002. H-NS oligomerization domain structure reveals the mechanism for high order self-association of the intact protein. J. Mol. Biol. 324841-850. [DOI] [PubMed] [Google Scholar]
- 11.Frankel, G., A. D. Phillips, I. Rosenshine, G. Dougan, J. B. Kaper, and S. Knutton. 1998. Enteropathogenic and enterohaemorrhagic Escherichia coli: more subversive elements. Mol. Microbiol. 30911-921. [DOI] [PubMed] [Google Scholar]
- 12.Friedberg, D., T. Umanski, Y. Fang, and I. Rosenshine. 1999. Hierarchy in the expression of the locus of enterocyte effacement genes of enteropathogenic Escherichia coli. Mol. Microbiol. 34941-952. [DOI] [PubMed] [Google Scholar]
- 13.Goldberg, M. D., M. Johnson, J. C. Hinton, and P. H. Williams. 2001. Role of the nucleoid-associated protein Fis in the regulation of virulence properties of enteropathogenic Escherichia coli. Mol. Microbiol. 41549-559. [DOI] [PubMed] [Google Scholar]
- 14.Grant, A. J., M. Farris, P. Alefounder, P. H. Williams, M. J. Woodward, and C. D. O'Connor. 2003. Co-ordination of pathogenicity island expression by the BipA GTPase in enteropathogenic Escherichia coli (EPEC). Mol. Microbiol. 48507-521. [DOI] [PubMed] [Google Scholar]
- 15.Kanamaru, K., K. Kanamaru, I. Tatsuno, T. Tobe, and C. Sasakawa. 2000. SdiA, an Escherichia coli homologue of quorum-sensing regulators, controls the expression of virulence factors in enterohaemorrhagic Escherichia coli O157:H7. Mol. Microbiol. 38805-816. [DOI] [PubMed] [Google Scholar]
- 16.Kaper, J. B., J. P. Nataro, and H. L. Mobley. 2004. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2123-140. [DOI] [PubMed] [Google Scholar]
- 17.Li, M., I. Rosenshine, S. L. Tung, X. H. Wang, D. Friedberg, C. L. Hew, and K. Y. Leung. 2004. Comparative proteomic analysis of extracellular proteins of enterohemorrhagic and enteropathogenic Escherichia coli strains and their ihf and ler mutants. Appl. Environ. Microbiol. 705274-5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McDaniel, T. K., K. G. Jarvis, M. S. Donnenberg, and J. B. Kaper. 1995. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 921664-1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McDaniel, T. K., and J. B. Kaper. 1997. A cloned pathogenicity island from enteropathogenic Escherichia coli confers the attaching and effacing phenotype on E. coli K-12. Mol. Microbiol. 23399-407. [DOI] [PubMed] [Google Scholar]
- 20.McGuffin, L. J., K. Bryson, and D. T. Jones. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16404-405. [DOI] [PubMed] [Google Scholar]
- 21.Mellies, J. L., S. J. Elliott, V. Sperandio, M. S. Donnenberg, and J. B. Kaper. 1999. The Per regulon of enteropathogenic Escherichia coli: identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler). Mol. Microbiol. 33296-306. [DOI] [PubMed] [Google Scholar]
- 22.Nataro, J. P., and J. B. Kaper. 1998. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11142-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schlosser-Silverman, E., M. Elgrably-Weiss, I. Rosenshine, R. Kohen, and S. Altuvia. 2000. Characterization of Escherichia coli DNA lesions generated within J774 macrophages. J. Bacteriol. 1825225-5230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin, S., M. P. Castanie-Cornet, J. W. Foster, J. A. Crawford, C. Brinkley, and J. B. Kaper. 2001. An activator of glutamate decarboxylase genes regulates the expression of enteropathogenic Escherichia coli virulence genes through control of the plasmid-encoded regulator, Per. Mol. Microbiol. 411133-1150. [DOI] [PubMed] [Google Scholar]
- 25.Sircili, M. P., M. Walters, L. R. Trabulsi, and V. Sperandio. 2004. Modulation of enteropathogenic Escherichia coli virulence by quorum sensing. Infect. Immun. 722329-2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spassky, A., S. Rimsky, H. Garreau, and H. Buc. 1984. H1a, an E. coli DNA-binding protein which accumulates in stationary phase, strongly compacts DNA in vitro. Nucleic Acids Res. 125321-5340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sperandio, V., J. L. Mellies, R. M. Delahay, G. Frankel, J. A. Crawford, W. Nguyen, and J. B. Kaper. 2000. Activation of enteropathogenic Escherichia coli (EPEC) LEE2 and LEE3 operons by Ler. Mol. Microbiol. 38781-793. [DOI] [PubMed] [Google Scholar]
- 28.Tendeng, C., and P. N. Bertin. 2003. H-NS in Gram-negative bacteria: a family of multifaceted proteins. Trends Microbiol. 11511-518. [DOI] [PubMed] [Google Scholar]
- 29.Thieffry, D., A. M. Huerta, E. Perez-Rueda, and J. Collado-Vides. 1998. From specific gene regulation to genomic networks: a global analysis of transcriptional regulation in Escherichia coli. Bioessays 20433-440. [DOI] [PubMed] [Google Scholar]
- 30.Ueguchi, C., C. Seto, T. Suzuki, and T. Mizuno. 1997. Clarification of the dimerization domain and its functional significance for the Escherichia coli nucleoid protein H-NS. J. Mol. Biol. 274145-151. [DOI] [PubMed] [Google Scholar]
- 31.Umanski, T., I. Rosenshine, and D. Friedberg. 2002. Thermoregulated expression of virulence genes in enteropathogenic Escherichia coli. Microbiology 1482735-2744. [DOI] [PubMed] [Google Scholar]
- 32.Williams, R. M., S. Rimsky, and H. Buc. 1996. Probing the structure, function, and interactions of the Escherichia coli H-NS and StpA proteins by using dominant negative derivatives. J. Bacteriol. 1784335-4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.