

Abstract
The dynamics of HIV infection have been studied in humans and in a variety of animal models. The standard model of infection has been used to estimate the basic reproductive ratio of the virus, calculated from the growth rate of virus in acute infection. This method has not been useful in studying the effects of vaccination, since, for the vaccines developed so far, early growth rates of virus do not differ between control and vaccinated animals. Here, we use the standard model of viral dynamics to derive the reproductive ratio from the peak viral load and nadir of target cell numbers in acute infection. We apply this method to data from studies of vaccination in SHIV and SIV infection and demonstrate that vaccination can reduce the reproductive ratio by 2.3- and 2-fold, respectively. This method allows the comparison of vaccination efficacies among different viral strains and animal models in vivo.
Human immunodeficiency virus (HIV) infects approximately 0.5% of the world population and is a major cause of morbidity and mortality worldwide. A vaccine for HIV is urgently required, and a variety of vaccine modalities have been tested in animal models of infection. A number of these studies have shown protection in monkey models of infection, although the ability of the vaccine to protect appears to vary with the viral strain and animal model used (8). The recent failure of a large vaccine study in humans (1) suggests that further understanding of the basic dynamics of infection and the impact of vaccination are required in order to understand the variable efficacies of vaccination in different infections.
The initial ability of HIV to propagate within the host is determined by the abundance of target cells (e.g., CD4+ T lymphocytes) the virus can infect in order to produce progeny, by the replicative capacity of the virus, and by how cytopathic it is to infected cells. At later stages of the disease, in addition to changes in the target cell availability, there may also be changes in virus-specific properties, such as the ability of the virus to reproduce and the survival of productively infected cells as a result of changes in immune pressure and viral evolution. These parameters may be variable among individuals and within one individual over time and affect the impact of vaccination. In order to compare the efficacies of vaccination strategies, we need a quantitative measure of the factors that influence virus replication.
In this work, we focus on the basic reproductive ratio (R0) as a measure of vaccine efficacy in the acute phase of infection. In epidemiology, R0 measures the potential for the spread of an epidemic and is defined as the average number of people infected by one infected individual in a susceptible population. If this number is below 1, the disease will not spread in a population. Therefore, the knowledge of R0 allows one to estimate the fraction of a population that needs to be vaccinated in order to eradicate the disease. Analogously, in host-pathogen dynamics, the basic reproductive ratio is defined as the number of infected cells generated by one infected cell during its lifetime at the start of infection, i.e., before any depletion of target cells. In order for the infection to spread within the host, this number has to be larger than 1; otherwise, the infection will be cleared before it has a chance to spread. The basic reproductive ratio will depend on the ability of virus to infect cells (“infectivity”), on the rate of virion production by infected cells, on the lifetime of infected cells, and on the rate at which free virus is cleared. The aim of vaccines is therefore to create virus-specific immunity that changes any of these parameters and thus decreases the basic reproductive ratio of the virus, preferably below 1 (so that infection does not spread beyond the initially infected cells). The importance of R0 in HIV vaccination, in order to assess how much protection a particular vaccine can achieve, has been stressed previously (11). Specifically, a variety of different animal models have been used to study HIV vaccination. They differ in a number of ways, particularly in the virulence and cell tropism of the virus. Comparison between models may be difficult because of differences in the underlying dynamics of infection. For example, it may be more difficult to prevent CD4+ T-cell depletion in a very virulent animal model than in other infections, because of the higher underlying growth rate. Thus, a vaccine may appear efficacious in one model but ineffective in another, despite the fact that it may have the same impact on viral growth. Measurement of R0 allows comparison of the effectiveness of different vaccines within a given animal model, as well as across different models. It could be a very useful and common standard to compare different vaccination protocols.
The basic reproductive ratio can be determined from the rate of exponential growth of virus in the initial period, during which the target cell levels are almost constant (14, 20, 29). However, in monkeys infected with the same type of simian immunodeficiency virus (SIV) there is in fact very little variation in the basic reproductive ratio determined from the initial exponential growth between controls and vaccinees, despite the variety of outcomes later in infection. Different factors identified as possible predictors of disease progression (29), such as the peak viral load, target cell nadir, decay rate of virus following peak viremia, set point viral load, and chronic target cell levels, all correlate poorly or not at all with the differences in the reproductive ratio determined from the primary exponential growth phase (29). In particular, it is not possible to assess the efficacy of cytotoxic-T-lymphocyte (CTL)-based vaccines on the basis of the basic reproductive ratio, because there is no effect of vaccination on viral growth before approximately 10 days postinfection, which is at the end of the exponential growth period (4, 6).
We have recently shown the existence of a strong correlation between the viral load at peak and the target cell depletion in the acute phase (7, 32). Using this correlation, one can show that vaccination results in the reduction of the peak viral load and of acute CD4+ T-cell depletion, thus improving the chronic-phase prognosis.
The dependence of the nadir in the CD4+ T-cell count on the viral peak can be obtained from the standard model of virus dynamics (7, 32). Here, we show that this relationship between the viral peak and the number of target cells 1 week after the peak (corresponding approximately to the minimum number of target cells in primary infection) is parameterized by the basic reproductive ratio of the virus. In other words, the decrease in the peak viral load leads to less target cell depletion at nadir, and both are a consequence of a lower basic reproductive ratio. Thus, we can in principle determine the basic reproductive ratio from experimental data on the viral peak and target cell nadir. We show that this relationship is indeed supported by experimental data from CXCR4-tropic simian-human immunodeficiency virus (SHIV) infection (for viral loads and CD4+ T-cell counts in peripheral blood) and for CCR5-tropic SIV infection data (for plasma viral loads and memory CD4+ T-cell depletion in the gut).
We show that the reproductive ratio estimated from the viral peak and the target cell nadir, which we call the “reproductive ratio at the peak,” is significantly lower than the basic reproductive ratio estimated from the exponential growth. In addition, we found that in vaccinated animals the reproductive ratio at the peak is on average twofold lower than in control animals. We attribute this difference to the cellular immune response appearing before the peak viral load, around day 10 of infection, and changing the properties of the virus and infected-cell dynamics (e.g., decreasing the lifetime of infected cells through the cytolytic function of CTLs or changing infectivity or virus production through the release of cytokines). Thus, we propose that the “reproductive ratio at the peak,” a measurement that includes information on both the viral peak and the target cell nadir, can be useful as a standard to compare vaccine protocols.
MATERIALS AND METHODS
In the CXCR4-tropic virus study, 35 rhesus macaques (Macaca mulatta) were vaccinated with a variety of regimens, consisting of SIV gag-containing plasmid DNA (with different adjuvants), modified vaccinia virus Ankara, and adenovirus type 5 vectors, as previously reported (27). Animals were challenged intravenously at 6 weeks (study A) or at 12 weeks (study B) after the final boost with 50 monkey infectious doses of SHIV-89.6P. Viral loads and CD4+ T-cell counts were monitored in peripheral blood every 2 to 4 days until 4 weeks after infection and then weekly. The results of the two studies were combined for the purpose of this work.
In the CCR5-tropic virus study, 20 rhesus macaques were challenged intravenously with 100 monkey infectious doses of uncloned SIVmac251. Six of these animals received prior vaccination with plasmid DNA encoding SIV envelope, Gag, and Pol, and were boosted with recombinant adenovirus encoding the same antigens. Plasma and jejunum tissue samples were collected at various time points by biopsy or at necropsy, and tissue CD4+ T-cell percentages and plasma viral loads were determined (17, 18).
RESULTS
Reproductive ratio in the initial exponential growth phase of disease.
The standard model of virus dynamics (15, 21) describes the relationships between the change in the number of uninfected cells, T, that are targets for the virus; infected cells, I; and free virus particles, V, in a given volume of blood or tissue.
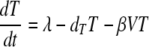 |
(1) |
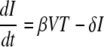 |
(2) |
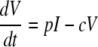 |
(3) |
The parameters λ, the production rate of new target cells, and dT, the loss rate of uninfected cells, describe the disease-free target cell dynamics. The disease-free equilibrium number of target cells is equal to T0, which is equal to λ/dT. The infectivity, β, characterizes the rate at which a virus particle infects a target cell, and δ is the death rate of infected cells; δ ≫ dT. Free virus is produced by infected cells at rate p and is cleared at rate c. In this model, the immune response is assumed to be constant, and its effects are contained in the virus parameters δ, c, β, and p. All the parameters are assumed to be constant in time (28). The basic reproductive ratio, R0, for this model is as follows (15, 21):
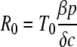 |
(4) |
The virus infection can spread only if R0 is >1. In this case, the viral load generally increases to the peak and then decays, in order to finally reach the steady-state value, V*. Target cells drop to the minimum and then partly recover and settle at the steady-state value, T*, which is equal to T0/R0. The standard model has been used to investigate different aspects of HIV/SIV/SHIV infection, such as the turnover of infected cells (7, 12, 31), the clearance rate of virus (23), and the “burst size” of infected cells (3, 13).
The basic reproductive ratio of a virus is determined from the initial rate of exponential expansion, r0, during which the number of target cells is approximately constant (14, 20, 25). Thus, if we know the initial exponential growth rate of a virus (r0 ☰ 1/V dV/dt) and the death rate of infected cells (δ), we can estimate R0:
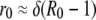 |
(5) |
In Fig. 1A, we show viral-load data during the first weeks of infection from the Shiver et al. study (27), in which 21 out of 35 rhesus macaques were vaccinated with seven different SIV gag-containing vaccines, and all 35 were challenged intravenously with CXCR4-tropic SHIV-89.6P. The initial exponential growth rate and the decay rate of virus after the peak were remarkably similar between monkeys, irrespective of whether they were vaccinated. The results of the statistical analysis are summarized in Fig. 1C and D and Table 1.
FIG. 1.

Dynamics of acute SHIV-89.6P infection. The time courses of the viral load (A) and CD4+ T-cell numbers (B) are shown over the first 40 days of SHIV infection for control (black lines) and vaccinated (red lines) animals. The initial viral growth rates (r0) (C) and viral decay rates (δ) (D) after the peak viral load of SHIV-89.6P do not differ significantly between controls (black dots) and vaccinees (red dots). However, the peak viral load (E) and the nadir CD4+ T-cell number (F) are significantly different between controls and vaccinees. In panels C to F, the gray rectangles represent the confidence intervals.
TABLE 1.
Summary of statisticsa
| Parameter |
r0 (per day)b
|
δ (per day)c
|
LogVPd
|
Tmin (no. of cells/μl)e
|
||||
|---|---|---|---|---|---|---|---|---|
| Control | Vaccinated | Control | Vaccinated | Control | Vaccinated | Control | Vaccinated | |
| Mean | 1.42 | 1.38 | 0.991 | 1.01 | 8.03 | 7.19 | 29.4 | 238 |
| SE | 0.055 | 0.043 | 0.058 | 0.046 | 0.12 | 0.13 | 13.7 | 48 |
| 95% CI | 1.30-1.54 | 1.29-1.47 | 0.866-1.12 | 0.913-1.10 | 7.75-8.28 | 6.92-7.49 | 1.13-57.6 | 137-339 |
For initial exponential growth rate, r0; decay rate of virus after the peak, δ; peak viral load, VP; and the target cell nadir, Tmin, in vaccinated and unvaccinated animals.
Mann-Whitney P value, 0.83.
Mann-Whitney P value, 0.90.
Mann-Whitney P value, 0.00018.
Mann-Whitney P value, 0.00015.
Equation 5 indicates how to measure R0 from the initial growth rate of virus and the death rate of infected cells. The initial growth rate of virus (r0) is directly estimated from the experimental data. The death rate of infected cells (δ) can be estimated from the rate of decline of the viral load after the peak in each animal. Using this approach, we estimate that R0 is 2.4 for vaccinated animals (95% confidence interval [CI], 2.3 to 2.6) and almost the same, 2.5, for control animals (95% CI, 2.3 to 2.7). The two values are not statistically different (Mann-Whitney, P = 0.391).
On the basis of the similarity in virus growth and decay rates between control and vaccinated animals, we (4, 6) and others (24) concluded that the cellular (CD8+ T-cell) immune response does not emerge before at least 10 days postinfection (reviewed in reference 5). This is why it has not been possible to assess the effects of vaccination from the reproductive ratio of the virus calculated from its growth rate within this initial period of 10 days.
Equation 5 considerably underestimates R0 because it neglects the delay between the time when a cell becomes infected and the time when it starts producing the virus. In a model with a fixed intracellular delay, τ (and assuming that all infected cells survive the delay period) (10), R0 is found from the initial growth rate as follows (15, 25):
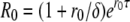 |
(6) |
With a delay of 1 day (22), we obtain an R0 of 9.9 for vaccinated animals (95% CI, 8.6 to 11) and an R0 of 11 for control animals (95% CI, 8.6 to 12), again not different for the two groups and consistent with previous studies of the basic reproductive ratio in SIV infection (20).
As demonstrated above, the traditional approach to measuring R0 in vaccinated animals shows no significant difference in R0 between controls and vaccinees, despite clear differences in infection outcomes. Thus, we aimed to develop a new measure of viral growth that can quantify vaccine effects.
Virus peak and target cell nadir in the standard model of virus dynamics.
In contrast to the exponential growth and decay rates of the viral load, the peak of the viral load and the nadir of CD4+ T-cells were significantly different between control and vaccinated animals (Fig. 1B, E, and F and Table 1). Since the peak viral load and level of depletion of CD4+ T cells were important factors that were clearly affected by vaccination and were correlated with outcome, we aimed to develop a measurement of viral growth and vaccine efficacy based on them. Obtaining an expression for the peak viral load or nadir target cell numbers is quite difficult using the standard model (equation 1 to equation 3), since they are dependent on a large number of other parameters. Thus, we needed to simplify the standard model to identify which parameters can be ignored because in practice they have negligible influence on the peak viral load and nadir targets. Before infection, target cells are at a constant level, which implies that the daily production (λ) and death (dTT0) are balanced, and it is thought that ∼1% of cells turn over in this way each day. Thus, early in infection, while there is very little depletion in target cell numbers (T ≈ T0) (Fig. 2), we can neglect those two terms in equation 1 because they balance each other. Around the peak of viremia, there is massive infection of target cells (βVT becomes very large), and up to 99% of cells disappear over a period of 1 to 2 weeks. During this period, the normal production and loss of target cells (λ and dT) are much smaller than the loss due to infection (i.e., βVT ≫ λ − dTT), and we can safely ignore the effects of normal turnover on the target cell number. That is, we can simplify the standard model to the following (7):
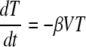 |
(7) |
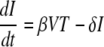 |
(8) |
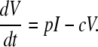 |
(9) |
In this reduced model, the viral load always vanishes at long times (i.e., infection is always cleared if there is no natural loss and replacement of target cells), with target cells steadily declining toward a low steady-state value, Tmin. However, we can further justify our approximation by noting that the profile of viral-load and target cell dynamics during primary infection (up to the target cell nadir, days 18 to 20 in Fig. 2) is only mildly affected. As expected, the exponential growth rate of the virus is the same as in the full standard model, and the viral load has a maximum, VP, with a value very close to the peak of the standard model (Fig. 2A).
FIG. 2.

A reduced model of viral dynamics. The time courses of virus growth and decay (A) and CD4+ T-cell (target cell) decline in primary infection (B) are shown, obtained by numerical integration of equation 1 to equation 3 and equation 7 to equation 9. (A) The peak viral loads for the reduced model (λ, dT = 0) and the standard model (λ = 10 cells/μl/day; dT = 0.01 day−1). (B) The nadir numbers of uninfected CD4+ T cells are indicated for the reduced model and the standard model. The total CD4+ T-cell number (infected plus uninfected) is also indicated for the standard model. The other parameters were as follows: β = 8 × 10−8 ml/copy/day, δ = 0.8 day−1, P = 103 μl/ml × 500 copies/cell/day, and c = 20/day. The initial inoculum was 50 copies/ml.
Figure 2 also shows that the steady-state target cell number, Tmin, in the reduced model is slightly lower, but still a very good approximation for the uninfected target cell nadir of the standard model in equation 1 to equation 3 if around 1% of CD4+ T cells turn over each day (i.e., for the parameters λ = 10 cells/μl/day and dT = 0.01/day [28] usually attributed to CD4+ T cells in HIV/SIV/SHIV infection). The condition for equation 7 to be a good approximation to the full model, that is, βVT ≫ λ − dTT, is violated only very close to the nadir of target cells, as the viral load decreases, and that is why we obtain a good estimate of the nadir from this approximation. This is true whenever the normal replacement or turnover of CD4+ T cells is relatively small.
Instead of uninfected target cells, the quantity that is usually measured experimentally is the total number of uninfected and infected cells (T + I). Figure 2B shows that the minimum of the total T-cell number is again very close to the minimum of target cells. Therefore, we can compare the experimental nadir target cell counts to the steady-state number of uninfected cells, Tmin, in the reduced model.
An advantage of the reduced system in equation 7 to equation 9 is that one can estimate VP and Tmin analytically. We shall use the analytical results as estimates for the viral peak and the target cell nadir in the full standard model.
The basic reproductive ratio of the reduced model, R0, is still given by equation 4 (with T0 being an arbitrary initial target cell number). The peak of viremia, as a function of R0, is as follows (see the Appendix for the derivation):
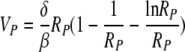 |
(10) |
which is valid for the reduced model (equation 7 to equation 9) and a good approximation for the standard model, with low target cell replacement and loss (equation 1 to equation 3).
Tmin is found as the solution of the equation (see the Appendix).
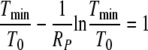 |
(11) |
The fraction of uninfected target cells at the nadir in the full standard model, approximately equal to Tmin/T0 in equation 11, depends only on the basic reproductive ratio, R0. The solution is illustrated in Fig. 3. For an R0 of >5, the value of Tmin/T0 is practically zero and the depletion at nadir is almost 100%. In Fig. 3, we plotted both Tmin/T0 (the ratio at the target cell nadir) and T*/T0 (the ratio in chronic infection, i.e., in the steady state, given by 1/R0) as functions of R0, showing that Tmin is always (for all R0) less than the steady-state target cell count, T*.
FIG. 3.
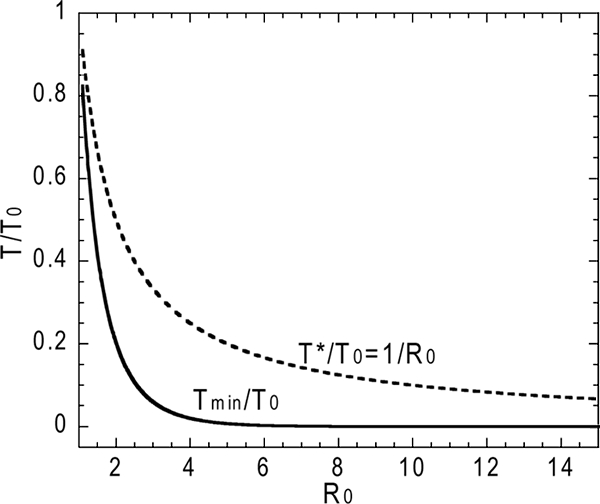
The target cell nadir, Tmin, is lower than the steady-state value, T*, for all values of R0.
Relationship between the virus peak and the target cell nadir in the model.
In the context of acute infection, i.e., the time between the virus peak and the target cell nadir, we shall call the expression in equation 4 the reproductive ratio at the peak (and use the symbol RP) instead of the basic reproductive ratio, in order to stress that the parameter values in this period may have changed from those of initial infection. Thus, one should replace the symbol R0 by RP in equation 10 and equation 11. For a very low target cell replacement rate λ and loss rate dT, the depletion (D) at the target cell nadir (D = 1 − Tmin/T0 [from equation 11]) depends only on the reproductive ratio at the peak, RP, since this is the only free parameter in the equation. The peak viral load (equation 10) depends on RP and, in addition, on the ratio δ/β. This means that, for viruses with different characteristic infectivities, β, or different cytotoxicities and cellular immune responses resulting in different death rates of infected cells, δ, the relationship between the virus peak and the target cell minimum will be described by a family of parallel sigmoid curves (Fig. 4A). RP varies along each curve with the characteristic ratio δ/β. Increases in infectivity shift the curve to the left, while decreases in infectivity shift it to the right.
FIG. 4.

(A) The curve describing the relationship between the logarithm of the peak viral load and target cell depletion at the nadir has a universal S shape and moves left or right only with the change in log(δ/β). The constant δ/β is assumed to be characteristic of the virus type. Points higher on the same curve have higher reproductive ratios at the peak RP. (B) The effect of lowering the virus peak depends on the basic reproductive ratio and the peak viral load (i.e., on the value of δ/β and the peak viral load, which are likely characteristic of a particular virus). For example, reducing the peak viral load by 0.5 log10 copies/ml has a negligible impact on CD4+ T-cell depletion in the shaded area to the right, because depletion remains near 100%. However, for an infection with a lower R0 and a lower peak viral load, the same reduction in the peak viral load reduces CD4+ T-cell depletion significantly (shaded area to left).
Reducing the reproductive ratio at the peak for a given virus decreases the peak viral load. The reduction of the peak by, say, 0.5 log unit will have little effect on target cell depletion for very high viral loads (when RP is high, there is a plateau at nearly 100% depletion [Fig. 4]), but the same reduction can significantly reduce CD4+ T-cell depletion and change the prognosis for lower viral loads (Fig. 4B).
The relationship between the peak viral load and the CD4+ T-cell nadir in experimental infection.
The analysis above provided a theoretical relationship between the peak viral load and the CD4+ T-cell nadir. We then investigated whether this relationship was observed in experimental models of HIV infection. As an illustration, we present the SHIV-89.6P data (27) for the peak of virus and the depletion at the CD4+ T-cell minimum. The advantage of considering a CXCR4-tropic virus is that it infects “all” CD4+ T cells, so that the target pool in peripheral blood is well defined and can be measured. In addition, for the total number of CD4+ T cells, it is safe to assume that the net replacement rate, λ, from the external source (thymus) and, consequently, the loss rate, dT, are low (i.e., that less than 1% of the normal uninfected CD4+ T-cell number is replaced from the thymus per day).
In Fig. 5A we show the measured values of depletion of CD4+ T cells at the nadir, defined as (1 − Tmin/T0) × 100%, against the peak viral load for the control and vaccinated animals. The theoretical relationship between the virus peak (equation 10) and the CD4+ nadir (Tmin in equation 11) for each value of the reproductive ratio RP depends on δ/β. We have shown in our earlier work that δ and β do not vary much between individual monkeys, whether vaccinated or not. Briefly, the death rate of infected cells is the main determinant of the slope of the viral load after the peak, and it does not differ between controls and vaccinees (Fig. 1A and D) (4, 6). Also, we have estimated the infectivity of SHIV-89.6P in rhesus macaques on the same data set and found that there is no significant difference between vaccinated and unvaccinated animals (7). As explained in Fig. 4A, the shape of the theoretical sigmoid curve, D(VP), cannot be changed by fitting, since it is parameterized only by RP. It is universal for all diseases that are well described by the standard model, with low replacement and loss rates of target cells. However, by shifting the curve along the x axis, we can determine the best-fit value of the factor δ/β that minimizes the sum-of-squares deviation of experimental data from the theoretical curve (Fig. 4A). Since the quantities described by both axes have errors, the squared deviation of each point is defined as the sum of squares of the deviations in x and y directions from the closest point on the theoretical curve (a form of type 2 regression). We found that the best-fit value of log(δ/β) is 7.53 (δ/β = 3.39 × 107 copies/ml) with a CI of 7.45 to 7.65 found by bootstrapping. The correlation coefficient (r2) was 0.985.
FIG. 5.

Fitting theoretical curves to experimental data. Best-fit curves for the relationship between CD4+ T-cell depletion at the nadir and the peak of virus (from equation 10 and equation 11) for SHIV-89.6P data (7, 27) (A) and for SIVmac251 data (B) (17, 18, 32). The solid lines are the best-fit curves obtained by optimization of the value of δ/β.
In our previous work (7), we estimated infectivity from the relationship between the peak viral load and the number of CD4+ T cells 1 week after the peak, using the experimental viral-load timeline and the first two equations of the reduced model, equation 7 and equation 8. The procedure required advance knowledge of the death rate of infected cells, δ, which we estimated from the maximum decay rate of the viral load after the peak as follows: δ = 0.84/day. This led to the best-fit average infectivity, β, of 4.4 × 10−8 ml copy−1 day−1 (95% CI, between 3.4 × 10−8 and 5.6 × 10−8 ml copy−1 day−1). Our present estimate of δ/β as 3.39 × 107 copies/ml with a δ of 0.84/day would give a lower best-fit infectivity of 2.48 × 10−8 ml copy−1 day−1 for the same death rate of infected cells. However, since the death rate of infected cells used in the previous work was the minimum estimate of the actual death rate, it is possible that both δ and our best-fit infectivity are higher.
Analyzing infection by CCR5-tropic viruses is complicated because the number of target cells (CCR5+ CD4+ T cells) is more difficult to measure. However, in the gastrointestinal tract, most CD4+ T cells express CCR5 coreceptor, so most gut CD4+ T cells are targets for CCR5-tropic viruses. In recent studies, Mattapallil et al. (17, 18) measured CD4+ depletion in the jejunum in 20 rhesus macaques infected with CCR5-tropic SIVmac251, 6 of which were previously vaccinated.
Nine animals were euthanized before the viral load reached the first peak, so they could not be analyzed. Figure 5B contains data points from the remaining 11 animals (7 control animals and 4 vaccinated animals). The longitudinal data for each animal contain only three results of gut biopsies with percentages of CD4+ T-cells out of all CD3+ lymphocytes, and the lowest percentage was always the last point. We considered this point to be close to the target cell nadir.
We assumed slow turnover of memory CD4+ T cells prior to infection, consistent with the lack of reconstitution of these cells during antiretroviral therapy (19). The decay rate of SIVmac251-infected cells has been independently estimated as a δ of 1.49/day (2). By means of the same procedure using equation 7, equation 8, and the experimental viral load, we estimated the infectivity as a β of 1.45 × 10−7 ml copy−1 day−1, so that δ/β was 1.02 × 107 (32).
The line corresponds to the value of log(δ/β), which is equal to 7.20 (δ/β = 1.58 × 107 copies/ml), which minimizes the sum-of-squares deviation of experimental data from the theoretical curve [the CI for log(δ/β) is 7.09 to 7.59; the correlation coefficient, r2, is 0.874). The scatter of the experimental points around the theoretical curve is much larger than for SHIV infection in Fig. 5A. One reason for the larger scatter is the infrequent sampling in the longitudinal data (each animal had only one biopsy before challenge and one postchallenge and a necropsy), which probably caused imprecision in the estimates of the minimum. One would need in principle several samples in the 2- to 3-week period after the peak in order to correctly estimate the nadir. Another reason for the scatter could be that the depletion was calculated from the change in the fraction of CD4+ T cells (out of all CD3+ cells) instead of the CD4+ T-cell count used in the model. The two ways of calculating depletion would be equivalent only if the number of CD3+ lymphocytes stayed constant during the infection. The increased scatter of experimental data compared to SHIV infection can also be observed in the curve fit in reference 32.
Vaccination reduces the reproductive ratio at the peak of the virus.
The fitting of the model to experimental data suggests that the theoretical curves are a good predictor of the relationship between the viral load and CD4+ T-cell depletion in SHIV/SIV infection. Thus, we went on to use this approach to estimate the impact of vaccination on the reproductive ratio at peak. We first applied this approach to the experimental data on SHIV89.6P infection. Figure 5A shows that vaccinated animals have on average significantly lower peak viral loads (vaccinated logVP = 7.19 [95% CI, between 6.92 and 7.47]; unvaccinated logVP = 8.03 [95% CI, 7.78 and 8.28]; Mann-Whitney, P = 0.0004) and lower CD4+ T-cell depletion at minimum (vaccinated Dmin = 81.7% [95% CI, 71.4 and 92.1%]; unvaccinated Dmin = 97.4% [95% CI, 95.2 and 99.7%]; Mann-Whitney, P = 0.0003), so we expect the average reproductive ratio at the peak to be lower in vaccinated animals. The results are summarized in Fig. 6.
FIG. 6.
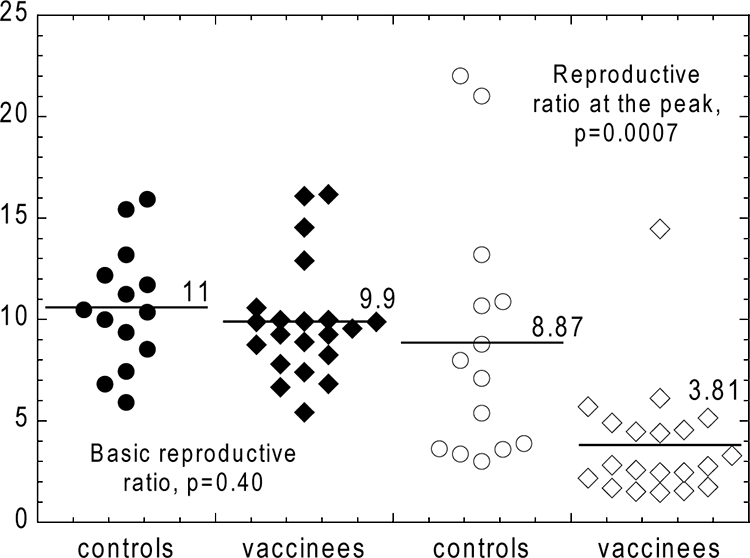
Measuring the impact of vaccination. The values of the basic reproductive ratio (R0) and the reproductive ratio at peak (RP) were estimated for SHIV-89.6P infection. Distributions of values of the basic reproductive ratio, R0, obtained from the initial exponential growth rate with a fixed intracellular delay of 1 day show no difference between control (solid circles) and vaccinated (solid diamonds) animals. The values of the peak reproductive ratio, RP, (estimated from the virus peak and the target cell nadir) are reduced approximately twofold in vaccinated animals (open diamonds) compared to controls (open circles).
The change in the reproductive ratio from R0 to RP is assumed to be the result of the change in the virus-dependent parameters due to the onset of cellular immunity a few days before the peak of the viral load, i.e., before significant depletion of target cells (5). The relationship between the virus peak and the CD4+ T-cell nadir in SHIV-89.6P is consistent with slow replacement of uninfected cells, so that in this case, the depletion at nadir can be considered to be completely determined by the peak reproductive ratio, RP, of the virus (equation 11). From the range of the nadir CD4+ T-cell depletions and peak viral loads, we can determine the range of the reproductive ratios at the peak of SHIV-89.6P in the infected animals in the Shiver et al. study (18). Therefore, we estimate the range of RP from the range of peak viral loads and the line of best fit in Fig. 5A, assuming δ/β = 107.53. The obtained range of RP is between 1.5 and 19.2. The peak reproductive ratio, RP, for vaccinated animals is 3.81, with a 95% CI between 2.3 and 4.6. For unvaccinated animals, RP is 8.87 with a 95% CI between 4.8 and 11.0. Vaccinated animals have on average a significantly lower peak reproductive ratio (Mann-Whitney, P = 0.0007).
Since the 21 animals in the Shiver study were vaccinated with seven different types of vaccines, all we can conclude is that, in general, the effect of vaccination is to reduce the peak reproductive ratio of the virus. The amount by which RP is reduced with respect to the average in control animals would reflect the effectiveness of a particular vaccine.
We obtained similar reduction in the reproductive ratio at the peak for SIVmac251 in the jejunum tissues of the macaques in the Mattapallil et al. study (17, 18). The vaccinated animals had an RP value of 1.8, and control animals had an RP value of 3.6; however, the difference did not reach significance (Mann-Whitney, P = 0.073) due to the small sample size (11 animals).
DISCUSSION
Vaccination trials have been used to compare the effectiveness of different vaccines in a number of monkey species, and using a wide variety of vaccine modalities. The efficacies of different vaccines can then be ranked by comparison of their abilities to preserve CD4+ T-cell numbers or reduce peak viral loads. Such comparisons can be misleading at times, since in viruses with a higher R0, it is much more difficult to prevent CD4 T-cell depletion than in viruses with low R0. Thus, the ideal metric for ranking vaccines would allow comparison of efficacies both within one infection model and across multiple infection models. Moreover, such a metric should also allow consideration of whether a given vaccine may be more effective in more virulent infections (with high R0) or less virulent infections, in order to predict which will perform best in HIV.
Previous approaches to comparing viral virulence have measured the basic reproductive ratios (R0) of different infection models. The basic reproductive ratio contains all model parameters in the initial exponential growth phase of the virus. While R0 may be useful in comparing different animal models of infection, it is not useful in quantifying the impact of vaccination. In particular, it does not reflect the effects of the cellular immune responses (or, consequently, the effects of vaccination) in primary infection, because in current vaccines these effects become evident only at the end of the early expansion period (4, 5).
In order to overcome this limitation, we used a modeling approach to develop a new method of estimating both the virulence of infection and the impact of vaccination, which we term the reproductive ratio at peak. We first showed that the relationship between the virus peak and the target cell nadir in the acute phase of the CXCR4-tropic SHIV infection and CCR5-tropic SIV infection is consistent with the standard model of virus dynamics. This relationship allowed us to estimate the decrease in reproductive capacity of the virus caused by vaccination. We have defined the reproductive ratio at the peak as the same function of the standard model parameters as in the basic reproductive ratio but evaluated in the period between the viral peak and the CD4+ T-cell nadir. In this period, CTL response is the main factor responsible for the change of parameters. We found that in both SHIV and SIV infections, vaccination on average reduced the reproductive ratio at the peak between 2 and 2.5 times. This was not sufficient to clear the virus. In order to suppress the infection during the first few weeks, vaccines should decrease the reproductive ratio below unity, i.e., approximately eightfold for SHIV and fourfold for SIV. In the absence of CTL responses, the basic and the peak reproductive ratios would probably be the same (this is in principle testable in macaques with anti-CD8 treatment). However, our method cannot determine specifically which parameters have changed as a result of the CTL response, only that the compound quantity RP has changed considerably as a result of vaccination.
The disadvantage of using the reproductive ratio at the peak as a measure of immune pressure is that the method requires knowledge of the target cell numbers, which is straightforward to measure in blood for CXCR4-tropic viruses but may require sampling of mucosal sites for CCR5-tropic viruses, like HIV (9). In the present study, we have also focused on CD4+ T cells as the major targets for HIV infection; however, macrophages and other cells may also play a minor role (26, 30). With this limitation, the method would be applicable to a variety of viral infections.
The basic reproductive ratio at peak permits comparisons of vaccine efficacies between studies performed in different animal models of infection. In addition, it also has the inherent threshold of unity in its definition. The idea of reducing the basic reproductive ratio of the virus below unity by inducing specific immune response has been present in the context of HIV vaccination for a long time (11). However, we believe that we present the first method that allows evaluation of the impact of vaccination using such a concept, in this case, the “reproductive ratio at the peak.”
Acknowledgments
This work was supported by the James S. McDonnell Foundation 21st Century Research Award/Studying Complex Systems and the Australian National Health and Medical Research Council. M.P.D. is a Sylvia and Charles Viertel Senior Medical Research Fellow. R.M.R. was supported by grant P20-RR18754 from the National Institutes of Health.
APPENDIX
Derivation of equation 10 for virus peak.
Let us assume that at the start of infection, t = 0, we have an initial target cell number T0 and an initial viral load V0, with an initial number of infected cells, I0, equal to 0. If the peak viral load, VP, occurs at time tP and the target and infected cell numbers at tP are TP and IP, respectively, then
 |
(A1) |
leads to
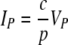 |
(A2) |
If we assume that V and I peak at the same time, which is true when c is ≫δI, then from
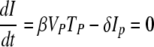 |
(A3) |
it follows that
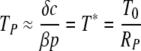 |
(A4) |
where T* is the steady-state target cell number in the full standard model, equation 1 to equation 3. The lag between the peak of infected cells and the viral load becomes negligible in the limit δ ≪ c. This is justified in the case of HIV/SIV/SHIV infection, where the parameter estimates are as follows: δ ≈ 1.0/day and c ≈ 20/day (16, 23).
Integrating
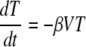 |
(A5) |
and equations A1 and A3 from t = 0 to t = tP and using equations A2 and A4, we get the following expression for the virus peak:
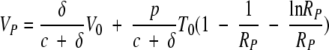 |
(A6) |
Since δ is ≪c and the initial virus concentration in blood, V0, is usually much lower than T0, the first term on the right-hand side of equation A6 can be neglected compared to the second term. The peak viral load then depends only on the constant parameters of the system (Fig. 7A). Since δ is ≪c, from equation A6 we obtain the approximate expression for the virus peak:
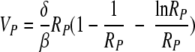 |
(A7) |
Derivation of equation 11 for the target cell minimum. In the infinite time limit, all three derivatives, equation 7 to equation 9, must vanish. This means that the steady-state values of both V and I are zero, while the steady-state value of target cells, Tmin, is undetermined.
FIG. A1.

The virus peak (A) and target cell nadir (B) do not depend on the amount of virus in the inoculum (i.e., on the initial conditions) unless it is greater than the peak. Increasing the initial viral load only causes the peak/nadir to appear earlier.
We can find Tmin by integration. We substitute the expression for the viral load from equation A5,
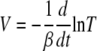 |
(A8) |
into the second term on the right-hand side of equation A1 and integrate over time from zero to infinity. Taking into account that the infection is always cleared, i.e., that the viral load vanishes in the long time limit, we obtain the following result:
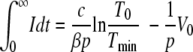 |
(A9) |
Next, we integrate both sides of equation A3 and substitute the expression A9 for the integral of infected cells. We note that the integral on the left-hand side of equation A3 vanishes at both limits (at t = 0 and at t = ∞), leading to
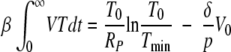 |
(A10) |
After substituting equation A10 into equation A5 and integrating from zero to infinity, we finally obtain the equation for Tmin/T0,
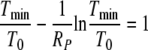 |
(A11) |
In deriving equation A11, we have again neglected the initial viral load in the inoculum (V0 ≈ 0).
Footnotes
Published ahead of print on 17 September 2008.
REFERENCES
- 1.Anonymous. 2007. HIV vaccine failure prompts Merck to halt trial. Nature 449390. [DOI] [PubMed] [Google Scholar]
- 2.Brandin, E., R. Thorstensson, S. Bonhoeffer, and J. Albert. 2006. Rapid viral decay in simian immunodeficiency virus-infected macaques receiving quadruple antiretroviral therapy. J. Virol. 809861-9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, H. Y., M. Di Mascio, A. S. Perelson, D. D. Ho, and L. Zhang. 2007. Determination of virus burst size in vivo using a single-cycle SIV in rhesus macaques. Proc. Natl. Acad. Sci. USA 10419079-19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Davenport, M. P., R. M. Ribeiro, and A. S. Perelson. 2004. Kinetics of virus-specific CD8+ T cells and the control of human immunodeficiency virus infection. J. Virol. 7810096-10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davenport, M. P., R. M. Ribeiro, L. Zhang, D. P. Wilson, and A. S. Perelson. 2007. Understanding the mechanisms and limitations of immune control of HIV. Immunol. Rev. 216164-175. [DOI] [PubMed] [Google Scholar]
- 6.Davenport, M. P., L. Zhang, A. Bagchi, A. Fridman, T. M. Fu, W. Schleif, J. W. Shiver, R. M. Ribeiro, and A. S. Perelson. 2005. High-potency human immunodeficiency virus vaccination leads to delayed and reduced CD8+ T-cell expansion but improved virus control. J. Virol. 7910059-10062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davenport, M. P., L. Zhang, J. W. Shiver, D. R. Casmiro, R. M. Ribeiro, and A. S. Perelson. 2006. Influence of peak viral load on the extent of CD4+ T-cell depletion in simian HIV infection. J. Acquir. Immune Defic. Syndr. 41259-265. [DOI] [PubMed] [Google Scholar]
- 8.Feinberg, M. B., and J. P. Moore. 2002. AIDS vaccine models: challenging challenge viruses. Nat. Med. 8207-210. [DOI] [PubMed] [Google Scholar]
- 9.Guadalupe, M., E. Reay, S. Sankaran, T. Prindiville, J. Flamm, A. McNeil, and S. Dandekar. 2003. Severe CD4+ T-cell depletion in gut lymphoid tissue during primary human immunodeficiency virus type 1 infection and substantial delay in restoration following highly active antiretroviral therapy. J. Virol. 7711708-11717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herz, A. V., S. Bonhoeffer, R. M. Anderson, R. M. May, and M. A. Nowak. 1996. Viral dynamics in vivo: limitations on estimates of intracellular delay and virus decay. Proc. Natl. Acad. Sci. USA 937247-7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ho, D. D., and Y. X. Huang. 2002. The HIV-1 vaccine race. Cell 110135-138. [DOI] [PubMed] [Google Scholar]
- 12.Ho, D. D., A. U. Neumann, A. S. Perelson, W. Chen, J. M. Leonard, and M. Markowitz. 1995. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373123-126. [DOI] [PubMed] [Google Scholar]
- 13.Hockett, R. D., J. M. Kilby, C. A. Derdeyn, M. S. Saag, M. Sillers, K. Squires, S. Chiz, M. A. Nowak, G. M. Shaw, and R. P. Bucy. 1999. Constant mean viral copy number per infected cell in tissues regardless of high, low, or undetectable plasma HIV RNA. J. Exp. Med. 1891545-1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Little, S. J., A. R. McLean, C. A. Spina, D. D. Richman, and D. V. Havlir. 1999. Viral dynamics of acute HIV-1 infection. J. Exp. Med. 190841-850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lloyd, A. L. 2001. The dependence of viral parameter estimates on the assumed viral life cycle: limitations of studies of viral load data. Proc. Biol. Sci. 268847-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Markowitz, M., M. Louie, A. Hurley, E. Sun, M. Di Mascio, A. S. Perelson, and D. D. Ho. 2003. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-cell decay in vivo. J. Virol. 775037-5038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattapallil, J. J., D. C. Douek, A. Buckler-White, D. Montefiori, N. L. Letvin, G. J. Nabel, and M. Roederer. 2006. Vaccination preserves CD4 memory T cells during acute simian immunodeficiency virus challenge. J. Exp. Med. 2031533-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattapallil, J. J., D. C. Douek, B. Hill, Y. Nishimura, M. Martin, and M. Roederer. 2005. Massive infection and loss of memory CD4+ T cells in multiple tissues during acute SIV infection. Nature 4341093-1097. [DOI] [PubMed] [Google Scholar]
- 19.Mehandru, S., M. A. Poles, K. Tenner-Racz, P. Jean-Pierre, V. Manuelli, P. Lopez, A. Shet, A. Low, H. Mohri, D. Boden, P. Racz, and M. Markowitz. 2006. Lack of mucosal immune reconstitution during prolonged treatment of acute and early HIV-1 infection. PLoS Med. 3e484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nowak, M. A., A. L. Lloyd, G. M. Vasquez, T. A. Wiltrout, L. M. Wahl, N. Bischofberger, J. Williams, A. Kinter, A. S. Fauci, V. M. Hirsch, and J. D. Lifson. 1997. Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 717518-7525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nowak, M. A., and R. M. May. 2000. Virus dynamics: Mathematical principles of immunology and virology. Oxford University Press, Oxford, United Kingdom.
- 22.Perelson, A. S., A. U. Neumann, M. Markowitz, J. M. Leonard, and D. D. Ho. 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science 2711582-1586. [DOI] [PubMed] [Google Scholar]
- 23.Ramratnam, B., S. Bonhoeffer, J. Binley, A. Hurley, L. Zhang, J. E. Mittler, M. Markowitz, J. P. Moore, A. S. Perelson, and D. D. Ho. 1999. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet 3541782-1785. [DOI] [PubMed] [Google Scholar]
- 24.Reynolds, M. R., E. Rakasz, P. J. Skinner, C. White, K. Abel, Z. M. Ma, L. Compton, G. Napoe, N. Wilson, C. J. Miller, A. Haase, and D. I. Watkins. 2005. CD8+ T-lymphocyte response to major immunodominant epitopes after vaginal exposure to simian immunodeficiency virus: too late and too little. J. Virol. 799228-9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ribeiro, R. M., N. M. Dixit, and A. S. Perelson. 2006. Modeling the in vivo growth rate of HIV: implications for vaccination, p. 231-246. In R. Paton and L. A. McNamara (ed.), Multidisciplinary approaches to theory in medicine. Elsevier, Amsterdam, The Netherlands.
- 26.Schacker, T., S. Little, E. Connick, K. Gebhard, Z. Q. Zhang, J. Krieger, J. Pryor, D. Havlir, J. K. Wong, R. T. Schooley, D. Richman, L. Corey, and A. T. Haase. 2001. Productive infection of T cells in lymphoid tissues during primary and early human immunodeficiency virus infection. J. Infect. Dis. 183555-562. [DOI] [PubMed] [Google Scholar]
- 27.Shiver, J. W., T. M. Fu, L. Chen, D. R. Casimiro, M. E. Davies, R. K. Evans, Z. Q. Zhang, A. J. Simon, W. L. Trigona, S. A. Dubey, L. Huang, V. A. Harris, R. S. Long, X. Liang, L. Handt, W. A. Schleif, L. Zhu, D. C. Freed, N. V. Persaud, L. Guan, K. S. Punt, A. Tang, M. Chen, K. A. Wilson, K. B. Collins, G. J. Heidecker, V. R. Fernandez, H. C. Perry, J. G. Joyce, K. M. Grimm, J. C. Cook, P. M. Keller, D. S. Kresock, H. Mach, R. D. Troutman, L. A. Isopi, D. M. Williams, Z. Xu, K. E. Bohannon, D. B. Volkin, D. C. Montefiori, A. Miura, G. R. Krivulka, M. A. Lifton, M. J. Kuroda, J. E. Schmitz, N. L. Letvin, M. J. Caulfield, A. J. Bett, R. Youil, D. C. Kaslow, and E. A. Emini. 2002. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature 415331-335. [DOI] [PubMed] [Google Scholar]
- 28.Stafford, M. A., L. Corey, Y. Cao, E. S. Daar, D. D. Ho, and A. S. Perelson. 2000. Modeling plasma virus concentration during primary HIV infection. J. Theor. Biol. 203285-301. [DOI] [PubMed] [Google Scholar]
- 29.Staprans, S. I., P. J. Dailey, A. Rosenthal, C. Horton, R. M. Grant, N. Lerche, and M. B. Feinberg. 1999. Simian immunodeficiency virus disease course is predicted by the extent of virus replication during primary infection. J. Virol. 734829-4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.van der Ende, M. E., M. Schutten, B. Raschdorff, G. Grossschupff, P. Racz, A. Osterhaus, and K. Tenner-Racz. 1999. CD4 T cells remain the major source of HIV-1 during end stage disease. AIDS 131015-1019. [DOI] [PubMed] [Google Scholar]
- 31.Wei, X., S. K. Ghosh, M. E. Taylor, V. A. Johnson, E. A. Emini, P. Deutsch, J. D. Lifson, S. Bonhoeffer, M. A. Nowak, B. H. Hahn, M. S. Saag, and G. M. Shaw. 1995. Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373117-122. [DOI] [PubMed] [Google Scholar]
- 32.Wilson, D. P., J. J. Mattapallil, M. D. Lay, L. Zhang, M. Roederer, and M. P. Davenport. 2007. Estimating the infectivity of CCR5-tropic simian immunodeficiency virus SIVmac251 in the gut. J. Virol. 818025-8029. [DOI] [PMC free article] [PubMed] [Google Scholar]